Paeds · nephrology-urology-fluids-and-electrolytes
Hypokalaemia and hyperkalaemia
Also known as Hypokalaemia · Hyperkalaemia · Potassium disorders · Dyskalaemia · Low potassium · High potassium · Potassium emergency
Fellowship guide to potassium disorders in children: hypokalaemia as a serum potassium below 3.5 and hyperkalaemia as above 5.5 mmol/L, the three-axis classification of intake, transcellular shift and renal or gut loss, the cardiac membrane electrophysiology behind peaked T waves and U waves, emergency management of hyperkalaemia with calcium gluconate, salbutamol and insulin-dextrose, safe intravenous potassium replacement for hypokalaemia, Bartter and Gitelman syndromes, and hypomagnesaemia as the cause of refractory hypokalaemia.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
Potassium is the dominant intracellular cation: about 98 percent of the body's store sits inside cells, and only a sliver lives in the small extracellular compartment the serum measures. This single fact explains almost everything about potassium disease. A child can lose hundreds of millimoles from the body and still show a near-normal serum level, because insulin, catecholamines and acid-base status keep shifting potassium back and forth across the cell membrane to defend that level. It also explains why a small shift out of cells — a little acidosis, a dose of digoxin, a fast-growing tumour breaking down — can move the serum potassium into the danger zone long before total-body potassium is high. [4] [1]
This page treats the two disorders as a mirror image. Hyperkalaemia is the faster killer — it stops the heart — so it gets the resuscitation framework: calcium, salbutamol, insulin-dextrose, removal. Hypokalaemia is the quieter danger — it weakens muscle and prolongs repolarisation — so it gets the replacement framework: oral versus intravenous, the rate limit, and magnesium. Both share the same organising logic: classify by the three axes, let the ECG set the tempo, and treat the cause while you treat the number. [1] [3]
Overview & Definition
Hypokalaemia is a serum potassium below 3.5 mmol/L. It is graded as mild (3.0 to 3.5), moderate (2.5 to 3.0) and severe (below 2.5); severe disease, or any level with symptoms or ECG changes, is an emergency. Hyperkalaemia is a serum potassium above 5.5 mmol/L. In the newborn the upper bound is slightly higher, but any level above 6.5 mmol/L, or any level with ECG changes at all, is a true emergency. Both thresholds are serum values, and both demand a repeat sample to exclude artefact before you act on an asymptomatic, mildly abnormal result. [1] [4]
The serum potassium is a concentration, not a content. It tells you how much potassium sits in the extracellular water at that instant, not how much the whole body holds. A child in diabetic ketoacidosis may walk in with a serum potassium of 5.0 mmol/L and a total-body deficit of 3 to 6 mmol/kg, because the acidosis has shifted intracellular potassium out into the blood while the urine has been washing it away for days. The number and the deficit point in opposite directions, and this is the trap that catches the unwary prescriber. [4] [3]
Classification
Every cause of dyskalaemia lives on one of three axes: intake (how much potassium enters the system), transcellular shift (how much moves between the intracellular and extracellular compartments), and output (how much the kidney or gut removes). The figure below lays the common paediatric causes onto these axes for both disorders. [4] [1]
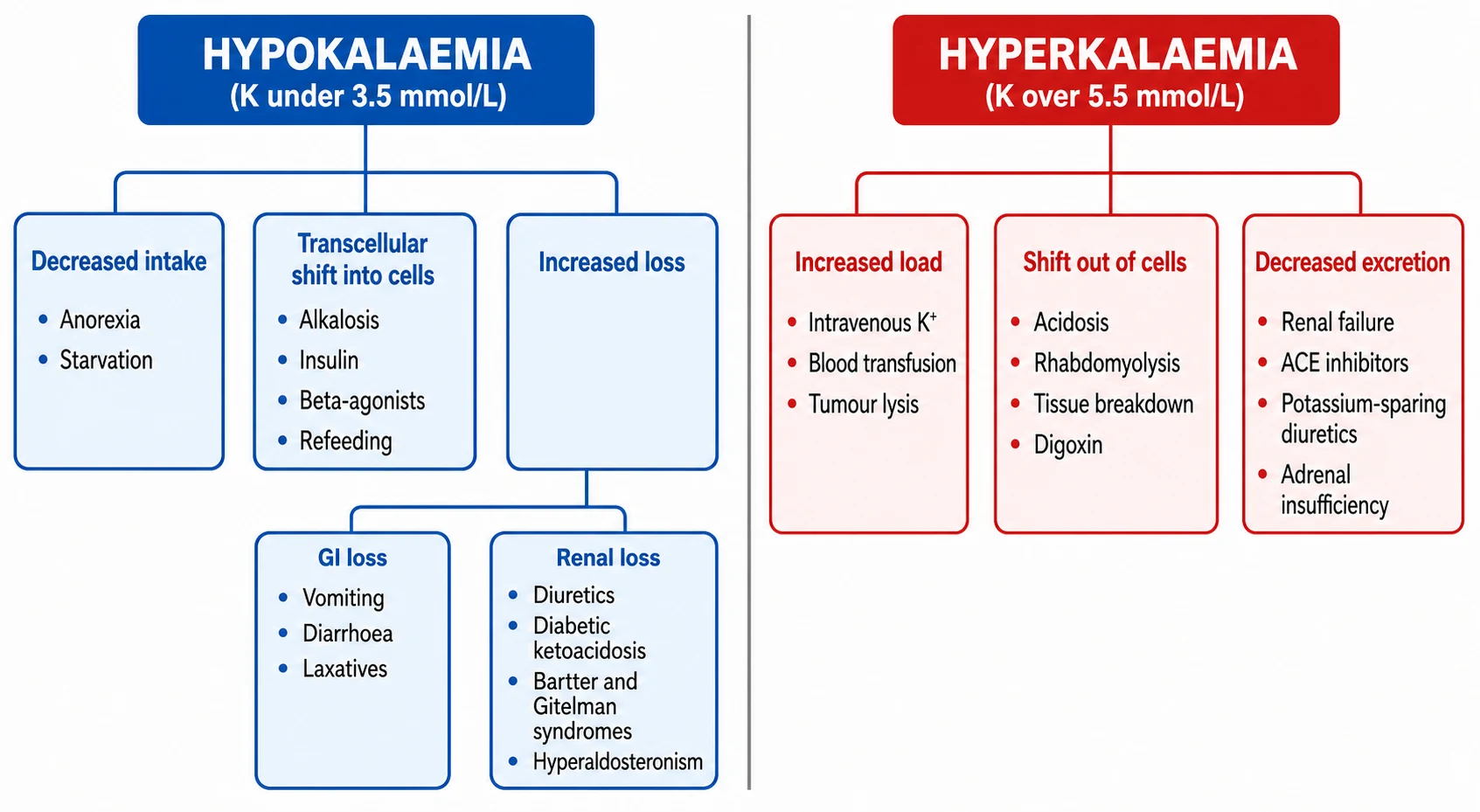
For hypokalaemia the decisive branch point is whether potassium is being lost through the kidney or somewhere else. A urine potassium above roughly 20 mmol/L (or a transtubular potassium gradient above about 3) points to renal wasting — diuretics, the osmotic diuresis of diabetic ketoacidosis, Bartter or Gitelman syndrome, hyperaldosteronism, or renal tubular acidosis. A low urine potassium points to extrarenal loss — vomiting, diarrhoea, laxative abuse — or to a shift into cells from alkalosis, insulin or refeeding. [4] [9]
For hyperkalaemia the same three axes apply. Increased load covers exogenous potassium (intravenous supplements, stored-blood transfusion, potassium-containing fluids) and endogenous release (tumour lysis, rhabdomyolysis, haemolysis). Shift out of cells covers metabolic acidosis, insulin deficiency, beta-blockade, digoxin toxicity and succinylcholine. Decreased excretion covers acute and chronic kidney disease, adrenal insufficiency, type 4 renal tubular acidosis, and the drugs that converge on the distal nephron — ACE inhibitors, angiotensin receptor blockers, potassium-sparing diuretics, NSAIDs and trimethoprim. [1] [4]
