Paeds · nephrology-urology-fluids-and-electrolytes
Congenital anomalies of the kidney and urinary tract
Also known as CAKUT · Congenital kidney and urinary tract anomalies · Multicystic dysplastic kidney · Renal dysplasia · Posterior urethral valves · Horseshoe kidney · Renal agenesis
Fellowship guide to congenital anomalies of the kidney and urinary tract, the CAKUT spectrum that accounts for the largest single cause of paediatric end-stage kidney disease. Covers the embryological basis in the ureteric bud and metanephric mesenchyme interaction, the classification into kidney parenchymal anomalies including multicystic dysplastic kidney and renal agenesis, ureteric anomalies, and bladder and urethral anomalies including posterior urethral valves, the antenatal and postnatal investigation pathway using ultrasound, MCUG, DMSA, and MAG3, the management from prophylactic antibiotics to surgical correction and nephrology surveillance, and the long-term risk of chronic kidney disease that every child with CAKUT carries.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
Overview & Definition
A newborn with a palpable abdominal mass, an antenatal finding of hydronephrosis, or a male infant with a poor urinary stream has the hallmark presentation of a congenital anomaly of the kidney and urinary tract. The group of structural malformations known by the acronym CAKUT spans the entire urinary tract from the renal parenchyma to the urethra, and it is the single largest cause of end-stage kidney disease in children worldwide. The common thread is a developmental error in kidney or urinary tract formation during embryogenesis, which manifests as malformation, obstruction, or dysplasia. [1]
The spectrum ranges from mild anomalies that cause no symptom and are found incidentally, to severe bilateral malformations that are fatal in the newborn period from pulmonary hypoplasia and renal failure. At one end is the unilateral multicystic dysplastic kidney that involutes over years and needs only surveillance, and at the other is bilateral renal agenesis, the Potter sequence, in which the absence of amniotic fluid prevents lung development and the newborn dies of respiratory failure within hours. Between these extremes lie the common and testable conditions: posterior urethral valves, renal hypoplasia, vesicoureteral reflux, and duplex collecting systems. [4]
The clinical importance of CAKUT lies in its cumulative burden. Individual anomalies may seem minor in isolation, but the risk of urinary tract infection, hypertension, and progressive renal scarring accumulates over childhood. Harambat and colleagues showed that CAKUT accounts for the largest proportion of paediatric chronic kidney disease in registry data, which is why every child with an antenatal renal finding needs structured postnatal evaluation and life-long nephrology surveillance. [10]
Classification
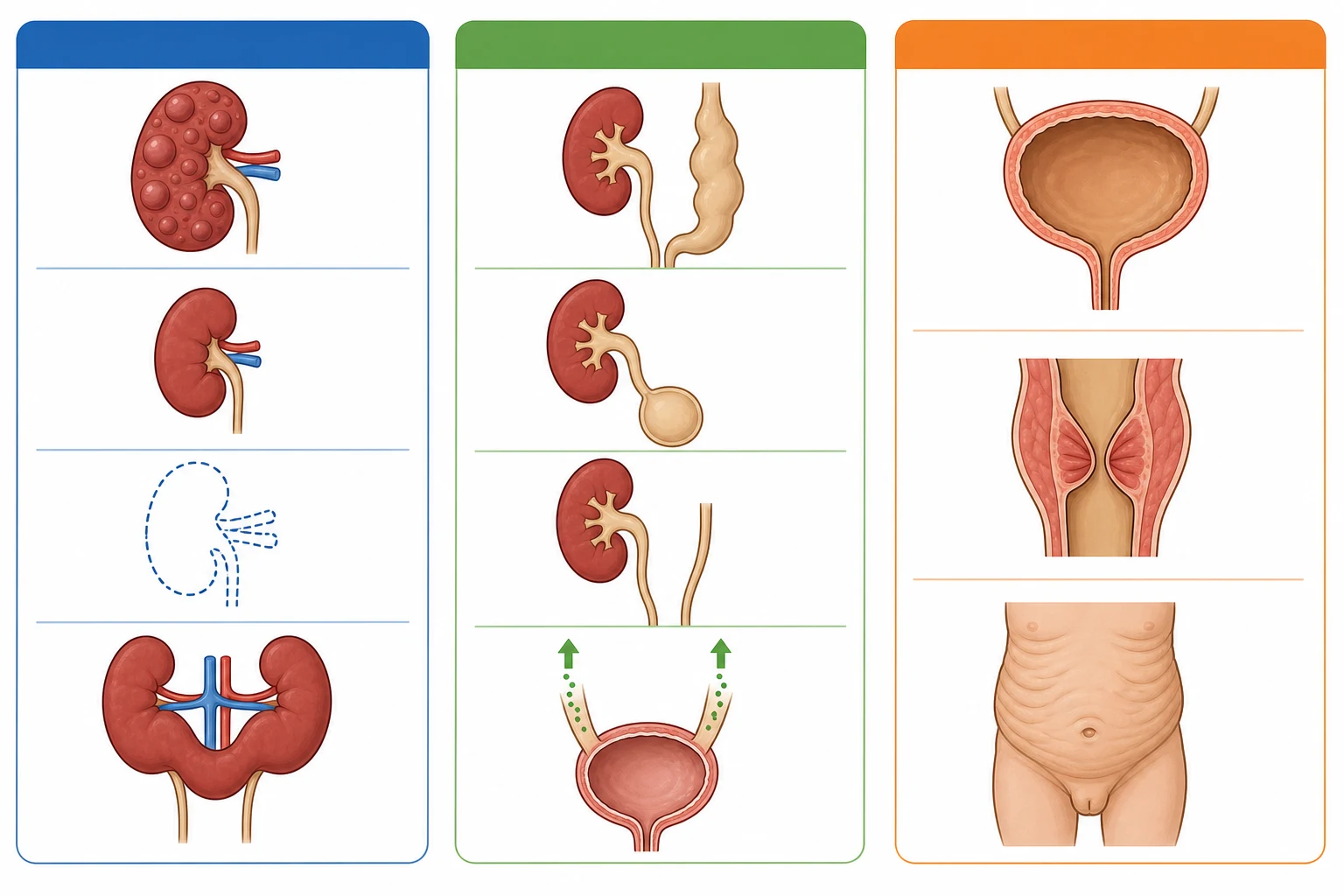
The classification follows the anatomical level of the malformation, because each level points to a different embryological mechanism, a different clinical presentation, and a different management pathway. Rodriguez organised the spectrum into three groups, and the system holds in clinical practice because it mirrors the investigation pathway from the renal ultrasound to the urethra. [4]
The first group is the kidney and parenchymal anomalies. Renal agenesis is the complete absence of one or both kidneys from failure of the ureteric bud to form. Renal hypoplasia is a kidney that is small but structurally intact, with a reduced nephron number. Renal dysplasia is a kidney with disorganized development, containing cartilage, cysts, and primitive tubules, and it is the pathological hallmark of the malformed kidney. The multicystic dysplastic kidney is a severe, non-functional dysplastic kidney replaced by cysts of varying size that do not connect, giving the classic grape-cluster appearance. Horseshoe kidney is the fusion of the lower poles across the midline, which lies lower in the abdomen because its ascent is arrested by the inferior mesenteric artery. [3]
The second group is the ureteric and collecting system anomalies. Vesicoureteral reflux is the retrograde flow of urine from the bladder into the ureter, caused by a short or absent intramural tunnel. Megaureter is a dilated ureter from obstruction, reflux, or a non-obstructive, non-refluxing cause. A ureterocele is a cystic dilatation of the intramural ureter within the bladder, which may obstruct the ipsilateral ureter or the bladder neck. A duplex collecting system has two pelvicalyceal systems draining one kidney, often with two ureters, and the upper-pole ureter typically inserts ectopically and distally, following the Weigert-Meyer rule. [3]
