Paeds · nephrology-urology-fluids-and-electrolytes
Haemolytic uraemic syndrome
Also known as HUS · STEC-HUS · Typical haemolytic uraemic syndrome · Atypical haemolytic uraemic syndrome · aHUS
Fellowship guide to paediatric haemolytic uraemic syndrome: the defining triad of microangiopathic haemolytic anaemia, thrombocytopenia, and acute kidney injury; the distinction between STEC-HUS (approximately 90 percent of cases, following Shiga toxin-producing E. coli) and atypical HUS from complement dysregulation; the critical point that supportive care is the mainstay for STEC-HUS while eculizumab is first-line for aHUS; and the need for long-term renal follow-up in all survivors.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
Overview & Definition
A previously well toddler who had bloody diarrhoea a week ago and now arrives pale, listless, and passing very little urine has haemolytic uraemic syndrome. The name describes the three things going wrong at once: red blood cells are being destroyed inside narrowed blood vessels (haemolytic anaemia), platelets are being consumed as clots form in those vessels (thrombocytopenia), and the vessels that are most affected are in the kidneys, where the clots block the filtering surface and shut down urine production (uraemia). This triad, caused by widespread thrombotic microangiopathy in small vessels, defines the syndrome regardless of the trigger. [1]
The trigger is usually an intestinal infection. About 90 percent of paediatric HUS follows gastroenteritis caused by Shiga toxin-producing Escherichia coli, most commonly the O157:H7 strain, and this form is called typical or STEC-HUS. A much smaller but far more dangerous group, accounting for 5 to 10 percent of cases, has no diarrhoeal prodrome and is driven by uncontrolled activation of the body's complement immune pathway. This is atypical HUS, and the distinction between the two is the single most consequential decision in the disease because atypical HUS requires urgent treatment with eculizumab, a complement-blocking drug, while typical STEC-HUS is managed with supportive care alone. [3]
Classification
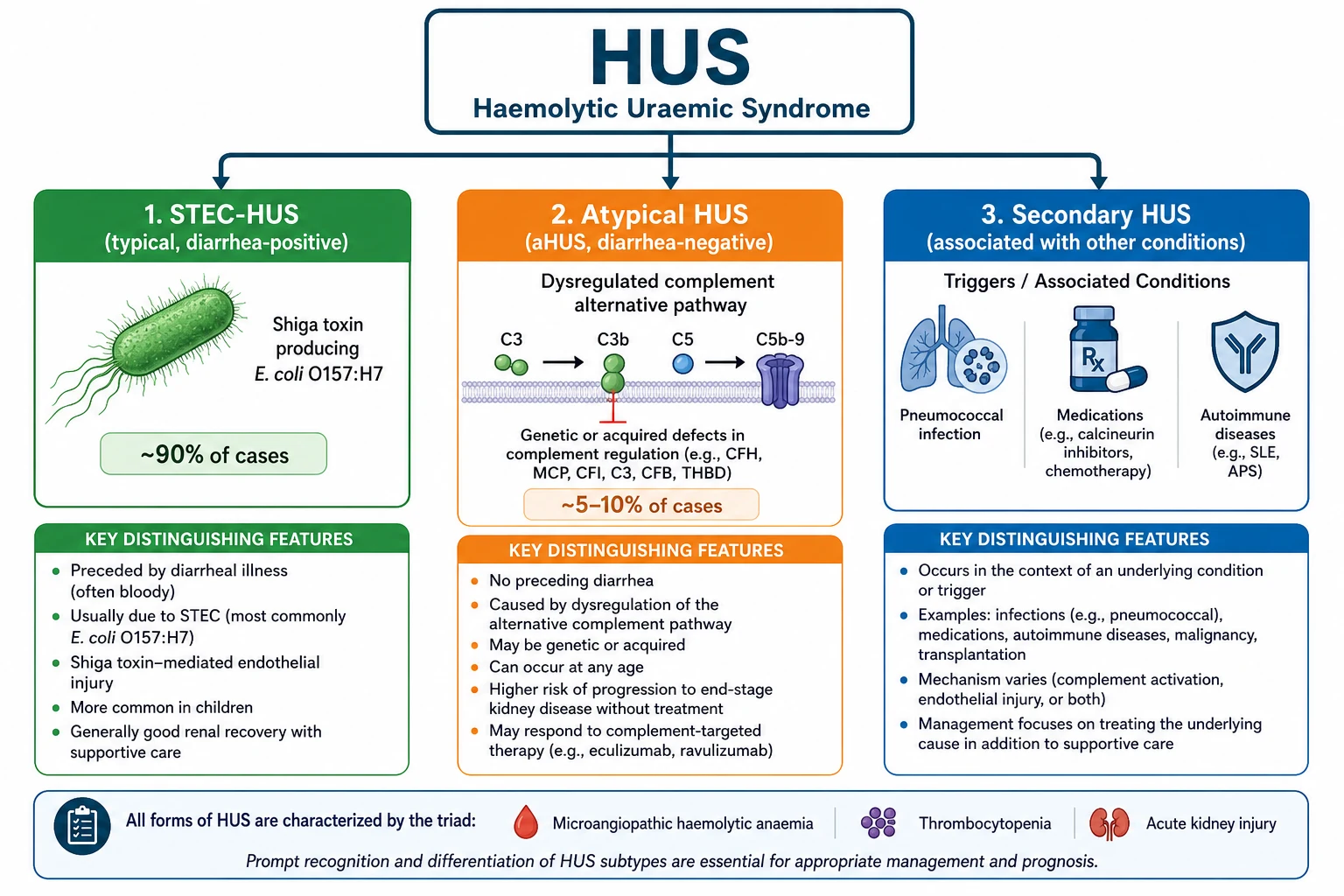
Classification serves one practical purpose: it separates the child who needs supportive care from the child who needs urgent complement blockade. The diagnostic label of HUS is confirmed by the triad, and the aetiological label determines the treatment. The first split is between diarrhoea-associated (typical) and diarrhoea-negative (atypical or secondary). A child with a clear history of bloody diarrhoea five to ten days before the onset of HUS almost certainly has STEC-HUS, which accounts for approximately 90 percent of all paediatric cases. A child with no diarrhoeal prodrome, a relapsing or familial pattern, or onset in the neonatal period raises the question of atypical HUS. [1]
[3]Secondary HUS is the third category, triggered by identifiable non-complement causes. Pneumococcal HUS follows invasive Streptococcus pneumoniae infection, where the bacterial enzyme neuraminidase exposes the Thomsen-Friedenreich antigen on red cells, triggering autoimmune haemolysis. Drug-induced HUS is associated with calcineurin inhibitors, quinine, and chemotherapeutic agents. Autoimmune conditions such as systemic lupus erythematosus and antiphospholipid syndrome, malignancy, and post-transplantation contexts can all produce secondary thrombotic microangiopathy. The practical point is that secondary HUS is managed by treating the underlying cause, not by eculizumab, though overlap exists. [7]
Epidemiology & Risk Factors
Haemolytic uraemic syndrome is the most common cause of community-acquired acute kidney injury in young children, with an incidence of approximately 1 to 2 per 100,000 children per year. The peak age is under five years, and the disease is slightly more common in summer when enteric infections peak. Approximately 5 to 15 percent of children who acquire STEC O157:H7 infection go on to develop HUS, making this organism one of the most virulent enteric pathogens encountered in paediatric practice. [1]