Hypokalaemia (K below 3.5 mmol/L)
- Shift into cells: alkalosis, insulin, beta-2 agonists, refeeding
- Renal loss (urine K high): diuretics, DKA, Bartter, Gitelman, hyperaldosteronism, RTA
- Extrarenal loss (urine K low): vomiting, diarrhoea, laxatives, sweating
- Decreased intake: anorexia, starvation, free-water loading
Hyperkalaemia (K above 5.5 mmol/L)
- Increased load: IV potassium, stored blood, tumour lysis, rhabdomyolysis
- Shift out of cells: acidosis, insulin lack, beta-blockers, digoxin, succinylcholine
- Decreased excretion: renal failure, ACE inhibitors, ARBs, spironolactone, Addison
- Pseudohyperkalaemia: haemolysed sample, tight tourniquet, marked leucocytosis
Epidemiology & Risk Factors
Dyskalaemia is among the commonest electrolyte emergencies in hospitalised children, and the risk concentrates in a few well-defined groups: children on diuretics, those with acute kidney injury, premature neonates, postoperative patients, and — above all — children in diabetic ketoacidosis. The shared thread is that all of these states disrupt one or more of the three axes simultaneously: the kidney that cannot excrete, the diuretic that wastes, the insulin that shifts, and the immature tubule that cannot keep up. [1] [4]
Diabetic ketoacidosis deserves special emphasis because it is the single commonest setting for clinically important hypokalaemia in children. Osmotic diuresis strips potassium through the urine, and the obligate excretion of ketoanions drags cations with it, so total-body potassium falls even though the presenting serum level is often high. Once insulin and intravenous fluids begin, potassium pours back into cells and the serum level can collapse within hours. Death from hypokalaemia during DKA treatment is one of the classic, preventable causes of DKA mortality, which is why every DKA protocol builds potassium replacement into the first bag of fluid. [4] [3]
Neonates, especially premature infants, are vulnerable to both ends of the spectrum. The distal tubule is immature, the extracellular fluid compartment is large, and a substantial intracellular-to-extracellular potassium shift occurs in the first days of life. Non-oliguric hyperkalaemia is a recognised entity in very preterm infants, while aggressive diuretic use or inadequate intake readily produces hypokalaemia on the neonatal unit. [3]
Pathophysiology
Potassium sets the resting membrane potential of every excitable cell, and the heart is where this matters most. The sodium-potassium ATPase on the cell membrane pumps three sodium ions out for every two potassium ions in, holding potassium high inside the cell and low outside. The gradient is what keeps the resting membrane polarised — negative on the inside, ready to fire. [4]
In hyperkalaemia the extracellular potassium rises, so the gradient across the membrane shrinks and the resting potential becomes less negative (closer to zero). This sounds like it should make the cell more excitable, but in fact it partially inactivates the fast sodium channels that carry the action potential upstroke. Conduction slows. The atria and ventricles depolarise more and more sluggishly, the repolarisation accelerates abnormally early, and the ECG traces the decline step by step: tall peaked T waves, then PR prolongation and loss of the P wave, then widening of the QRS, then a sine-wave fusion of all the complexes, and finally ventricular fibrillation or asystole. [2] [4]
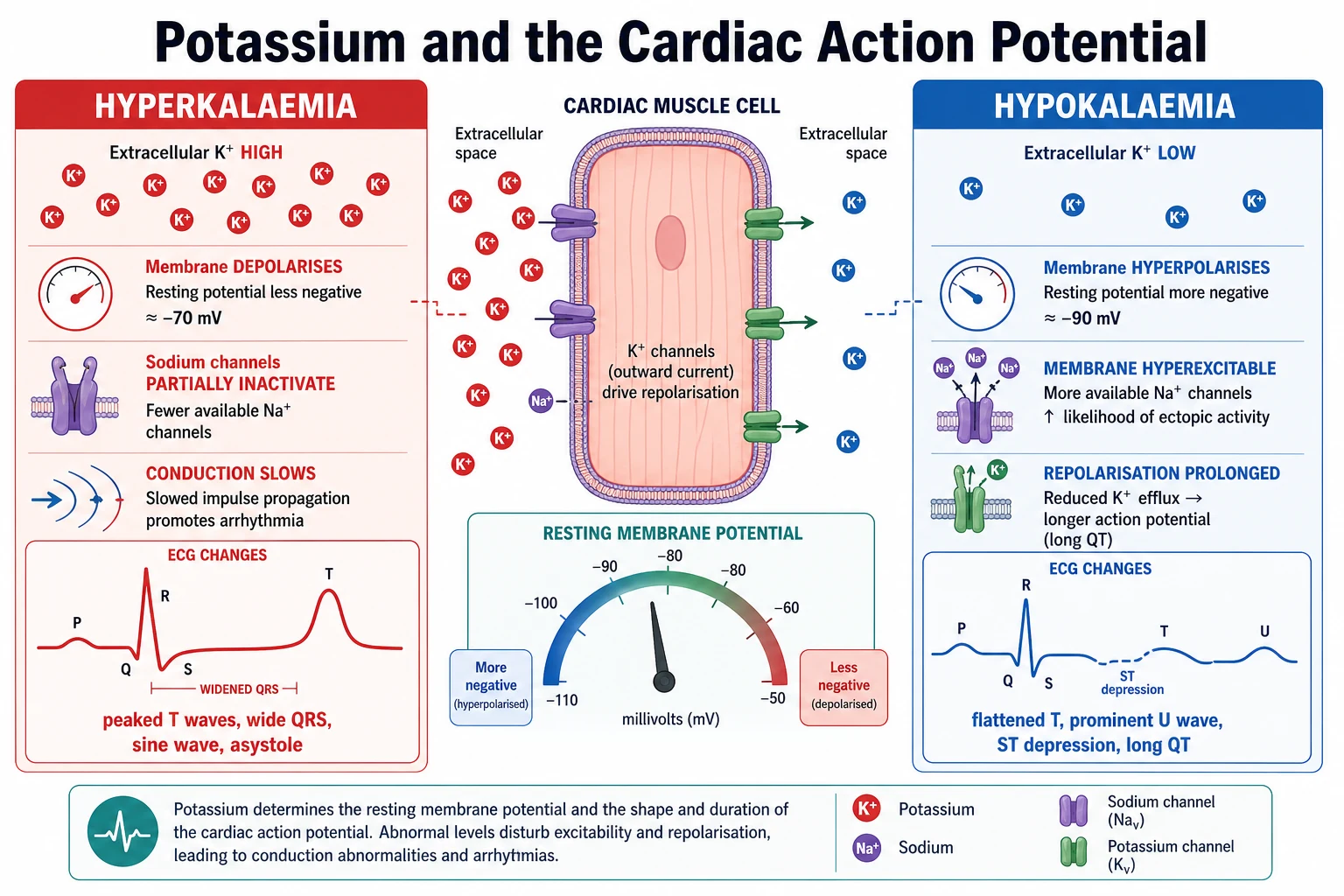
In hypokalaemia the opposite happens. The gradient steepens, the resting potential becomes more negative (hyperpolarised), and repolarisation is prolonged. The T wave flattens and inverts, the ST segment sags, and a new wave — the U wave — appears after the T wave, representing delayed repolarisation of the Purkinje and mid-myocardial fibres. The effective QT interval lengthens (more accurately, a long QU), which sets up the conditions for re-entry and torsades de pointes. At the same time the skeletal and smooth muscle weaken, because a more negative resting potential takes the membrane further from the threshold for contraction, producing the proximal weakness, ileus and urinary retention of severe disease. [4] [11]