The third group is the bladder and urethral anomalies. Posterior urethral valves are membrane-like folds in the male posterior urethra that obstruct urinary outflow, the most common cause of lower urinary tract obstruction in male infants. Prune belly syndrome, the triad of absent abdominal musculature, cryptorchidism, and a dilated urinary tract, is a rare but distinctive syndrome. Neurogenic bladder from spinal cord lesions such as myelomeningocele causes a functional obstruction and storage failure. [9]
Parenchymal
- Renal agenesis, hypoplasia, dysplasia
- Multicystic dysplastic kidney (grape-cluster cysts)
- Horseshoe kidney (fused lower poles)
- Embryology: ureteric bud and metanephric mesenchyme interaction
Ureteric
- Vesicoureteral reflux (short intramural tunnel)
- Megaureter (obstructive, refluxing, or non-refluxing)
- Ureterocele (cystic intramural dilatation)
- Duplex system (Weigert-Meyer rule)
Bladder and urethral
- Posterior urethral valves (most common male obstruction)
- Prune belly syndrome (absent abdominal wall)
- Neurogenic bladder (myelomeningocele)
- High back-pressure and renal dysplasia risk
Epidemiology & Risk Factors
CAKUT is the most common group of congenital malformations detected by antenatal ultrasound, with a birth prevalence of 3 to 6 per 1000 live births. The widespread use of routine mid-trimester anomaly scanning means that the majority of cases are now identified before birth, which has shifted the clinical encounter from the acute presentation of a sick neonate to the planned postnatal evaluation of a well-appeasing newborn. Lee and colleagues showed in a meta-analysis that antenatal hydronephrosis predicts a postnatal CAKUT in a significant proportion of cases, with the risk proportional to the degree of dilation. [7]
The most important risk factor for a child with CAKUT is the degree of bilateral involvement, because the total functioning nephron mass determines the lifetime trajectory. Bilateral renal agenesis is fatal, bilateral dysplasia leads to early CKD, and a solitary functioning kidney from unilateral agenesis or multicystic dysplastic kidney has a more subtle but real risk of hyperfiltration injury over decades. The second key risk factor is the presence of lower urinary tract obstruction, because the back-pressure from posterior urethral valves or a neurogenic bladder damages the developing kidneys in utero and produces the most severe renal dysplasia. [8]
Genetics contributes to the risk. Kolvenbach and Hildebrandt showed that monogenic causes are found in a substantial minority of CAKUT cases, and copy number variants and polygenic effects account for further cases. A family history of CAKUT, parental consanguinity, and syndromic features all increase the yield of genetic testing. Rao and colleagues demonstrated in a cohort of over 1000 Chinese children that pathogenic variants in known CAKUT genes were identified in a meaningful proportion, which supports the role of genetic testing in the workup of bilateral or syndromic disease. [2]
Aboriginal and Torres Strait Islander children and other Indigenous populations carry a higher background burden of CKD and renal dysplasia, which compounds the long-term risk. Maternal diabetes, teratogenic drug exposure, and oligohydramnios from any cause are additional risk factors that increase the likelihood and severity of CAKUT in the developing fetus. [2]
Pathophysiology
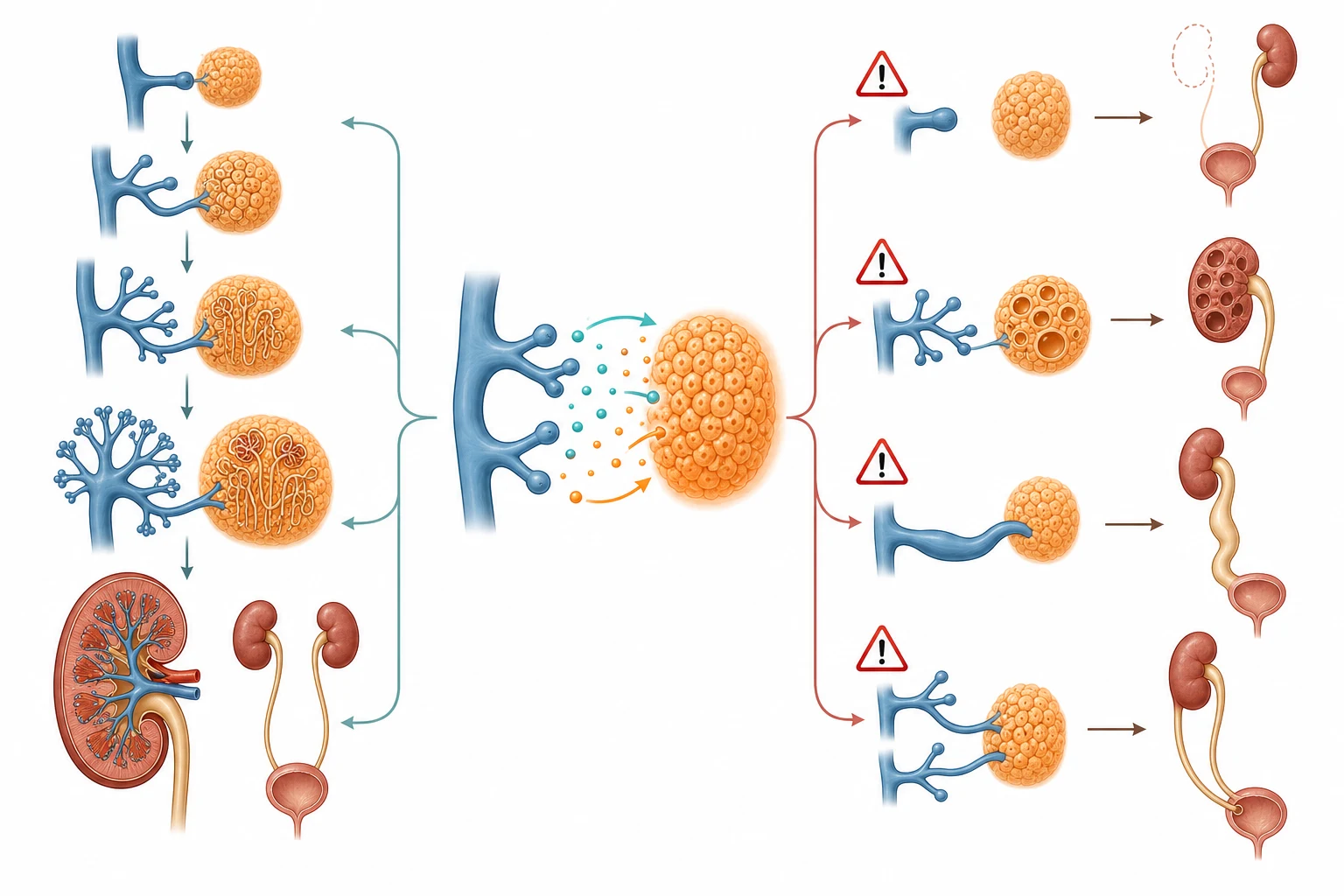
The kidney forms from the reciprocal interaction of two embryonic structures, the ureteric bud and the metanephric mesenchyme, and every form of CAKUT can be traced to a disruption of this interaction at a specific stage. The ureteric bud grows out from the Wolffian duct and invades the metanephric mesenchyme around the fifth week of gestation. The bud branches repeatedly to form the collecting system, the renal pelvis, the calyces, and the collecting ducts, while the mesenchyme differentiates into the nephron, the glomerulus, and the tubule. The two structures signal to each other through molecular pathways including GDNF and its receptor RET, BMP4, and sonic hedgehog, and a disruption in any of these produces a distinct malformation. [3]
Failure of the ureteric bud to form produces renal agenesis, because the mesenchyme does not receive the inductive signal and undergoes apoptosis. Failure of the bud to branch correctly produces renal dysplasia, in which the kidney contains disorganised tissue including cartilage and primitive ducts rather than functional nephrons. Brockwell and colleagues reviewed the molecular pathways in detail and showed that mutations in the GDNF-RET pathway and in transcription factors including PAX2, HNF1B, and EYA1 are among the best characterised genetic causes. [3]