Several factors increase the likelihood that a STEC infection will progress to HUS. Young age, especially under five years, is the strongest host factor. The use of antimotility agents such as loperamide prolongs gut carriage of the organism and may increase toxin absorption. Antibiotic therapy during STEC infection has been associated in some studies with an increased risk of HUS, possibly because certain antibiotics induce bacteriophage lysis and release of Shiga toxin, though this remains debated. A high peripheral white cell count at presentation is a marker of more severe inflammation and a predictor of progression. The 2011 German outbreak caused by E. coli O104:H4 demonstrated that adult patients and those with certain strains can have much higher rates of neurological complications and severe disease than typical childhood O157:H7 infections. [11]
Atypical HUS is rare, with an estimated incidence of 2 per million population per year. It is caused by pathogenic variants in genes regulating the alternative complement pathway, including complement factor H (the most common, found in 20 to 30 percent of aHUS cases), factor I, membrane cofactor protein (CD46), factor B, and C3. Anti-factor H autoantibodies account for approximately 5 to 10 percent of aHUS, especially in children. Without treatment, aHUS carries a mortality or end-stage kidney disease rate of 50 to 70 percent, and even with plasma-based therapy the long-term renal prognosis was poor before the advent of eculizumab. [2]
Pathophysiology
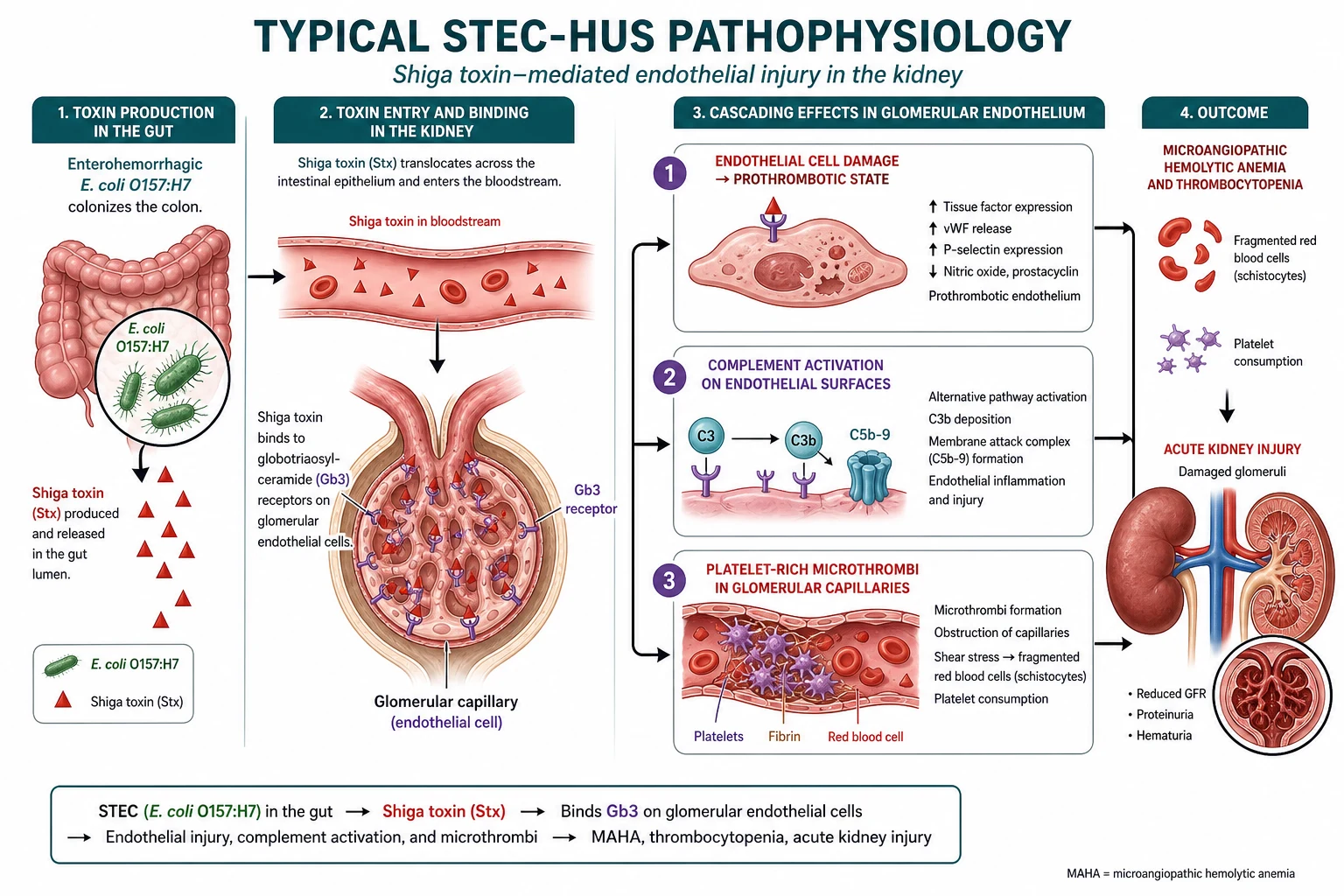
The mechanism of STEC-HUS is a chain reaction that begins in the gut and ends in the glomerulus. Shiga toxin-producing E. coli colonise the intestine and release Shiga toxin, which crosses the damaged gut epithelium and enters the systemic circulation. The toxin has a high affinity for globotriaosylceramide (Gb3) receptors, which are densely expressed on glomerular endothelial cells, tubular epithelial cells, and mesangial cells. When Shiga toxin binds to these cells it triggers direct cytotoxic injury, upregulates tissue factor expression, and activates complement on the endothelial surface. [4]
Once the glomerular endothelium is injured, a prothrombotic cascade unfolds. Exposed subendothelium and tissue factor activate platelets, which adhere and aggregate at the sites of injury. Fibrin deposits form, creating microthrombi in the glomerular capillaries and arterioles. These microthrombi have two direct consequences: they narrow the capillary lumen, reducing the filtration surface area and causing acute kidney injury, and they mechanically shear red blood cells as they try to squeeze past, producing the fragmented schistocytes that are the morphological hallmark of microangiopathic haemolytic anaemia. Platelets are consumed in the process, causing thrombocytopenia. The triad is complete. [1]
GB3
Complement activation plays a role in both STEC-HUS and aHUS, but with a critical difference. In STEC-HUS, Shiga toxin activates complement on the endothelial surface as a secondary phenomenon, contributing to the thrombotic cascade but not being the primary driver. In aHUS, complement dysregulation is the primary and sole mechanism. Loss-of-function variants in complement regulators such as factor H and factor I, or gain-of-function variants in complement activators such as factor B and C3, remove the brakes on the alternative pathway. Uncontrolled complement activation (C3 convertase) proceeds to the terminal pathway (C5 to C9, the membrane attack complex), which damages endothelial cells and triggers the same thrombotic microangiopathy. This is why blocking C5 with eculizumab is so effective in aHUS: it cuts the terminal pathway while leaving the upstream C3 opsonisation intact. [2]
In Australia and New Zealand, the public health response to STEC O157:H7 is robust, with mandatory notification and food safety surveillance. Access to eculizumab for aHUS is available through specialist paediatric nephrology centres, though it requires funding approval given its high cost. The IPNA (International Pediatric Nephrology Association) consensus recommendations align with the 2016 international consensus for aHUS management. [3]