The transcellular shift is reversible and fast. Insulin activates the sodium-potassium ATPase and pushes potassium into cells within minutes — this is the basis of insulin-dextrose therapy. Beta-2 agonists such as salbutamol do the same through cyclic AMP. Alkalosis shifts potassium in; acidosis shifts it out, and in organic acidoses (lactic, ketoacidosis) the shift is less than in mineral acidosis because the organic anion follows potassium into the cell. Digoxin poisons the sodium-potassium ATPase directly, releasing intracellular potassium — which is why digoxin toxicity is one of the few causes of hyperkalaemia that is itself diagnostic. [4] [5]
Clinical Presentation
Hypokalaemia presents with weakness. It begins in the proximal leg muscles — difficulty rising from sitting, climbing stairs — and progresses in severe disease to flaccid paralysis and respiratory muscle involvement. Smooth muscle is affected too: ileus, abdominal distension, constipation and urinary retention are classic. Deep tendon reflexes may be diminished. The child may also describe cramps, paraesthesiae and muscle pain. Infants and young children present non-specifically — poor feeding, lethargy, hypotonia — and the potassium is found on routine bloods. [4] [9]
Hyperkalaemia is treacherous because it is often clinically silent until it kills. Muscle weakness and paraesthesiae are described but unreliable prodromes. The real presentation is cardiac: palpitations, syncope, or sudden collapse, and on the monitor a tachyarrhythmia, a bradyarrhythmia, conduction block, or asystole. This is why the ECG, not the history, drives the tempo of hyperkalaemia management. [2] [4]
The history discriminates the cause. Ask about vomiting, diarrhoea and laxative use (extrarenal loss); diuretics, bronchodilators and insulin (drug effects); polyuria, polydipsia and weight loss (diabetes and tubulopathies); recent chemotherapy or a rapidly enlarging mass (tumour lysis); salt craving, fatigue and hyperpigmentation (adrenal insufficiency); and a family history of periodic paralysis, deafness with tubulopathy (Bartter type 4), or consanguinity. Examination adds the volume status (depletion in vomiting and diuretic use; overload in renal failure), blood pressure (high in mineralocorticoid excess, low in adrenal insufficiency), and any syndrome features. [1] [9]
Differential Diagnosis
For hypokalaemia, branch first on the urine potassium. Renal loss (high urine potassium) covers the loop and thiazide diuretics, the osmotic diuresis of diabetic ketoacidosis, the inherited salt-wasting tubulopathies Bartter and Gitelman syndromes, primary and secondary hyperaldosteronism, Cushing syndrome, and renal tubular acidosis types 1 and 2. Extrarenal loss (low urine potassium) covers vomiting (where the renal potassium loss is secondary to aldosterone-driven sodium retention, not the primary cause), diarrhoea, laxative and enema abuse, and profuse sweating. Shift into cells covers alkalosis, insulin excess, beta-2 agonist therapy, the refeeding syndrome, and familial periodic paralysis. [4] [9]
For hyperkalaemia, branch on the three axes. Increased load: exogenous potassium (intravenous supplements, potassium-containing fluid, stored-blood transfusion), tumour lysis syndrome, rhabdomyolysis, and severe haemolysis. Shift out of cells: metabolic acidosis (especially inorganic), insulin deficiency, beta-blockade, digoxin toxicity, succinylcholine, and rapid transfusion of old blood. Decreased excretion: acute kidney injury, chronic kidney disease, adrenal insufficiency (Addisonian crisis), type 4 renal tubular acidosis, and the distal-nephron drugs — ACE inhibitors, angiotensin receptor blockers, potassium-sparing diuretics (spironolactone, amiloride, triamterene), NSAIDs, and trimethoprim. [1] [4]
Bartter and Gitelman syndromes are the two inherited hypokalaemic tubulopathies that examiners love, and they are separated by calcium and magnesium. Both cause salt wasting, hypokalaemic metabolic alkalosis and normal-to-low blood pressure. Bartter syndrome is a defect of the thick ascending limb (mimicking a loop diuretic), presents earlier (often antenatally or in infancy with polyhydrosramnios, prematurity and failure to thrive), and shows hypercalciuria with a risk of nephrocalcinosis. Gitelman syndrome is a defect of the distal convoluted tubule (mimicking a thiazide), presents later (adolescence) with tetany, and shows hypomagnesaemia and hypocalciuria. That single calcium direction is the tie-breaker. [9] [10]
Bartter syndrome
- Thick ascending limb defect (loop-of-Henle, mimics furosemide)
- Onset antenatal or infancy: polyhydramnios, prematurity, failure to thrive
- Hypokalaemic metabolic alkalosis, normal or low blood pressure
- Hypercalciuria — risk of nephrocalcinosis
Gitelman syndrome
- Distal convoluted tubule defect (mimics thiazide diuretic)