Abnormal bud positioning produces ureteric anomalies. If the bud originates too cranially, the ureteric orifice is more cranial and lateral in the bladder, producing vesicoureteral reflux from a short intramural tunnel. If the bud originates too caudally, the ureteric orifice is ectopic and distal, producing an obstructing ureterocele or an ectopic ureter, and this is the basis of the Weigert-Meyer rule in duplex systems. The molecular basis of posterior urethral valves is less well understood, but the pathological consequence is clear: the valve tissue obstructs urinary outflow, the back-pressure dilates the bladder and the upper tracts, and the developing kidneys suffer dysplasia in utero. [2]
The downstream consequence of any CAKUT that reduces the functioning nephron mass is hyperfiltration injury in the remaining nephrons. The single-nephron GFR rises to compensate, which causes glomerular hypertension and sclerosis, and the process is self-perpetuating. Matsell and colleagues showed that children with a solitary functioning kidney from renal agenesis have different outcomes from those with a multicystic dysplastic kidney, because the dysplastic kidney may retain some anomalous tissue that produces renin and hypertension. This hyperfiltration-injury paradigm explains why even a unilateral anomaly carries a long-term risk of CKD. [5]
Clinical Presentation
The presentation of CAKUT has changed with the era of routine antenatal ultrasound. Most children are now identified before birth by a mid-trimester scan showing hydronephrosis, a renal cyst, oligohydramnios, or an absent or enlarged bladder, and the postnatal evaluation is planned rather than emergent. The minority who present after birth do so with a palpable abdominal mass in the newborn period, a febrile urinary tract infection in infancy, or, later, with hypertension, failure to thrive, or an incidental finding on imaging performed for another reason. [12]
Antenatal hydronephrosis is the gateway finding. Lee and colleagues showed in their meta-analysis that the risk of a postnatal CAKUT is proportional to the degree of antenatal pelvic dilation, with mild dilation resolving in most cases and severe dilation carrying a high risk of obstruction or reflux. The Society for Fetal Urology grading system, from grade 1 with isolated pelvic dilation to grade 5 with cortical thinning, guides the intensity of the postnatal evaluation. [7]
The male neonate with posterior urethral valves presents a distinctive picture. The bladder is palpable and distended, the abdominal wall may show visible distension or the prune-belly appearance, and the urinary stream is poor. The more severe cases present at birth with respiratory distress from pulmonary hypoplasia caused by the oligohydramnios in utero, which is the Potter sequence. The less severe cases present in infancy with recurrent urinary tract infection, poor growth, or a declining renal function. The clinical clue is always a male infant with a palpable bladder or a poor urinary stream. [9]
Modes of presentation for CAKUT across the age spectrum
Antenatal hydronephrosis on mid-trimester ultrasound, the most common mode
Neonatal palpable abdominal mass from a distended bladder or enlarged kidney
Respiratory distress in a male newborn from pulmonary hypoplasia and Potter sequence
Febrile urinary tract infection in infancy from reflux or obstruction
Hypertension, failure to thrive, or declining renal function in an older child
Incidental finding on imaging performed for an unrelated reason
The multicystic dysplastic kidney is most often detected antenatally as a multicystic renal mass and presents postnatally as a unilateral flank mass or an incidental ultrasound finding. Bilateral multicystic dysplastic kidney is rare and usually fatal from respiratory and renal failure. The horseshoe kidney is often asymptomatic and found incidentally, but it may present with a urinary tract infection from an associated ureteropelvic junction obstruction or with haematuria from trauma. [6]
Differential Diagnosis
The first distinction is between a CAKUT and an inherited cystic kidney disease, because the management and the genetic counselling differ. Autosomal recessive polycystic kidney disease presents in the newborn period with bilateral enlarged echogenic kidneys and may be confused with bilateral dysplasia, but the family history, the pattern of inheritance, and the liver fibrosis distinguish it. Autosomal dominant polycystic kidney disease is usually asymptomatic in childhood and shows bilateral renal cysts in a child with an affected parent. [4]