Clinical Presentation
The classic story of STEC-HUS is a child under five years who has had diarrhoea, often bloody, for several days, followed approximately one week later by a rapid onset of pallor, lethargy, and reduced urine output. The parents may describe the urine as dark or tea-coloured, and may have noticed bruising or petechiae. The key temporal feature is the gap: the diarrhoea may be resolving when the haematological and renal features emerge, and it is this separation that distinguishes HUS from simple gastroenteritis. [1]
The tempo can vary. Some children develop the full triad over 24 to 48 hours, while others evolve over a week. Pallor reflects the haemolytic anaemia, which can be severe with haemoglobin falling below 60 g per litre. Jaundice from unconjugated bilirubin may be visible. Petechiae, bruising, and mucosal bleeding reflect thrombocytopenia, though major bleeding is uncommon because the thrombocytopenia is usually moderate (platelets 30 to 100 times 10 to the 9 per litre). Oliguria or anuria signals the acute kidney injury, and fluid retention produces oedema, hypertension, and sometimes respiratory distress from pulmonary oedema. [5]
Neurological involvement occurs in 20 to 40 percent of children with STEC-HUS and is the strongest predictor of a poor outcome. The spectrum ranges from irritability and drowsiness to seizures, coma, and stroke. The mechanism is multifactorial: electrolyte disturbances (hyponatraemia, hypocalcaemia), hypertension-related encephalopathy, microvascular thrombosis in the central nervous system, and direct effects of Shiga toxin on neuronal tissue. A child with HUS who develops seizures or altered consciousness needs urgent neurological assessment and imaging, and this is the group in which eculizumab is most often considered for STEC-HUS despite the lack of definitive trial evidence. [8]
Atypical HUS presents differently. There is no diarrhoeal prodrome, the onset may be insidious, and the disease tends to be relapsing or progressive. A family history of HUS, early onset (including neonatal), or recurrence after apparent recovery should raise suspicion for aHUS. These children need urgent complement testing and genetic analysis, and eculizumab should be started as soon as the diagnosis is suspected without waiting for the results. [3]
Differential Diagnosis
The first task when a child presents with haemolytic anaemia, thrombocytopenia, and renal failure is to confirm that this is HUS and not another thrombotic microangiopathy. The most important alternative is thrombotic thrombocytopenic purpura (TTP), which produces the same microangiopathic haemolytic anaemia and thrombocytopenia but is caused by severe deficiency of ADAMTS13, the enzyme that cleaves ultra-large von Willebrand factor multimers. TTP presents with predominantly neurological rather than renal involvement, no diarrhoeal prodrome, and an ADAMTS13 activity below 10 percent. The distinction is urgent because TTP requires immediate plasma exchange, which is not routinely indicated for STEC-HUS. [6]
[6]Disseminated intravascular coagulation (DIC) is the other critical exclusion. DIC also produces schistocytes and thrombocytopenia, but unlike HUS it deranges the coagulation cascade: the prothrombin time and activated partial thromboplastin time are prolonged, fibrinogen is low, and D-dimer is markedly elevated. In HUS the coagulation studies are normal because the thrombosis is localised to the microvasculature and does not consume clotting factors systemically. This single difference (normal coagulation in HUS, abnormal in DIC) is a high-yield discriminator that examiners test repeatedly. [1]
Other conditions to consider include malignant hypertension, which can cause thrombotic microangiopathy with very high blood pressure and hypertensive retinopathy, and which requires aggressive blood pressure control rather than complement blockade. Autoimmune haemolytic anaemia (Evans syndrome) has a positive direct antiglobulin test, unlike the Coombs-negative haemolysis of HUS. Systemic lupus erythematosus and antiphospholipid syndrome should be considered in older children and adolescents presenting with thrombotic microangiopathy. Sepsis, particularly with organisms causing disseminated infection, can mimic HUS. [7]