- Onset later: adolescence or young adulthood, often with tetany
- Hypokalaemic metabolic alkalosis, normal or low blood pressure
- Hypomagnesaemia and hypocalciuria — the calcium tie-breaker
The KDIGO Controversies Conference consensus on Gitelman syndrome (PMID 28003083) underpins international management. ANZ and UK practice follow this consensus for magnesium and potassium supplementation targets and for transition planning, while individual units apply slightly different intravenous potassium concentration and rate conventions — always confirm the local paediatric protocol before prescribing. [10]
Clinical & Bedside Assessment
Begin with airway, breathing and circulation, and attach a cardiac monitor immediately. The first question in any dyskalaemia is not "what is the cause" but "is this child about to arrest", and the answer is on the ECG. Obtain a 12-lead tracing early and look for the signatures: in hyperkalaemia, tall peaked (tented) T waves that narrow at the base, then PR prolongation and flattening or disappearance of the P wave, then widening of the QRS, then a sine wave; in hypokalaemia, flattened or inverted T waves, ST depression, and a U wave appearing after the T wave. [2] [4]
Assess volume status at the bedside, because it decides the route and rate of any replacement. A child who is dry (cool peripheries, prolonged capillary refill, tachycardia, low venous pressure) needs volume before and alongside potassium; a child who is overloaded (as in renal failure) needs the potassium brought down, not a fluid load. Palpate the abdomen for ileus or bladder distension in hypokalaemia, and test the deep tendon reflexes and muscle power. [1] [3]
Take a focused history aimed at the three axes. Run through the medication list — diuretics, ACE inhibitors, angiotensin receptor blockers, spironolactone, beta-agonists, insulin, digoxin, trimethoprim, NSAIDs. Ask about gastrointestinal losses, recent chemotherapy, endocrine symptoms (polyuria, polydipsia, weight loss, salt craving, pigmentation), dietary fads, and a family history of tubulopathy or periodic paralysis. In parallel, screen for the precipitating emergency — diabetic ketoacidosis, tumour lysis, Addisonian crisis, sepsis with renal failure — because treating the level without the cause will fail. [1] [9]
Investigations
The core panel is the same for both disorders and can be drawn from one cannula: a repeat serum potassium (to exclude pseudohyperkalaemia from haemolysis or a tight tourniquet), sodium, urea and creatinine, a venous or arterial blood gas for pH, bicarbonate and base excess, magnesium, calcium, phosphate, glucose, and a 12-lead ECG. Send the gas immediately — the pH and bicarbonate tell you about the shift axis and flag DKA, renal failure and lactic acidosis within seconds. [1] [2]
SHIFT
For hypokalaemia, a urine potassium (or the transtubular potassium gradient) splits renal from extrarenal loss. A spot urine potassium above about 20 mmol/L, or a transtubular potassium gradient above roughly 3, indicates the kidney is the culprit. Follow with plasma renin and aldosterone to separate the tubulopathies (high renin, high aldosterone, normal or low blood pressure) from mineralocorticoid excess (low renin, low aldosterone, high blood pressure), and check the magnesium — because hypomagnesaemia is both a cause and the reason replacement fails. [9] [4]
For hyperkalaemia, targeted tests follow the screen: creatine kinase for rhabdomyolysis, urate, phosphate and lactate dehydrogenase for tumour lysis, cortisol and a short synacthen test where adrenal insufficiency is possible, and a digoxin level where the picture fits. A normal or high serum potassium in diabetic ketoacidosis is never reassuring — total-body potassium is depleted, and the fall begins the moment insulin starts. [1] [3]
Management — Resuscitation
Severe hyperkalaemia with ECG changes is a cardiac emergency, and the first drug is not one that lowers potassium — it is calcium. Intravenous 10 percent calcium gluconate at 0.5 mL/kg (a maximum of about 10 mL, which is roughly 2.3 mmol of calcium) given slowly over 5 minutes with continuous cardiac monitoring raises the threshold of the cardiac membrane and restores conduction within minutes. It does not touch the potassium level, so it must be followed by a shifting agent. If the ECG is still abnormal the dose may be repeated. Calcium chloride is an alternative in arrest or via a central line, but calcium gluconate is preferred peripherally because it is less vesicant if it extravasates. [2] [3]