The second distinction is between a true obstruction and a transient dilation. Physiologic or transient hydronephrosis is common on antenatal ultrasound and resolves in the first months of life, but the clinician cannot assume this without a postnatal evaluation. The meta-analysis by Lee and colleagues showed that even mild antenatal hydronephrosis carries a risk of postnatal pathology, so the threshold for postnatal investigation is low. Ureteropelvic junction obstruction, from a congenital narrowing at the pelviureteric junction, is the most common cause of true obstruction and is distinguished from reflux by the MAG3 renogram. [7]
CAKUT
- Structural malformation from embryological error
- Most common cause of paediatric ESKD
- Examples: PUV, MCDK, VUR, duplex systems
- Antenatal hydronephrosis is the gateway finding
Inherited cystic disease
- ADPKD: bilateral cysts, autosomal dominant, affected parent
- ARPKD: bilateral enlarged echogenic kidneys, liver fibrosis
- Genetic testing confirms the diagnosis
- Distinct from dysplasia on histopathology
Transient dilation
- Physiologic hydronephrosis, resolves postnatally
- No structural malformation or obstruction
- Low-grade antenatal finding
- Postnatal ultrasound confirms resolution
The third distinction is between a CAKUT and a syndromic association. CAKUT occurs as part of many syndromes, including the branchio-oto-renal syndrome, the renal-coloboma syndrome from PAX2 mutations, the Townes-Brocks syndrome from SALL1 mutations, and the HNF1B-related maturity-onset diabetes of the young type 5 with renal cysts. A dysmorphic child with a renal anomaly warrants a genetics evaluation, because the renal finding may be the presenting feature of a multisystem syndrome. [2]
Clinical & Bedside Assessment
The bedside assessment of a child with suspected CAKUT answers three questions: is the child in immediate danger from renal failure or sepsis, what is the structural anomaly, and what is the renal function. The airway, breathing, and circulation are assessed first, because the neonate with severe bilateral CAKUT may present with respiratory distress from pulmonary hypoplasia or with shock from urosepsis. The abdomen is examined for a palpable mass, which may be a distended bladder from posterior urethral valves, a multicystic dysplastic kidney, or a hydronephrotic kidney. [12]
The blood pressure is measured in every child with a renal anomaly, because hypertension is both a complication of CAKUT and a sign of renal dysplasia. The growth parameters are plotted, because failure to thrive is a feature of chronic kidney disease from bilateral dysplasia or obstructive uropathy. The external genitalia are examined, because posterior urethral valves occur only in males and syndromic CAKUT may have genital anomalies. The spine is examined for the stigmata of occult spinal dysraphism, because a neurogenic bladder from a tethered cord is a treatable cause of upper tract damage. [1]
The history should include the antenatal findings, because the severity and the progression of antenatal hydronephrosis guide the urgency of the postnatal evaluation. A family history of CAKUT, kidney disease, hearing loss, or diabetes raises the possibility of a monogenic or syndromic cause. The maternal history of diabetes, drug exposure, or oligohydramnios provides additional context. A history of a febrile urinary tract infection in infancy is a red flag for vesicoureteral reflux or obstruction and warrants imaging. [2]
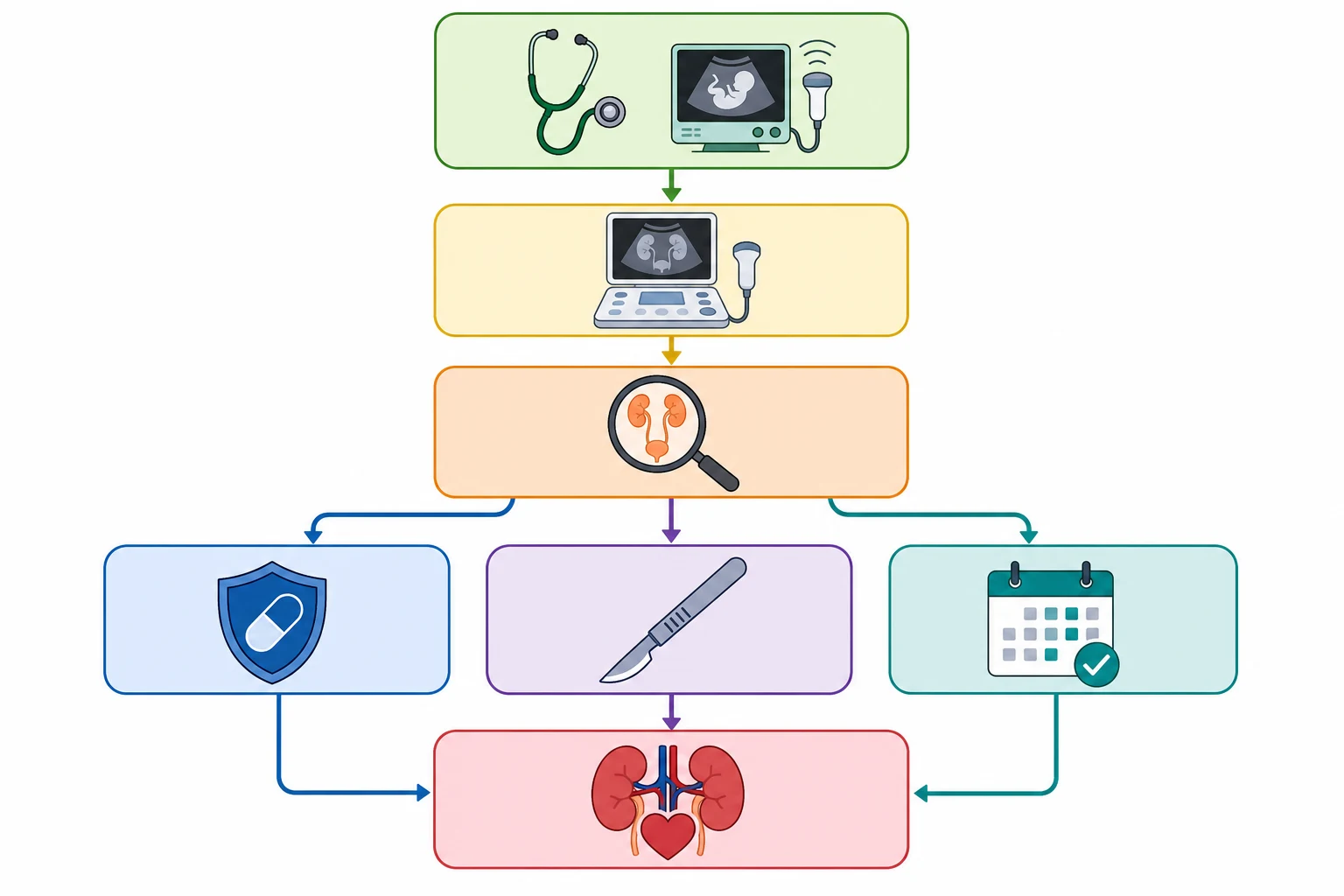
Investigations
The investigation pathway for CAKUT is sequential and image-led, with each test answering a specific question. The first test is always the postnatal renal ultrasound, which confirms the antenatal finding, characterises the anatomy, and grades the hydronephrosis. The timing matters, because the neonatal dehydration in the first 48 hours can mask hydronephrosis, so the ultrasound is repeated at 5 to 7 days of life unless the antenatal finding was severe enough to warrant immediate imaging. [12]
The micturating cystourethrogram, or MCUG, is the gold standard for detecting vesicoureteral reflux and posterior urethral valves. It involves catheterising the bladder, filling it with contrast, and imaging during voiding, and it shows the reflux grade from 1 to 5 and the dilated posterior urethra of valves. The MCUG is invasive and carries a risk of urinary tract infection, so it is reserved for children with a high pre-test probability of reflux or obstruction, and prophylactic antibiotics are given around the procedure. [12]