Clinical & Bedside Assessment
Assessment runs in parallel with resuscitation and targets three questions: how severe is the organ dysfunction, what is the likely cause, and what complications are present. The history establishes the timeline and the prodrome: ask about the onset, character (bloody or watery), and duration of diarrhoea, the interval between diarrhoea and the current presentation, any antibiotic or antimotility agent use, and the urine output. A careful dietary and exposure history may identify the source of STEC, such as undercooked ground beef, unpasteurised milk, or contact with farm animals. [1]
Examination focuses on the triad and its consequences. Pallor and jaundice assess the severity of haemolysis. Skin examination looks for petechiae, purpura, and bruising. Abdominal examination may reveal tenderness from ongoing colitis or, rarely, an appendicitis-like presentation. Cardiovascular examination checks for signs of volume overload: raised jugular venous pressure, gallop rhythm, crackles on auscultation, and peripheral oedema. Blood pressure measurement is mandatory because hypertension is common, driven by renin-mediated vasoconstriction from renal ischaemia and by volume overload, and can be severe enough to cause encephalopathy. [5]
Neurological examination is essential in every child with HUS. Look for altered consciousness, irritability, focal deficits, and signs of raised intracranial pressure. Seizures, even if self-limited, are a marker of severe disease and warrant urgent imaging. A child who is drowsy or has had a seizure in the context of HUS needs to be in a high-dependency or intensive care setting. Fluid balance assessment is one of the most error-prone elements: the child may be intravascularly depleted from gastroenteritis while simultaneously volume-overloaded from oliguric renal failure, and the net status determines whether fluids are needed or restricted. [1]
Investigations
The diagnosis rests on demonstrating the triad with laboratory evidence and confirming or excluding the cause. Blood tests confirm microangiopathic haemolytic anaemia: low haemoglobin, elevated lactate dehydrogenase, undetectable or markedly reduced haptoglobin, elevated unconjugated bilirubin, and schistocytes on the peripheral blood film. Schistocytes (fragmented red cells, helmet cells) are the morphological signature, typically present at at least 1 percent of red cells. The direct antiglobulin (Coombs) test is negative, which is essential for distinguishing HUS from autoimmune haemolysis. [1]
Coagulation studies are normal in HUS, and this is a critical discriminator. The prothrombin time, activated partial thromboplastin time, fibrinogen, and D-dimer should be normal or only mildly abnormal. If the coagulation profile is significantly deranged, think of DIC rather than HUS. Renal function tests show elevated creatinine and urea, and the severity varies widely from mild acute kidney injury to anuric renal failure requiring dialysis. Urinalysis shows haematuria and proteinuria. Electrolyte disturbances are common: hyperkalaemia, metabolic acidosis, hyponatraemia, hypocalcaemia, and hyperphosphataemia. [5]
Stool should be cultured for E. coli O157:H7 on sorbitol-MacConkey agar, and Shiga toxin should be detected by PCR or enzyme immunoassay. However, stool studies are frequently negative by the time HUS develops because the organism may have been cleared, so a negative stool culture does not exclude STEC-HUS. For atypical HUS, complement studies are essential: C3 (low in 30 to 50 percent of aHUS but may be normal), C4 (usually normal), factor H, factor I, and anti-factor H antibodies. Genetic testing for complement gene variants is performed in specialist centres but should not delay treatment. ADAMTS13 activity should be measured to exclude TTP if the presentation is atypical. [3]
Management — Resuscitation
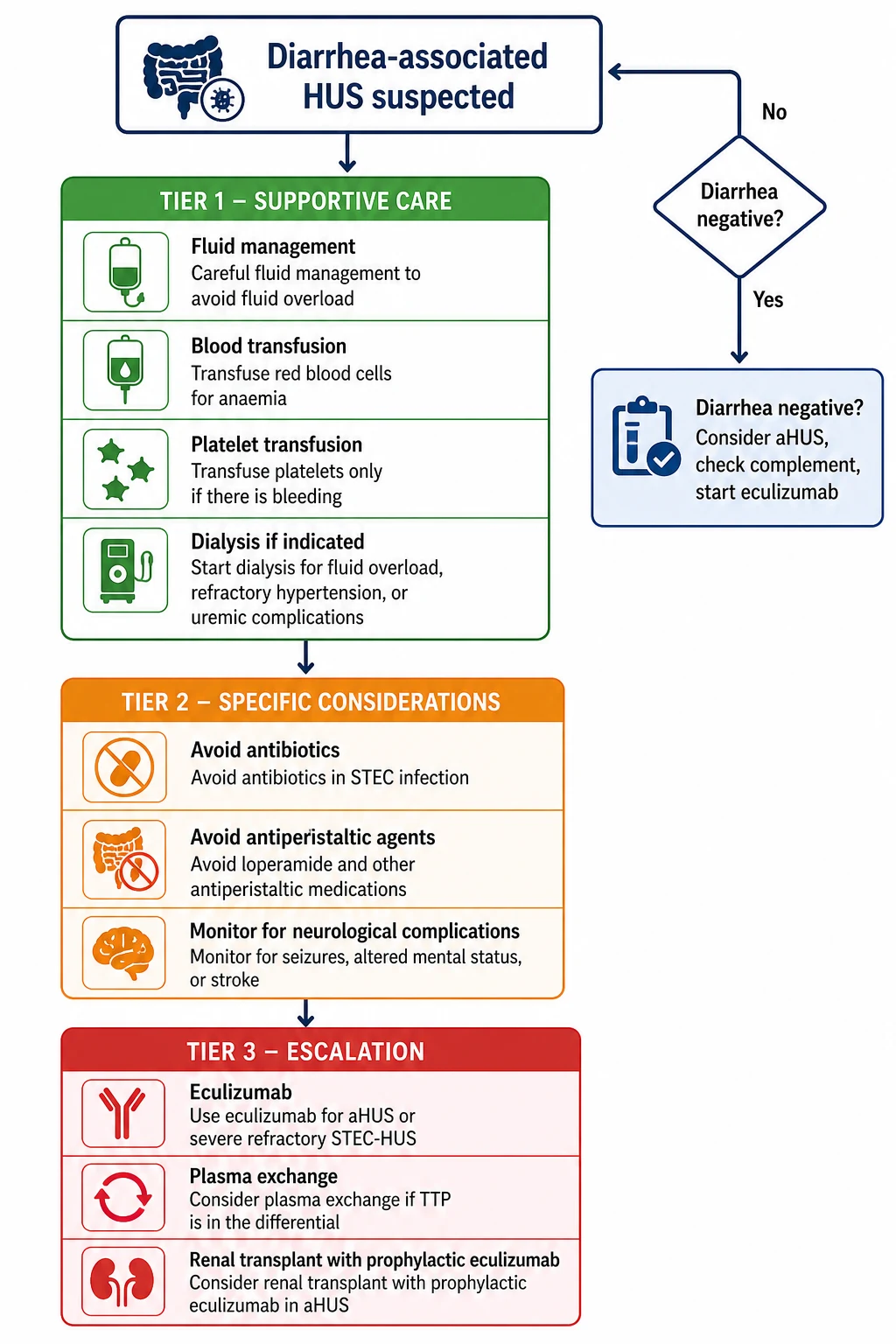
