Paeds · nephrology-urology-fluids-and-electrolytes
IgA nephropathy and IgA vasculitis nephritis
Also known as IgA nephropathy · Berger disease · IgA vasculitis · Henoch-Schonlein purpura · IgA vasculitis nephritis
Fellowship guide to IgA nephropathy and IgA vasculitis nephritis: the most common primary glomerulonephritis worldwide, defined by dominant mesangial IgA deposition and classified by the Oxford MEST-C score; IgA vasculitis as the most common childhood vasculitis whose renal lesion is histologically identical; the shared multi-hit pathogenesis centred on galactose-deficient IgA1; the distinction of synpharyngitic haematuria from post-infectious glomerulonephritis by normal C3 and a short latency; and the treatment ladder from ACE inhibitor or ARB supportive care to corticosteroids, targeted-release budesonide, and immunosuppression for crescentic disease.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
Overview & Definition
A teenager who notices dark, smoky urine the morning after a sore throat, or a five-year-old who arrives with a purpuric rash on the shins and blood in the urine, both have the same underlying problem: immune complexes built from a defective antibody called IgA are settling in the filtering surface of the kidney and injuring it. When that process is confined to the kidney it is called IgA nephropathy, the most common primary glomerulonephritis in the world. When the same IgA complexes also land in the skin, gut, and joints it is called IgA vasculitis (the modern name for Henoch-Schonlein purpura), and when it scars the kidney it is called IgA vasculitis nephritis. The renal lesion in the two conditions is histologically identical. [8]
The link between the two is the central teaching point. Both are driven by a faulty IgA molecule whose sugar chains are incomplete — galactose-deficient IgA1 (Gd-IgA1). The body makes antibodies against this abnormal molecule, the resulting immune complexes circulate, and they lodge wherever they can, above all in the glomerular mesangium. Whether the child presents with isolated kidney disease or with a full systemic vasculitis depends on where those complexes deposit, not on a different mechanism. [11]
Classification
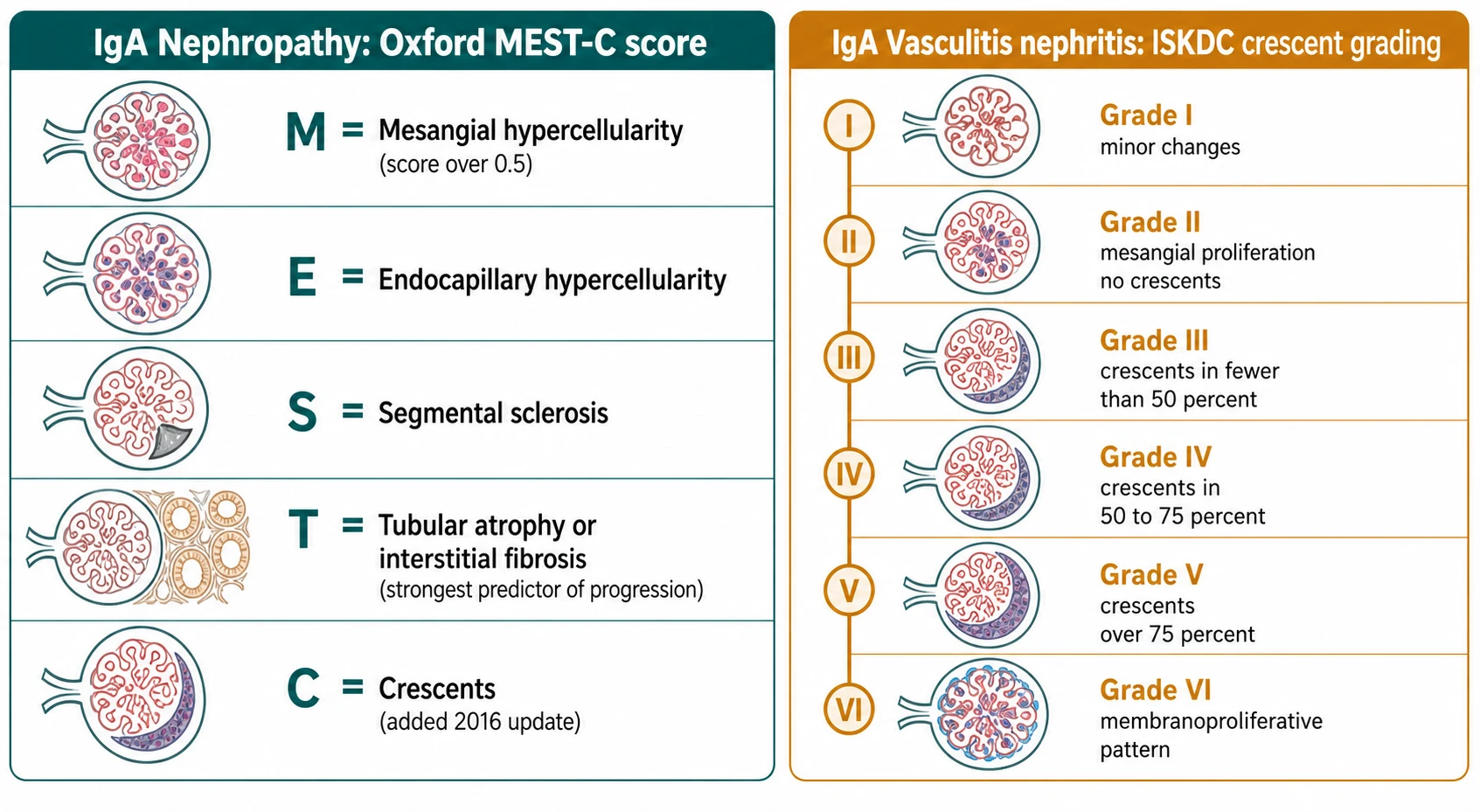
Classification answers two separate questions: how badly scarred is the kidney already, and how active is the inflammation now. The Oxford MEST-C score tackles the first for IgA nephropathy. Each letter is a scored biopsy feature, and together they predict how likely the kidney is to fail over the coming years. M is mesangial hypercellularity, E is endocapillary hypercellularity, S is segmental sclerosis, T is tubular atrophy or interstitial fibrosis, and C is crescents. Of these, T is the strongest predictor of long-term loss of function, and C — which was added in the 2016 update after a multicentre study confirmed that crescents carry independent prognostic weight — flags active, potentially treatable inflammation. [2]