The DMSA scan, or dimercaptosuccinic acid scintigraphy, is a static cortical scan that maps the functioning renal cortex and detects scars, dysplasia, and the split renal function between the two kidneys. It is performed months after any acute infection to avoid confusion with acute pyelonephritis, and it is essential for the long-term surveillance of the child with reflux or obstruction. The MAG3 renogram, or mercaptoacetyltriglycine scan, is a dynamic scan that shows the uptake, drainage, and split function of each kidney, and it is the definitive test for ureteropelvic junction obstruction and for monitoring the drainage after surgical correction. [7]
Blood tests assess the renal function and the complications. The serum creatinine and the estimated GFR are the baseline markers, though the neonatal creatinine reflects the maternal creatinine for the first days of life and must be interpreted with care. The electrolytes, the acid-base balance, and the full blood count screen for CKD and infection. Urinalysis and urine culture are essential in any febrile child, because a urinary tract infection may be the first sign of a CAKUT. Genetic testing is indicated for bilateral or syndromic CAKUT, and the yield is highest when there is a family history or parental consanguinity. [11]
Management — Resuscitation
The resuscitation priorities in the neonate with CAKUT are to secure the airway in the child with pulmonary hypoplasia, to decompress any obstruction, and to prevent infection. The neonate with posterior urethral valves and respiratory distress from the Potter sequence needs immediate respiratory support, and the obstruction is decompressed with a urethral catheter as soon as the airway is stable. The catheter is the first therapeutic intervention in any obstructive uropathy, because the relief of the back-pressure improves the renal perfusion and the renal function. [9]
The neonate with bilateral renal dysplasia or severe obstructive uropathy may present with metabolic acidosis, hyperkalaemia, or fluid overload from renal failure. These biochemical disturbances are managed with the standard neonatal AKI protocols: correction of hyperkalaemia, management of acidosis, and fluid restriction if overloaded. The nephrology team is involved early for the child with significant renal impairment, and the renal replacement therapy pathway is established if the disturbance is severe. [9]
Prophylactic antibiotic for obstructive uropathy or high-grade reflux
Dose
Trimethoprim 2 mg per kg at night
Prophylactic antibiotics are started at diagnosis for any child with a high probability of reflux or obstruction, to prevent urinary tract infection and renal scarring while the definitive investigations are completed. The agent of first choice is trimethoprim at 2 mg per kg at night, or nitrofurantoin or cephalexin in the neonate. The antibiotics are continued until the MCUG confirms or excludes reflux and obstruction, and they are maintained long-term in the child with high-grade reflux until spontaneous resolution or surgical correction. [12]
Management — Definitive & Stepwise
[12]The definitive management is determined by the specific anomaly, the degree of renal function, and the age of the child. The overarching principles are to relieve any obstruction, to prevent infection, to preserve the renal function, and to monitor for the long-term complications of CKD. Each anomaly has a distinct management pathway, and the decisions are made in a multidisciplinary team of paediatric nephrologists, urologists, radiologists, and geneticists. [1]
Posterior urethral valves are managed by endoscopic valve ablation once the neonate is stable. The catheter is maintained until the ablation, and the upper tracts are reassessed by ultrasound afterward. The child with persistent dilation or a declining renal function after ablation may need a temporary urinary diversion such as a vesicostomy. The long-term management focuses on the bladder function, because the valve bladder is a thick-walled, low-compliance bladder that stores poorly and may cause upper tract damage despite the valve ablation. Clean intermittent catheterisation and anticholinergic medication are used for the poorly compliant bladder. [9]
The multicystic dysplastic kidney is managed conservatively in almost all cases. Aslam and Watson showed in their long-term cohort that the majority of unilateral multicystic dysplastic kidneys involute over years, and the risk of hypertension and malignancy is low. The nephrectomy is reserved for the rare case of a very large mass, persistent hypertension, or a concern for malignancy. The child undergoes regular ultrasound surveillance for involution, blood pressure monitoring, and an assessment of the contralateral kidney, because the contralateral kidney may have reflux or ureteropelvic junction obstruction. [6]