The overriding principle of STEC-HUS management is that there is no specific therapy that reliably alters the disease course, so meticulous supportive care is the treatment. Resuscitation begins with airway, breathing, and circulation, with particular attention to fluid and electrolyte balance, which is the most challenging and consequential aspect of management. Children are often volume-depleted from the prodromal gastroenteritis but simultaneously at risk of fluid overload from acute kidney injury, so the clinician must assess volume status carefully and resuscitate with isotonic crystalloid in aliquots while monitoring response. [1]
Fluid management requires precision. A child who is volume-depleted and still passing urine may need cautious rehydration with isotonic saline, but a child who is oliguric or anuric must be fluid-restricted to insensible losses plus urine output, because any excess accumulates and causes pulmonary and cerebral oedema. Daily weights, strict input-output charts, and central venous pressure monitoring in severe cases guide therapy. Hyperkalaemia is common and dangerous: treatment includes calcium gluconate for cardiac stabilisation, insulin-dextrose and salbutamol for intracellular potassium shift, and sodium bicarbonate if metabolic acidosis is present. Refractory hyperkalaemia, severe metabolic acidosis, or fluid overload unresponsive to diuretics are indications for renal replacement therapy. [5]
Anaemia is treated with packed red cell transfusion when the haemoglobin falls below 60 to 70 g per litre. Transfusions should be given cautiously because rapid expansion of intravascular volume can precipitate or worsen hypertension and pulmonary oedema. Platelet transfusion is reserved for active bleeding, before invasive procedures such as central line insertion, or if the platelet count falls below 10 times 10 to the 9 per litre with bleeding risk. Routine platelet transfusion is avoided because it may fuel the thrombotic process by adding substrate to the microvascular thrombi. [1]
Hypertension is managed with calcium channel blockers such as amlodipine (0.1 to 0.2 mg per kg per dose, maximum 10 mg, once daily or divided) or nifedipine, beta-blockers, or ACE inhibitors in selected cases. Severe hypertension may require intravenous antihypertensives such as labetalol (0.25 to 1 mg per kg per hour infusion) or nicardipine (0.5 to 1 microgram per kg per minute infusion). Seizures are treated with standard anticonvulsants, and any child with neurological involvement needs urgent imaging to exclude stroke or cerebral oedema. Peritoneal dialysis is the preferred mode of renal replacement therapy in young children, though haemodialysis or continuous renal replacement therapy may be used depending on the clinical context and local expertise. [5]
Management — Definitive & Stepwise
Definitive management diverges sharply based on the aetiology. For STEC-HUS, supportive care through the acute episode is the standard of care, and most children recover renal function over days to weeks. No specific therapy has been proven to alter the course of typical STEC-HUS in randomised controlled trials. The Cochrane review of interventions for HUS found insufficient evidence for antibiotics, plasma exchange, or eculizumab in STEC-HUS, and the approach remains one of vigilant supportive care and complication management. [1]
Confirm HUS triad
Assess for diarrhoeal prodrome
Supportive care for all
If aHUS suspected
Eculizumab regimen
Antibiotics should generally be avoided in STEC infection because several studies have associated their use with an increased risk of HUS, possibly by inducing bacteriophage-mediated Shiga toxin release. However, this remains controversial, and antibiotics may be necessary if there is a concomitant bacterial infection or if the child is severely septic. The evidence from systematic reviews is mixed, but the prevailing recommendation in paediatric practice is to avoid routine antibiotic therapy in STEC gastroenteritis. Antimotility agents such as loperamide are contraindicated because they prolong gut carriage and may increase toxin absorption. [1]