Immediately after calcium, shift potassium into cells. Nebulised salbutamol 2.5 mg for a child under 25 kg and 5 mg for a child over 25 kg lowers the serum potassium within 30 minutes by activating the beta-2 receptor and driving the sodium-potassium ATPase. The evidence base in children, from the Kemper and McClure studies, shows a reliable fall; salbutamol is fast, cheap and well tolerated, and is usually combined with insulin-dextrose rather than given alone. [5] [6]
The second shifting agent is intravenous insulin 0.1 unit/kg with glucose 0.5 g/kg (for example 5 mL/kg of 10 percent dextrose), which begins to lower potassium within 15 minutes and works for several hours. Monitor blood glucose every 15 to 30 minutes for at least two hours afterwards, because iatrogenic hypoglycaemia is common — occurring in roughly 10 to 20 percent of treatments — and can be severe. The glucose dose has risen over time precisely to prevent this complication. [7] [8]
Calcium gluconate 10% (hyperkalaemia with ECG changes)
Dose
0.5 mL/kg
Salbutamol (to shift potassium into cells)
Dose
Nebulised 2.5 mg if under 25 kg; 5 mg if over 25 kg
For severe symptomatic hypokalaemia the resuscitation is controlled intravenous potassium chloride. The cardinal rule is that potassium must never be given as an undiluted bolus and never faster than the rate limit: a maximum of 0.2 mmol/kg/hour via a peripheral line, and a maximum of 0.4 mmol/kg/hour via a central line with continuous cardiac monitoring and an infusion pump. The concentration should not exceed about 40 mmol/L in a peripheral line. Use an infusion pump, never a free-running drip, and reassess the serum potassium frequently. Correct magnesium first if it is low, because the potassium will not be retained until magnesium is restored. [1] [4]
Management — Definitive & Stepwise
The definitive ladder for hyperkalaemia has four rungs, applied in order. First, stabilise the myocardium with calcium gluconate when the ECG is abnormal. Second, shift potassium into cells with salbutamol and insulin-dextrose. Third, remove potassium from the body — consider dialysis for refractory hyperkalaemia, ongoing potassium load, or renal failure with oliguria or fluid overload. Cation-exchange resins are slow (working over many hours) and are not adequate as sole emergency therapy; sodium zirconium cyclosilicate and patiromer are newer agents with a limited paediatric evidence base. Fourth, identify and remove the precipitant — stop the offending drug, treat the tumour lysis, manage the renal failure, give hydrocortisone for adrenal crisis — and monitor the potassium and glucose serially. [1] [2]
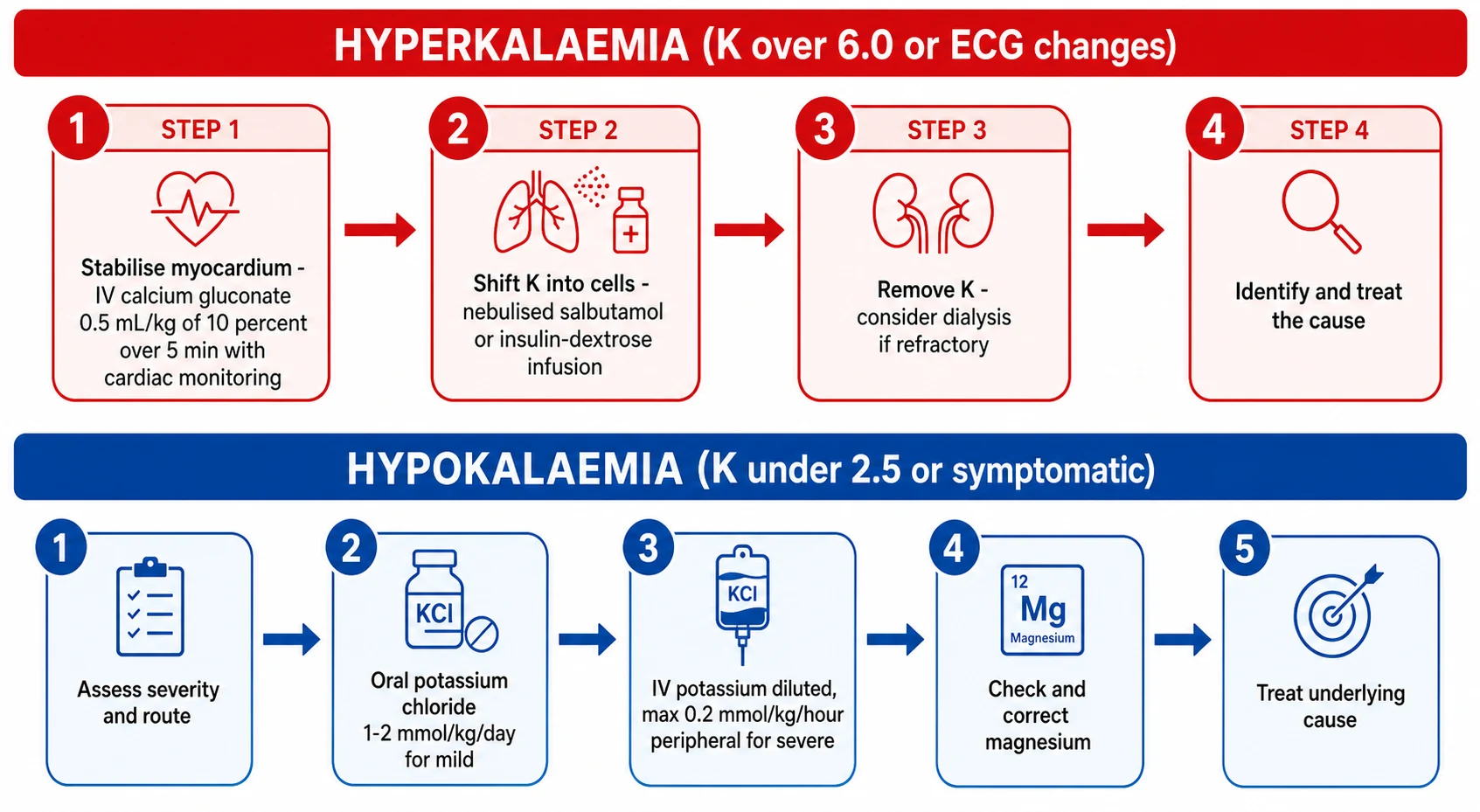
The definitive ladder for hypokalaemia is simpler but no less disciplined. Assess severity and route: mild, asymptomatic hypokalaemia is managed orally; severe or symptomatic disease, or a child who cannot absorb orally, needs intravenous replacement. Oral potassium chloride is given at 1 to 2 mmol/kg/day in divided doses; it is irritating to the gut, so slow-release preparations and divided dosing help. Intravenous potassium chloride at the rate limit above is reserved for severe or symptomatic disease. Correct magnesium — 25 to 50 mg/kg of magnesium sulfate — before or alongside potassium, because hypokalaemia is refractory while magnesium is low. Finally, treat the underlying cause: stop the diuretic or change it, manage the diarrhoea, treat the tubulopathy with supplements, and remove the offending drug. [4] [9]
Hyperkalaemia with ECG changes — the four-rung ladder
Stabilise the myocardium: IV calcium gluconate 0.5 mL/kg of 10% over 5 min with cardiac monitoring
Shift K into cells: nebulised salbutamol (2.5 mg under 25 kg, 5 mg over 25 kg) AND insulin 0.1 unit/kg with glucose 0.5 g/kg
Remove K: consider dialysis if refractory, ongoing load, or renal failure with fluid overload
Treat the cause and monitor: serial potassium and glucose every 1 to 2 hours
Severe hypokalaemia — the replacement ladder
Assess severity and route; secure cardiac monitoring
Correct hypomagnesaemia first (magnesium sulfate 25 to 50 mg/kg)
Oral potassium chloride 1 to 2 mmol/kg/day for mild disease
IV potassium chloride, max 0.2 mmol/kg/hour peripheral (0.4 central), infusion pump only
Treat the underlying cause and monitor serial potassium
Specific Subtypes & Scenarios