Oxybutynin for the poorly compliant valve bladder
Dose
0.2 to 0.4 mg per kg per dose, two to three times daily
The solitary functioning kidney, whether from renal agenesis or a multicystic dysplastic kidney, is managed with lifelong surveillance. Matsell and colleagues showed that the outcomes differ between agenesis and multicystic dysplastic kidney, and both groups need monitoring for hyperfiltration injury, hypertension, and proteinuria. The surveillance includes annual blood pressure, urinalysis for proteinuria, and a periodic serum creatinine, with an ACE inhibitor or an angiotensin receptor blocker for the child who develops hypertension or proteinuria. [5]
Vesicoureteral reflux is managed on the basis of the grade and the clinical context. Low-grade reflux resolves spontaneously in most children and is managed with prophylactic antibiotics and surveillance. High-grade reflux, recurrent febrile infection despite prophylaxis, or progressive renal scarring may warrant endoscopic correction or ureteric reimplantation. The horseshoe kidney needs no specific treatment unless there is an associated obstruction, but the child is counselled to seek medical attention for abdominal trauma, because the fused kidney is more susceptible to injury. [1]
Specific Subtypes & Scenarios
Posterior urethral valves deserve a dedicated discussion because they are the most severe and the most testable form of CAKUT. They occur only in males, with an incidence of approximately 1 in 4000 to 8000 live male births. The valves are membrane-like folds in the posterior urethra that obstruct the urinary outflow, and the severity of the obstruction determines the phenotype. The most severe cases present in the newborn period with the Potter sequence and pulmonary hypoplasia, and the mildest cases present in childhood with a poor urinary stream or recurrent infection. [9]
Robinson and colleagues reported the population-based long-term kidney outcomes in children with posterior urethral valves. They found that a substantial proportion of children progress to CKD over childhood and adolescence, and the early predictors of poor outcome include the nadir creatinine in the first year, the presence of bilateral renal dysplasia, and the bladder dysfunction after ablation. The long-term management requires a coordinated approach from the urology and the nephrology teams, with attention to the bladder function, the renal function, and the growth. [8]
The multicystic dysplastic kidney is the second most testable entity. The classic ultrasound appearance is a non-renaliform mass of cysts of varying size that do not connect, which distinguishes it from the hydronephrosis of ureteropelvic junction obstruction in which the cysts communicate with the renal pelvis. The natural history, as documented by Aslam and Watson, is one of progressive involution in the majority of cases over the first decade. The risk of hypertension and of Wilms tumour is low, which is why the nephrectomy is no longer routine. The child is monitored with regular ultrasound and blood pressure checks. [6]
FILMS for the differential of an antenatal cystic renal mass
Renal agenesis may be unilateral or bilateral. Unilateral renal agenesis is compatible with a normal life, though the solitary kidney carries a risk of hyperfiltration injury and hypertension. Bilateral renal agenesis is the Potter sequence, which is fatal from pulmonary hypoplasia and renal failure in the newborn period. The horseshoe kidney is found in approximately 1 in 400 individuals and is often an incidental finding, though it may present with infection, obstruction, or trauma. The duplex collecting system is common and may be associated with an ectopic ureter, a ureterocele, or reflux to the lower moiety. [3]
[1]Complications & Pitfalls
The complications of CAKUT are the direct consequences of the reduced nephron mass and the disordered urinary tract, and they unfold over years to decades. Chronic kidney disease is the most significant, and the trajectory is determined by the initial nephron endowment, the degree of ongoing injury from infection and obstruction, and the hyperfiltration injury in the remaining nephrons. Harambat and colleagues showed that CAKUT accounts for the largest single cause of paediatric CKD in registry data, and the progression may accelerate through childhood and adolescence. [10]