[2]The crescent question is important enough that it changed the classification. The 2017 multicentre study by Haas and colleagues showed that crescents predict a faster loss of kidney function independently of the other Oxford features, which is why they were formalised as the C score in the 2016 update. Before that, crescents were noted but not weighted. [3]
IgA vasculitis nephritis is classified differently, by the older ISKDC system, because the key question there is how many glomeruli are already being destroyed by crescents. The grade runs from I (minor changes) through II (mesangial proliferation with no crescents), III (crescents in fewer than 50 percent of glomeruli), IV (crescents in 50 to 75 percent), V (crescents in over 75 percent), to VI (a membranoproliferative pattern). The systemic disease itself is classified by the EULAR, PRINTO and PRES Ankara 2008 criteria, which require palpable purpura or petechiae plus at least one of diffuse abdominal pain, arthritis or arthralgia, renal involvement, or histopathology showing IgA deposition. [6]
Epidemiology & Risk Factors
IgA nephropathy is the most common primary glomerulonephritis worldwide, with a biopsy-detected incidence of approximately 25 to 40 per million population per year. The true prevalence is higher because many people with mild microscopic haematuria are never biopsied. It is uncommon before the age of five, becomes detectable from late childhood, and peaks in the second and third decades, with a male predominance and notably higher detection in East Asian populations, which reflects both genuine prevalence and a lower biopsy threshold in those health systems. [8]
IgA vasculitis is the most common vasculitis of childhood, with an incidence of approximately 10 to 20 per 100,000 children per year. It peaks at age four to six years, is slightly more common in boys, and clusters in the winter months after upper respiratory infections. Renal involvement — IgA vasculitis nephritis — develops in approximately 20 to 50 percent of children, almost always within the first four weeks of the rash, and is the feature that determines long-term outcome. [7]
The strongest risk markers for a poor renal outcome are clinical, not demographic. Sustained proteinuria, especially over 1 g per day, is the single most powerful predictor of progression, followed by hypertension at presentation, a reduced estimated GFR at biopsy, and adverse Oxford features — above all the T score and the presence of crescents. The updated International IgA Nephropathy Prediction Tool, validated in children, integrates these into a single estimated risk of losing half of kidney function over five years. [10]