Plasma exchange has not been shown to improve outcomes in STEC-HUS and is not routinely recommended. It was explored based on the rationale that removing Shiga toxin or replacing complement factors might help, but the 2012 Lancet analysis of the German O104:H4 outbreak found no clear benefit. In contrast, plasma exchange may be used in TTP and is sometimes used in aHUS as a bridge to eculizumab, though eculizumab has superseded plasma therapy as first-line for aHUS. [11]
Eculizumab is the definitive therapy for aHUS. It is a humanised monoclonal antibody that binds complement C5, preventing its cleavage to C5a and C5b and blocking formation of the membrane attack complex (C5b to C9). The standard dosing for adults and children over 40 kg is 900 mg intravenously weekly for four doses, then 1200 mg every two weeks. For children under 40 kg, dosing is weight-based: 600 mg weekly for two doses (5 to 10 kg), 600 mg every two weeks for maintenance; 900 mg every two weeks for maintenance (10 to 20 kg); and so on according to the prescribing information. Before starting eculizumab, the patient must receive meningococcal vaccination (both conjugated ACWY and serogroup B) and prophylactic penicillin, because terminal complement blockade increases susceptibility to Neisseria meningitidis infection. The international consensus recommends starting eculizumab as soon as aHUS is suspected and TTP has been excluded, without waiting for genetic test results, because delay worsens renal outcomes. [3]
Eculizumab
Dose
Adults and children over 40 kg: 900 mg IV weekly for 4 doses, then 1200 mg every 2 weeks. Children 5 to 10 kg: 300 mg weekly then 300 mg every 2 weeks. Children 10 to 20 kg: 600 mg every 2 weeks. Children 20 to 30 kg: 600 to 900 mg loading then every 2 weeks. Children 30 to 40 kg: 900 mg then 1200 mg every 2 weeks.
The role of eculizumab in STEC-HUS is one of the most debated topics in paediatric nephrology. Case series and the 2011 German outbreak experience suggested possible benefit in the most severe cases with neurological involvement, but systematic reviews have shown inconsistent results. The 2024 systematic review by de Zwart and colleagues concluded that evidence is insufficient to recommend routine eculizumab in STEC-HUS, but it may be considered in children with severe neurological involvement or refractory disease after discussion with a paediatric nephrologist. [10]
Specific Subtypes & Scenarios
STEC-HUS is the most common and best-understood subtype, with a generally favourable prognosis under supportive care. The typical O157:H7-associated form follows a predictable course: acute gastroenteritis, onset of the triad five to ten days later, a peak of organ dysfunction in the first week, and gradual recovery over two to three weeks. Approximately 60 to 70 percent of children recover renal function in the acute phase, though long-term sequelae remain a concern. [5]
Atypical HUS is the subtype that has been transformed by eculizumab. Before complement blockade, aHUS had mortality and end-stage kidney disease rates approaching 50 to 70 percent, and post-transplant recurrence rates exceeding 80 percent. With eculizumab started early, many patients achieve partial or complete renal recovery, and prophylactic use at transplantation prevents recurrence. The clinical phenotype depends on the genetic background: patients with combined complement gene mutations, as described by Bresin and colleagues, have more severe disease and worse outcomes than those with single variants. Anti-factor H antibody-associated aHUS, common in children, may be managed with a combination of eculizumab and immunosuppression (rituximab or cyclophosphamide) to suppress antibody production. [12]
International aHUS Registry — eculizumab outcomes
Key finding
In the international aHUS registry, eculizumab treatment was associated with improved renal outcomes, with 70 to 80 percent of patients achieving platelet normalisation and many showing improvement in renal function. Earlier initiation of eculizumab (within 7 days of presentation) was associated with better renal recovery.
Practice change
Eculizumab should be started as soon as aHUS is suspected and TTP excluded, without waiting for genetic results, to maximise the chance of renal recovery.