Hypertension is common in children with CAKUT and may arise from the dysplastic kidney, the hyperfiltration injury, or the volume overload from CKD. It is often asymptomatic and is detected only by routine blood pressure monitoring, which is why every child with CAKUT has the blood pressure measured at every visit. Recurrent urinary tract infection causes renal scarring, which compounds the existing dysplasia and accelerates the CKD progression, and it is the rationale for the prophylactic antibiotics and the prompt treatment of any confirmed infection. [5]
The classic pitfalls are diagnostic and prognostic. Failing to investigate an antenatal hydronephrosis, on the assumption that it will resolve, delays the diagnosis of an obstruction or reflux and allows preventable renal scarring. Passing a urethral catheter in a male neonate with a palpable bladder, rather than assuming it is a normal variation, prevents the renal damage from undiagnosed posterior urethral valves. Underestimating the long-term CKD risk in a child with a unilateral anomaly, on the assumption that a single kidney is always normal, misses the window for the nephrology surveillance that detects the early hyperfiltration injury. [5]
Prognosis & Disposition
The prognosis depends on the specific anomaly, the bilaterality, the renal function at presentation, and the trajectory. The child with a unilateral multicystic dysplastic kidney and a normal contralateral kidney has an excellent prognosis, with the majority of dysplastic kidneys involuting and the child living a normal life with a single kidney. The child with bilateral renal dysplasia or severe posterior urethral valves has a guarded prognosis, with a substantial risk of progressive CKD and the eventual need for renal replacement therapy. [6]
The long-term data for posterior urethral valves are the most concerning. Robinson and colleagues showed in their population-based cohort that a significant proportion of boys with valves progress to CKD or end-stage kidney disease over childhood and adolescence, and the rate of progression is influenced by the early renal function, the bladder management, and the ongoing infection prevention. Caione and Nappo emphasised that the valve ablation is the beginning rather than the end of the management, and the long-term surveillance for the bladder dysfunction and the CKD is essential. [8]
Disposition is to a paediatric nephrology and urology service for every child with a confirmed CAKUT, with the intensity of follow-up tailored to the anomaly and the renal function. The child with a resolved transient hydronephrosis may be discharged, but the child with a unilateral dysplastic kidney, a solitary functioning kidney, or posterior urethral valves needs life-long surveillance. The transition to adult nephrology care is planned in adolescence, because the CKD risk persists into adulthood and the young adult with CAKUT needs ongoing blood pressure monitoring, renal function surveillance, and counselling on the reproductive and the genetic implications. [1]
Special Populations
The fetus with an antenatal diagnosis of CAKUT is the fastest-growing special population, because the near-universal antenatal ultrasound has shifted the encounter to before birth. The antenatal team involves the maternal-fetal medicine specialist, the paediatric nephrologist, and the paediatric urologist in the counselling of the family, and the severity of the finding guides the plan for the place and the mode of delivery. The most severe cases, such as bilateral renal agenesis, may be managed with palliative care, while the cases with a reversible obstruction, such as posterior urethral valves, may be considered for fetal intervention in selected centres. [12]
Children with syndromic CAKUT need a coordinated genetics and multidisciplinary evaluation. The branchio-oto-renal syndrome, the renal-coloboma syndrome from PAX2, the HNF1B-related cystic kidney disease, and the Townes-Brocks syndrome from SALL1 are among the best characterised, and each has a distinct renal phenotype and extrarenal manifestation. The genetic testing is guided by the clinical picture and the family history, and the results inform the counselling of the family and the surveillance of the siblings. [2]
Aboriginal and Torres Strait Islander children and children from lower-resource settings present a different challenge. The higher background burden of CKD and the reduced access to antenatal ultrasound and specialist services compound the risk, and the late presentation with an advanced CKD is more common. The emphasis in these settings is on the early detection through the routine antenatal ultrasound, the prompt postnatal evaluation of any finding, and the culturally appropriate counselling and surveillance. [10]