Pathophysiology
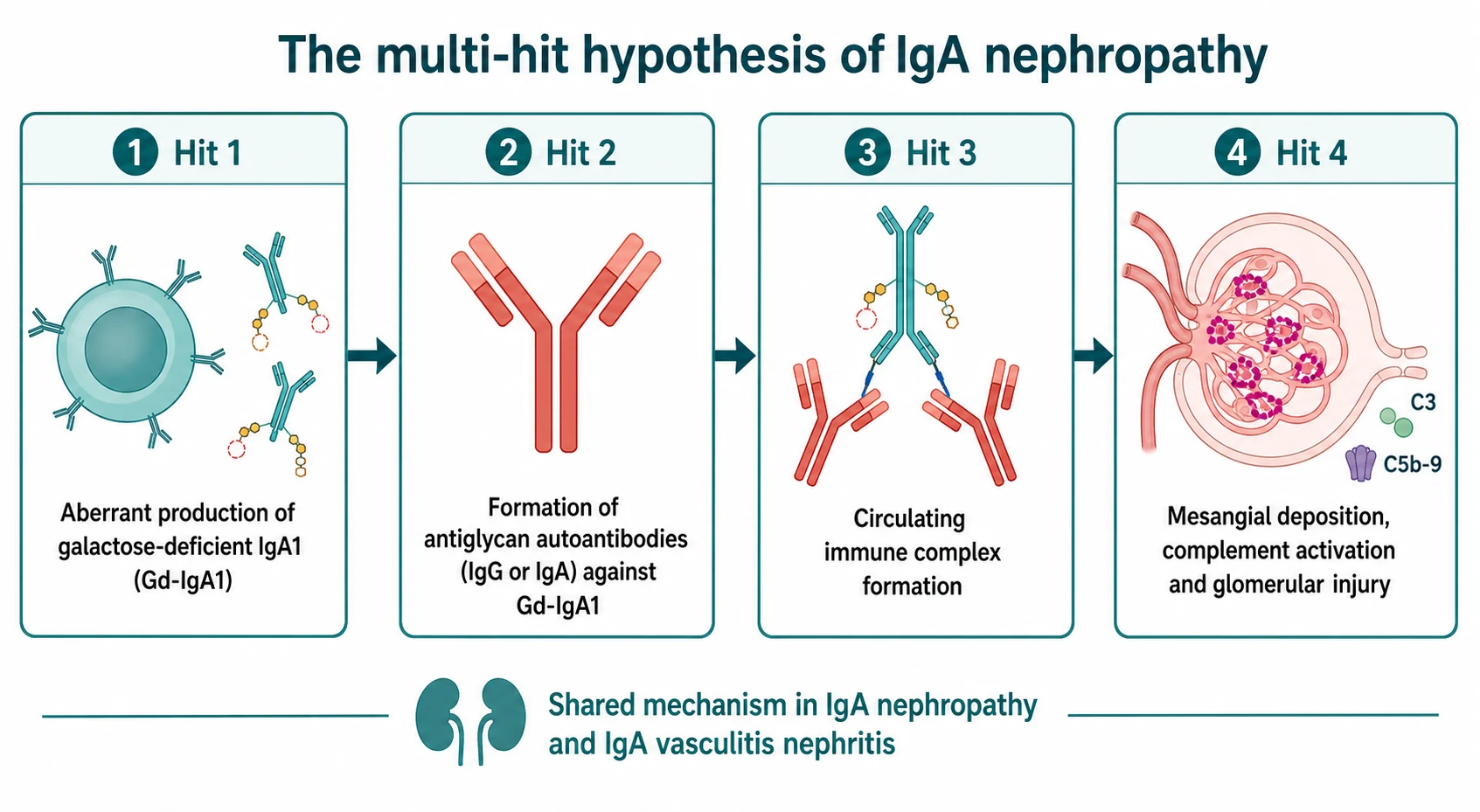
The mechanism is best understood as a cascade of four hits, and grasping the cascade matters because each hit is now a drug target. The story begins with a slightly mis-made antibody. The hinge region of IgA1 carries sugar chains, and in these patients the O-linked sugars are incompletely built — the molecule is galactose-deficient IgA1, or Gd-IgA1. [11]
Hit two is that the immune system does not tolerate this abnormal molecule. It makes autoantibodies, mainly of the IgG and IgA classes, that recognise the exposed sugar residues on Gd-IgA1 as foreign. Suzuki and colleagues showed that the same antiglycan autoantibodies are present in IgA vasculitis nephritis as in IgA nephropathy, which is the molecular proof that the two diseases are one process. [11]
MEST
Hit three is that the Gd-IgA1 and its autoantibodies bind together to form large circulating immune complexes. These are large and poorly cleared by the liver, so they persist in the blood. Hit four is deposition: the complexes lodge in the glomerular mesangium, where they activate mesangial cells and switch on complement, mainly through the alternative and lectin pathways. Complement activation drives generation of C3 and the membrane attack complex C5b to 9, the mesangial cells proliferate and overproduce matrix, and the result is the haematuria, proteinuria, and slowly progressive scarring that define the disease. In IgA vasculitis the identical complexes also deposit in skin, gut, and joint capillaries, producing the palpable purpura, abdominal pain, and arthritis. [8]
The clinically useful consequence of this mechanism is the drug pipeline. Reducing the production of Gd-IgA1 (targeted-release budesonide), blocking the autoantibodies (B-cell modulators such as atacicept and sibeprenlimab), and inhibiting complement (avacopan, iptacopan) all attack a specific hit in the cascade. [5]
Clinical Presentation
The classic presentation of IgA nephropathy is painless macroscopic haematuria that appears at the same time as a mucosal infection — a sore throat or a gastrointestinal illness — and the timing is the discriminator examiners love. The haematuria is synpharyngitic, meaning it appears within one to two days of the infection, whereas post-infectious glomerulonephritis waits one to two weeks. This single distinction, made at the bedside from the history, is the most reliable way to separate the two before any blood test returns. [8]