Pneumococcal HUS is a distinct subtype triggered by invasive Streptococcus pneumoniae infection. The bacterial enzyme neuraminidase cleaves sialic acid residues from the surface of red blood cells and other cells, exposing the cryptic Thomsen-Friedenreich (T) antigen. Most adult plasma contains anti-T antibodies, which bind the exposed antigen and trigger autoimmune haemolysis. The critical management point is that blood products (which contain plasma) should be avoided or washed, because transfusing plasma-containing products worsens the haemolysis by adding more anti-T antibodies. Pneumococcal HUS has a higher mortality rate than STEC-HUS. [7]
The 2011 German O104:H4 outbreak was the largest recorded HUS outbreak and provided unique insights. Caused by an enteroaggregative Shiga toxin-producing E. coli that had acquired the Stx2 phage, it affected predominantly adults rather than children and had unusually high rates of neurological complications (seizures, coma) and a mortality rate of approximately 1 percent overall but higher in severe cases. The outbreak drove the exploration of eculizumab in severe STEC-HUS, though controlled evidence remained lacking. [11]
Complications & Pitfalls
The acute complications of HUS span multiple organ systems and are the direct causes of mortality. Severe anaemia requiring transfusion is almost universal. Fluid overload with pulmonary oedema, hypertension with encephalopathy, hyperkalaemia with cardiac arrhythmia, and metabolic acidosis are the immediate dangers of acute kidney injury. Neurological complications including seizures, stroke, and coma occur in 20 to 40 percent of STEC-HUS cases and predict a worse prognosis. Pancreatic involvement can cause hyperglycaemia or frank diabetes mellitus, and hepatic dysfunction with transaminitis is common. [5]
Long-term renal sequelae are common and under-recognised. Even children who appear to recover fully in the acute phase have a 25 to 40 percent risk of long-term complications including proteinuria, hypertension, and reduced glomerular filtration rate. A subset of 5 to 10 percent progresses to end-stage kidney disease, sometimes years after the initial episode. This is why Rosales and colleagues emphasised the need for indefinite follow-up: the sequelae of HUS are late-emerging, and a child who looks well at discharge may develop renal disease years later. [5]
The most dangerous pitfall in atypical HUS is delay. Waiting for genetic test results before starting eculizumab can cost the child irreversible renal damage. The international consensus is clear: if aHUS is clinically suspected and TTP has been excluded, start eculizumab immediately. Similarly, failing to screen for aHUS in a child with diarrhoea-negative HUS, or in a child who relapses after apparent recovery, delays the diagnosis of a treatable condition. Giving plasma products in pneumococcal HUS, prescribing antibiotics or antimotility agents in STEC infection, and underestimating the need for long-term renal follow-up are all avoidable errors. [3]
Prognosis & Disposition
STEC-HUS has a generally good prognosis in children, with overall mortality of 3 to 5 percent in the modern intensive care era. Approximately 60 to 70 percent of children achieve full renal recovery in the acute phase, but the remaining 25 to 40 percent have long-term renal sequelae, and 5 to 10 percent progress to end-stage kidney disease. Predictors of a poor outcome include prolonged oligoanuria, high peak creatinine, severe hypertension, neurological involvement, and the need for prolonged dialysis. Children who require dialysis for more than 10 to 14 days have a substantially worse long-term prognosis. [5]
Severity
Mild STEC-HUS
No dialysis, no neurological involvement, rapid recovery. Prognosis excellent. Needs annual renal follow-up.
Severity
Moderate STEC-HUS
Requires dialysis for days, mild neurological symptoms. Prognosis good but 25 to 40 percent risk of long-term renal sequelae.
Severity
Severe STEC-HUS or aHUS
Prolonged dialysis, seizures, stroke, or aHUS diagnosis. High risk of ESKD or death without eculizumab for aHUS. Needs specialist centre.
Atypical HUS has a fundamentally different prognosis. Without treatment, mortality and end-stage kidney disease rates approach 50 to 70 percent. With eculizumab started early, outcomes have been transformed: many patients achieve renal recovery, and prophylactic eculizumab at transplantation prevents recurrence. The response depends on the degree of irreversible renal damage at the time eculizumab is started, which is why early initiation is critical. Patients with complement factor H or factor I variants have the highest recurrence risk and the worst untreated prognosis. [6]
Disposition depends on severity. All children with HUS should be admitted to hospital, ideally under a paediatric nephrology service, and those with severe disease (oliguric renal failure, neurological involvement, severe hypertension, fluid overload) should be in a paediatric intensive care unit with access to dialysis. Transfer to a tertiary centre should be arranged early, particularly if dialysis or eculizumab is likely to be needed. At discharge, every HUS survivor needs a plan for long-term nephrology follow-up with annual blood pressure measurement and urinalysis for life, because of the late-emerging sequelae. [5]
Special Populations
Children under five years are the highest-risk group for STEC-HUS and account for the majority of cases. The immature glomerular endothelium may be more susceptible to Shiga toxin injury due to higher Gb3 receptor density. Infants under one year with HUS deserve particular scrutiny for atypical or metabolic causes, including diacylglycerol kinase epsilon mutations which cause a recessive form of aHUS that presents in infancy and can mimic STEC-HUS. [3]
Children with inherited complement abnormalities have a lifelong risk of relapse and need genetic counselling. A first episode of HUS in a child with a family history, early onset, relapsing course, or diarrhoea-negative presentation should trigger complement screening. The 2013 study by Bresin and colleagues showed that combined complement gene mutations, found in approximately 10 percent of aHUS patients, confer a more severe phenotype with earlier onset and worse outcomes, reinforcing the need for thorough genetic evaluation. [12]
aHUS recurrence after renal transplantation is a critical consideration for children who progress to end-stage kidney disease. Without prophylactic eculizumab, recurrence rates exceed 80 percent in carriers of factor H or factor I mutations, and recurrent disease typically destroys the graft rapidly. This made living-related kidney donation relatively contraindicated in aHUS before the eculizumab era, because a related donor may share the same complement variant and develop aHUS after the stress of donation. Prophylactic eculizumab, started immediately before or at transplantation, has transformed post-transplant outcomes and is now the standard of care. [3]