The young adult transitioning from paediatric to adult care is the final special population. The CAKUT does not resolve, and the CKD risk persists and may accelerate in adulthood. The transition plan addresses the blood pressure monitoring, the renal function surveillance, the reproductive and the genetic counselling, and the coordination between the paediatric and the adult nephrology services. [1]
Evidence, Guidelines & Regional Differences
The evidence base for CAKUT spans the embryology, the genetics, the epidemiology, and the long-term outcomes. Murugapoopathy and Gupta provided the modern primer, organising the CAKUT spectrum and the clinical approach. Kolvenbach and Hildebrandt reviewed the genetics and the pathogenesis, showing that monogenic causes account for a substantial minority and that the GDNF-RET pathway and the transcription factors PAX2, HNF1B, and EYA1 are the best characterised. Brockwell and colleagues provided the comprehensive pathophysiology review, linking the molecular mechanisms to the clinical phenotypes. [3]
The long-term outcome data are the most practice-changing. The population-based cohort study by Robinson and colleagues showed that a significant proportion of boys with posterior urethral valves progress to CKD, which has intensified the surveillance and the bladder management. The study by Matsell and colleagues showed that the solitary functioning kidney from agenesis has a different outcome from the multicystic dysplastic kidney, which has refined the surveillance strategy. The meta-analysis by Lee and colleagues on antenatal hydronephrosis established the risk stratification that guides the postnatal evaluation. [8]
Murugapoopathy 2020
- CJASN primer on the CAKUT spectrum and the clinical approach
- The modern organising framework for the topic
Kolvenbach 2023
- Nature Reviews Nephrology on the genetics and pathogenesis
- Monogenic causes, GDNF-RET pathway, transcription factors
Lee 2006
- Pediatrics meta-analysis on antenatal hydronephrosis
- Risk stratification for the postnatal evaluation
Robinson 2024
- JASN population-based cohort on long-term PUV outcomes
- Substantial CKD progression in childhood and adolescence
The regional differences centre on the access to antenatal ultrasound and specialist care. In the high-income settings, the near-universal antenatal detection and the standardised postnatal pathway allow the early intervention and the long-term surveillance. In the lower-resource settings, the later presentation with an advanced CKD or a severe infection is more common, and the emphasis shifts to the prevention and the prompt treatment of the reversible causes. The international guidelines on the investigation of antenatal hydronephrosis, including the consensus from the Society for Fetal Urology, are applicable across the settings, with the intensity of the evaluation tailored to the local resources. [12]
Exam Pearls
ABC for the postnatal evaluation of an antenatal renal finding
References
- [1]Murugapoopathy V, Gupta IR A Primer on Congenital Anomalies of the Kidneys and Urinary Tracts (CAKUT). Clin J Am Soc Nephrol, 2020.PMID 32188635
- [2]Kolvenbach CM, Shril S, Hildebrandt F The genetics and pathogenesis of CAKUT. Nat Rev Nephrol, 2023.PMID 37524861
- [3]Brockwell M, Hergenrother S, Satariano M, Patel V Pathophysiology of Congenital Anomalies of the Kidney and Urinary Tract: A Comprehensive Review. Cells, 2024.PMID 39594614
- [4]Rodriguez MM Congenital Anomalies of the Kidney and the Urinary Tract (CAKUT). Fetal Pediatr Pathol, 2014.PMID 25313840
- [5]Matsell DG, Bao C, Po White T, Yu W, Parveen S, Acal L, Tasyurek SE Outcomes of solitary functioning kidneys-renal agenesis is different than multicystic dysplastic kidney disease. Pediatr Nephrol, 2021.PMID 33954810
- [6]Aslam M, Watson AR, Trent and Anglia MCDK Study Group Unilateral multicystic dysplastic kidney: long term outcomes. Arch Dis Child, 2006.PMID 16754654
- [7]Lee RS, Cendron M, Kinnamon DD, Nguyen HT Antenatal hydronephrosis as a predictor of postnatal outcome: a meta-analysis. Pediatrics, 2006.PMID 16882811
- [8]Robinson CH, Rickard M, Jeyakumar N, de Carvalho M, Lorenzo AJ, Farhat WA, et al Long-Term Kidney Outcomes in Children with Posterior Urethral Valves: A Population-Based Cohort Study. J Am Soc Nephrol, 2024.PMID 39167453
- [9]Caione P, Nappo SG Posterior urethral valves: long-term outcome. Pediatr Surg Int, 2011.PMID 21748651
- [10]Harambat J, van Stralen KJ, Kim JJ, Tizard EJ Epidemiology of chronic kidney disease in children. Pediatr Nephrol, 2012.PMID 21713524
- [11]Rao J, Liu X, Mao J, Tang H, Li Y, Shen Q, et al Genetic spectrum of renal disease for 1001 Chinese children based on a multicenter registration system. Clin Genet, 2019.PMID 31328266
- [12]Liu DB, Armstrong WR 3rd, Maizels M Hydronephrosis: prenatal and postnatal evaluation and management. Clin Perinatol, 2014.PMID 25155734