Diabetic ketoacidosis is the scenario where hypokalaemia kills, and it does so during treatment. Total-body potassium is depleted by the osmotic diuresis, yet the serum level at presentation is normal or high because acidosis has shifted potassium out of cells. Once insulin starts, potassium pours back into cells and the serum falls. Standard paediatric DKA protocols therefore add potassium to the intravenous fluids once the serum potassium falls below 5.5 mmol/L and urine output is established — typically 40 mmol/L of potassium chloride in the replacement fluid, equivalent to roughly 0.1 mmol/kg/hour. Anticipate the fall, check the potassium every two to four hours, and never give insulin without having a plan for potassium. [4] [3]
Tumour lysis syndrome is the hyperkalaemia of rapid cell turnover. Children with high-burden Burkitt lymphoma or T-cell acute lymphoblastic leukaemia release huge quantities of intracellular contents when chemotherapy begins, producing acute hyperkalaemia, hyperphosphataemia, hypocalcaemia and acute kidney injury within 12 to 72 hours. Prophylaxis is aggressive hydration and rasburicase (for high-risk disease) or allopurinol, and the hyperkalaemia is managed with the standard ladder, with a low threshold for dialysis when renal failure supervenes. [1]
Bartter and Gitelman syndromes are the chronic hypokalaemic tubulopathies. Both are managed with lifelong oral potassium and magnesium supplementation, together with agents that blunt the secondary hyperaldosteronism and hyperprostaglandinaemia — potassium-sparing diuretics (amiloride, spironolactone) and, in Bartter, prostaglandin synthetase inhibitors such as indometacin. Gitelman particularly needs generous magnesium replacement to control tetany and to allow potassium retention. Growth, blood pressure, renal function and bone density require long-term surveillance, and transition to adult nephrology care is part of the plan. [9] [10]
Renal tubular acidosis straddles the two disorders. Distal (type 1) and proximal (type 2) RTA produce hypokalaemia through bicarbonate wasting and secondary hyperaldosteronism; type 4 RTA produces hyperkalaemia through hypoaldosteronism or aldosterone resistance. The plasma bicarbonate and the urine pH, together with the potassium, separate them and guide alkali and mineralocorticoid therapy. [4]
Digoxin toxicity is the one hyperkalaemia where the treatment is not the standard ladder but the antidote. Digoxin poisons the sodium-potassium ATPase and releases intracellular potassium, so the serum potassium is itself a measure of severity. Digoxin-specific antibody fragment (Fab) binds the drug, reverses the toxicity, and lowers the potassium as the ATPase recovers — and is indicated for life-threatening arrhythmia, hyperkalaemia, or a known large ingestion. Calcium is traditionally avoided in digoxin toxicity because of concern over "stone heart", though this remains debated; the definitive step is Fab. [1] [4]
Complications & Pitfalls
The cardinal pitfall in hyperkalaemia is treating the number without the ECG, or — equally dangerous — withholding calcium while waiting for a repeat result when the tracing already shows a widened QRS. The ECG determines the urgency, and calcium is safe, fast and reversible, so when in doubt give it. The mirror-image pitfall is dismissing a markedly high level as artefact because the child looks well; the ECG is an imperfect predictor of severe hyperkalaemia, so a normal tracing never excuses ignoring a dangerous number. [11] [2]
The cardinal pitfall in hypokalaemia is the rate. Intravenous potassium given too fast causes fatal ventricular arrhythmia — this is why there is a hard rate limit, why an infusion pump is mandatory, and why undiluted bolus potassium is never given. The second pitfall is failing to correct magnesium: hypokalaemia will simply not resolve while magnesium is low, because low magnesium disinhibits the ROMK channel in the distal nephron and allows continued potassium secretion. Every refractory hypokalaemia is hypomagnesaemia until proven otherwise. [4] [9]
The third pitfall, and a growing one, is iatrogenic hypoglycaemia after insulin-dextrose. It is common — in the order of 10 to 20 percent of treatments — and can be severe, particularly in small children and in those who have not eaten. Prevent it with an adequate glucose dose (0.5 g/kg or more) and frequent blood glucose monitoring for at least two hours afterwards. The fourth pitfall is pseudohyperkalaemia both ways: failing to exclude it leads to unnecessary treatment of a phantom level, while dismissing a true emergency as a clotted sample costs the child the window in which calcium would have worked. [7] [8]
Prognosis & Disposition