Pregnant adolescents and young adults with a history of aHUS are at high risk of disease activation during pregnancy, particularly in the third trimester and postpartum. Complement activation is physiologically upregulated in pregnancy, and this can trigger aHUS in women with complement gene variants. Prophylactic eculizumab may be considered in high-risk pregnancies, and close monitoring is essential. [2]
Evidence, Guidelines & Regional Differences
The 2016 international consensus on aHUS management (Loirat et al) is the current landmark guideline for complement-mediated HUS. It recommends eculizumab as first-line therapy for aHUS, started as soon as TTP is excluded by a normal or near-normal ADAMTS13 activity, without waiting for genetic test results. The consensus provides a structured diagnostic algorithm for differentiating STEC-HUS from aHUS and from secondary TMA, and a treatment algorithm that prioritises early complement blockade. [3]
For STEC-HUS, the evidence base is limited by the lack of randomised controlled trials of specific therapies. The Cochrane review of interventions for HUS (Michael et al, 2009) found insufficient evidence for any specific therapy including plasma exchange, eculizumab, or antibiotics. Systematic reviews of eculizumab in STEC-HUS (2024) have shown inconsistent results, with possible benefit only in the most severe cases with neurological involvement. The German O104:H4 outbreak of 2011, the largest recorded, drove much of the current interest in eculizumab for severe STEC-HUS, but controlled evidence remains lacking. [10]
Regional differences exist in the availability and funding of eculizumab, which is one of the most expensive drugs in medicine. In high-income settings (ANZ, UK, North America, Europe), eculizumab is funded through national specialist programmes for aHUS. In resource-limited settings, access may be restricted, and the approach to aHUS may rely more heavily on plasma therapy despite its inferiority. The IPNA (International Pediatric Nephrology Association) provides guidance that is adapted to regional capacity. In terms of STEC prevention, food safety standards and public health surveillance vary: countries with robust meat inspection and food safety programmes (such as Australia and New Zealand) have lower rates of O157:H7 infection than regions where unpasteurised dairy and undercooked beef are more common. [3]
Exam Pearls
HUS is defined by the triad of microangiopathic haemolytic anaemia, thrombocytopenia, and acute kidney injury. STEC-HUS accounts for approximately 90 percent of cases, follows Shiga toxin-producing E. coli (most commonly O157:H7) after a 5 to 10 day prodromal bloody diarrhoea, and peaks under age five. Schistocytes on the blood film are the morphological hallmark. Coagulation studies are normal in HUS, and this is the key discriminator from DIC where PT, APTT, fibrinogen, and D-dimer are all deranged. ADAMTS13 activity below 10 percent distinguishes TTP from HUS. [1]
Eculizumab is the definitive therapy for aHUS. It blocks complement C5, preventing formation of the membrane attack complex. The adult and over-40 kg dose is 900 mg intravenously weekly for four doses, then 1200 mg every two weeks. Children under 40 kg use weight-based dosing. Before starting, the child must receive meningococcal vaccination (both conjugated ACWY and serogroup B) and prophylactic penicillin, because terminal complement blockade increases susceptibility to Neisseria meningitidis infection. Start eculizumab as soon as aHUS is suspected and TTP is excluded, without waiting for genetic results. [3]
Antibiotics should be avoided in STEC infection because they may increase Shiga toxin release via bacteriophage lysis. Antimotility agents (loperamide) are contraindicated because they prolong gut carriage. Platelet transfusion is avoided unless there is active bleeding or a procedure is planned, because it may worsen the thrombotic process. Approximately 25 to 40 percent of STEC-HUS survivors have long-term renal sequelae (proteinuria, hypertension, reduced GFR), and 5 to 10 percent progress to end-stage kidney disease, so all survivors need indefinite nephrology follow-up. aHUS recurrence after renal transplant exceeds 80 percent without prophylactic eculizumab. [5]
References
- [1]Tarr PI, Gordon CA, Chandler WL Shiga-toxin-producing Escherichia coli and haemolytic uraemic syndrome. Lancet, 2005.PMID 15781103
- [2]Noris M, Remuzzi G Atypical hemolytic-uremic syndrome. N Engl J Med, 2009.PMID 19846853
- [3]Loirat C, Fakhouri F, Ariceta G, Besbas N, Bitzan M, Bjerre A, et al An international consensus approach to the management of atypical hemolytic uremic syndrome in children. Pediatr Nephrol, 2016.PMID 25859752
- [4]Keir LS, Saleem MA Current evidence for the role of complement in the pathogenesis of Shiga toxin haemolytic uraemic syndrome. Pediatr Nephrol, 2014.PMID 23843163
- [5]Rosales A, Hofer J, Zimmerhackl LB, Jungraithmayr TC, Riedl M, Giner T, et al Need for long-term follow-up in enterohemorrhagic Escherichia coli-associated hemolytic uremic syndrome due to late-emerging sequelae. Clin Infect Dis, 2012.PMID 22412065
- [6]Schaefer F, Ardissino G, Ariceta G, Fakhouri F, Scully M, Isbel N, et al Clinical and genetic predictors of atypical hemolytic uremic syndrome phenotype and outcome. Kidney Int, 2018.PMID 29907460
- [7]Le Clech A, Simon-Tillaux N, Provot F, Delmas Y, Karras A, Kvetnansky R, et al Atypical and secondary hemolytic uremic syndromes have a distinct presentation and no common genetic risk factors. Kidney Int, 2019.PMID 30982675
- [8]Lapeyraque AL, Malina M, Fremeaux-Bacchi V, Boppel T, Kirschfink M, Oualha M, et al Eculizumab in severe Shiga-toxin-associated HUS. N Engl J Med, 2011.PMID 21612462
- [9]Percheron L, Gramada R, Tellier S, Salomon R, Kwon T, Baudouin V, et al Eculizumab treatment in severe pediatric STEC-HUS: a multicenter retrospective study. Pediatr Nephrol, 2018.PMID 29572749
- [10]de Zwart PL, Mueller TF, Spartà G, Luyckx VA Eculizumab in Shiga toxin-producing Escherichia coli hemolytic uremic syndrome: a systematic review. Pediatr Nephrol, 2024.PMID 38057431
- [11]Frank C, Werber D, Cramer JP, Askar M, Faber M, an der Heiden M, et al Epidemic profile of Shiga-toxin-producing Escherichia coli O104:H4 outbreak in Germany. N Engl J Med, 2011.PMID 21696328
- [12]Bresin E, Rurali E, Caprioli J, Sanchez-Corral P, Fremeaux-Bacchi V, Rodriguez de Cordoba S, et al Combined complement gene mutations in atypical hemolytic uremic syndrome influence clinical phenotype. J Am Soc Nephrol, 2013.PMID 23431077