Prognosis in dyskalaemia is governed by four things: the severity of the level, the speed of change, the presence of ECG changes, and the reversibility of the cause. A child whose potassium has climbed from 4.0 to 7.0 over an hour is at far greater risk than one who lives chronically at 6.0, because the myocardium has not adapted. Mortality in hyperkalaemia is driven by cardiac arrest, which is why early ECG and calcium are the highest-yield interventions in all of acute paediatric electrolyte medicine. Morbidity in hypokalaemia is driven by arrhythmia, respiratory muscle weakness, and ileus. [1] [4]
Admit to a high-dependency or intensive care setting any child with ECG changes, severe dyskalaemia (potassium above 6.5 or below 2.5 mmol/L), a child receiving intravenous potassium at or near the rate limit, and any child who needs dialysis. The stable child, after correction, goes to the ward with serial monitoring while the cause is addressed. On discharge, identify and treat the cause, adjust or stop offending drugs, arrange follow-up electrolytes, and for chronic tubulopathies set up the supplementation regimen and the surveillance plan. Give the family a clear safety-net: the warning signs (palpitations, weakness, collapse), the importance of medication adherence, and when to return. [9] [3]
Special Populations
Neonates, especially premature infants, are a law unto themselves. The distal tubule is immature, the glomerular filtration rate is low, and a large intracellular-to-extracellular potassium shift occurs in the first days of life. Non-oliguric hyperkalaemia in the very preterm infant can cause cardiac arrhythmia and is managed with shifting agents and, if needed, exchange transfusion or peritoneal dialysis; potassium handling improves as the tubule matures. At the other end, aggressive diuretic use or inadequate intake readily produces hypokalaemia on the neonatal unit. All potassium therapy in neonates is weight-based, with the rate limit applied per kilogram and cardiac monitoring throughout. [3]
Chronic kidney disease and dialysis children live at constant risk of hyperkalaemia, because the excretion axis is permanently impaired. Management is dietary potassium restriction, avoidance of constipation (which increases colonic potassium absorption), careful use of potassium-binding agents, and an agreed plan for when the dialysis is due. Intercurrent illness, a missed dialysis session, or an ACE inhibitor started for renoprotection can all tip these children into an emergency. [1]
Children with complex chronic conditions are often on multiple potassium-altering drugs at once — a diuretic for heart failure, an ACE inhibitor for renal protection, a beta-agonist for wheeze. Each drug moves the potassium in a predictable direction, but in combination they can produce unpredictable swings. A medication review is part of every assessment, and changes should be made one at a time with follow-up electrolytes. [9]
Inherited tubulopathies (Bartter, Gitelman) require lifelong supplementation, monitoring of calcium and magnesium (and, in Bartter, renal ultrasound for nephrocalcinosis), attention to growth and bone health, and a structured transition to adult nephrology care. Pregnancy in Gitelman syndrome demands particular attention to magnesium, as hypomagnesaemia can worsen and affect both mother and fetus. [10]
Evidence, Guidelines & Regional Differences
The evidence base for the paediatric hyperkalaemia ladder is older and smaller than the protocols imply, and most doses are extrapolated from adult practice tempered by physiology. The salbutamol evidence in children rests on small but consistent studies: Kemper and colleagues showed that a short salbutamol infusion effectively lowered potassium in children with acute hyperkalaemia, and McClure and colleagues demonstrated that both intravenous and nebulised salbutamol work. Salbutamol is now first-line for the shift step in most paediatric protocols, usually combined with insulin-dextrose for additive effect. [5] [6]
The insulin-dextrose evidence carries an important safety signal. Crnobrnja and colleagues quantified the association between insulin-dextrose treatment and hypoglycaemia in hyperkalaemic patients, finding a substantial incidence that has driven protocols to increase the glucose dose and to mandate frequent monitoring. The lesson, echoed in Moussavi and Koyfman's emergency-clinician review, is that the glucose must be generous and the monitoring relentless. [7] [8]
The most counter-intuitive evidence concerns the ECG itself. Assadi and colleagues found that electrocardiography was unreliable to detect potentially lethal hyperkalaemia in non-dialysis chronic kidney disease patients — the ECG often failed to show the expected changes even at dangerous levels. The practical consequence is dual: use the ECG to escalate urgency when it is abnormal, but never use a normal ECG to reassure yourself out of treating a severely high potassium. [11]
The KDIGO Controversies Conference on Gitelman syndrome (PMID 28003083) provides the international consensus on inherited hypokalaemic tubulopathy management. ANZ resuscitation council guidance, UK NICE and RCPCH pathways, and the Advanced Paediatric Life Support manual give broadly concordant drug-dose conventions for the hyperkalaemia ladder but differ in detail (for example the exact calcium gluconate maximum and the preferred insulin-to-glucose ratio). Always confirm the local paediatric protocol before prescribing, and document the guideline you are following. [10] [2]
The evidence is weakest in three places: the optimal insulin-to-glucose ratio in small children, the role of cation-exchange resins and the newer gastrointestinal potassium binders in acute paediatric treatment, and the threshold for dialysis versus continued medical therapy. These remain largely extrapolated from adult practice, and a specialist nephrology and intensive-care discussion is appropriate in the refractory case. [1] [4]
Exam Pearls
Memorise the hyperkalaemia ECG sequence as a countdown, because the examiner will ask it in order: tall peaked (tented) T waves first, then PR prolongation and loss of the P wave, then widening of the QRS, then the sine wave, and finally ventricular fibrillation or asystole. The hypokalaemia ECG is its mirror image: flattened or inverted T waves, ST depression, a prominent U wave after the T, and an apparent long QT that is really a long QU and a torsades risk. [4] [11]
The calcium-first rule is examiner gold. In hyperkalaemia with ECG changes, calcium gluconate stabilises the membrane within minutes but does not lower the potassium, so a shifting agent must follow. Examiners test the distinction: calcium is the firefighter, salbutamol and insulin-dextrose are the plumbers. Know both doses and both time-to-effect. [2] [5]
The refractory-hypokalaemia answer is always magnesium. Low magnesium disinhibits the ROMK channel and increases distal potassium secretion, so the kidney keeps wasting potassium no matter how much you replace. Fix the magnesium and the potassium will stay. This is one of the most reliable "one right answer" stems in paediatric electrolyte exams. [9] [4]
Finally, the Bartter-versus-Gitelman tie-breaker: Bartter is hypercalciuria, Gitelman is hypocalciuria and hypomagnesaemia. Both are hypokalaemic metabolic alkalosis with normal or low blood pressure; the calcium direction is what separates them at viva. Add that Bartter mimics a loop diuretic (earlier, antenatal, severe) and Gitelman mimics a thiazide (later, adolescent, tetany) and you have answered every question on the topic. [9] [10]
References
- [1]Zieg J; Ghose S; Raina R Electrolyte disorders related emergencies in children. BMC Nephrol, 2024.PMID 39215244
- [2]Rubens M; Kanaris C Fifteen-minute consultation: Emergency management of children presenting with hyperkalaemia. Arch Dis Child Educ Pract Ed, 2022.PMID 34344762
- [3]Masilamani K; van der Voort J The management of acute hyperkalaemia in neonates and children. Arch Dis Child, 2012.PMID 21920871
- [4]Viera AJ; Wouk N Potassium Disorders: Hypokalemia and Hyperkalemia. Am Fam Physician, 2015.PMID 26371733
- [5]Kemper MJ; Harps E; Hellwege HH; Müller-Wiefel DE Effective treatment of acute hyperkalaemia in childhood by short-term infusion of salbutamol. Eur J Pediatr, 1996.PMID 8789768
- [6]McClure RJ; Prasad VK; Brocklebank JT Treatment of hyperkalaemia using intravenous and nebulised salbutamol. Arch Dis Child, 1994.PMID 8129434
- [7]Moussavi K; Fitter S; Gabrielson SW; Koyfman A Management of Hyperkalemia With Insulin and Glucose: Pearls for the Emergency Clinician. J Emerg Med, 2019.PMID 31084947
- [8]Crnobrnja L; Metlapalli M; Jiang C; Govinna M The Association of Insulin-dextrose Treatment with Hypoglycemia in Patients with Hyperkalemia. Sci Rep, 2020.PMID 33328554
- [9]Fulchiero R; Seo-Mayer P Bartter Syndrome and Gitelman Syndrome. Pediatr Clin North Am, 2019.PMID 30454738
- [10]Blanchard A; Bockenhauer D; Bolignano D; Calò LA Gitelman syndrome: consensus and guidance from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference. Kidney Int, 2017.PMID 28003083
- [11]Assadi F; Mazaheri M; Rad EM Electrocardiography is Unreliable to Detect Potential Lethal Hyperkalemia in Patients with Non-dialysis Chronic Kidney Disease. Pediatr Cardiol, 2022.PMID 35389084