Many children never see blood in the urine and are detected instead when screening finds persistent microscopic haematuria with or without proteinuria. A smaller group presents with a full nephritic syndrome — haematuria, hypertension, oedema, and renal impairment — or with nephrotic-range proteinuria. The rare but dangerous presentation is rapidly progressive glomerulonephritis, in which kidney function declines over weeks and biopsy shows many crescents. [1]
IgA vasculitis presents with a tetrad, and the rash is the anchor. The mandatory feature is palpable purpura over the lower limbs and buttocks, typically sparing the trunk and upper limbs. The other three are arthritis or arthralgia (especially of the ankles and knees), diffuse colicky abdominal pain, and renal involvement. Abdominal pain occasionally precedes the rash by days and can mimic a surgical abdomen or intussusception. Renal involvement shows as microscopic or macroscopic haematuria with variable proteinuria, and it declares itself within the first four weeks of the rash in the great majority of children who develop it. [6]
Differential Diagnosis
For a child with macroscopic haematuria, the first fork is between IgA nephropathy and post-infectious glomerulonephritis. Both produce dark urine after an infection, but post-infectious disease has a one to two week latent interval, an elevated ASO titre, and a low C3. IgA nephropathy has a one to two day interval and a normal C3. If the C3 is low, think post-infectious disease, lupus nephritis, or membranoproliferative glomerulonephritis — not IgA nephropathy. [8]
[8]For persistent isolated microscopic haematuria, the key differentials are thin basement membrane nephropathy (a family history of haematuria and normal C3, with thin glomerular basement membranes on electron microscopy) and Alport syndrome (haematuria with sensorineural deafness and ocular signs, usually X-linked). Urological causes — stones, hypercalciuria, and rarely tumour — must be excluded when the haematuria is painless and episodic, particularly when it is non-glomerular (isomorphic red cells, no proteinuria). [1]
For IgA vasculitis, the purpura must be distinguished from the purpura of sepsis and meningococcaemia (which is rapidly progressive, tender, and accompanied by sepsis physiology), immune thrombocytopenic purpura (isolated low platelets, no arthritis or abdominal pain), and other small-vessel vasculitides such as ANCA-associated vasculitis and urticarial vasculitis. The IgA deposition on biopsy and the clinical tetrad are the distinguishing features. When crescentic IgA disease is found, the histological differential is ANCA-associated pauci-immune glomerulonephritis (little or no immune deposition) and anti-glomerular basement membrane disease (linear IgG deposition). [7]
Clinical & Bedside Assessment
The history should fix the timing of the haematuria against any preceding infection, because that single relationship is the most discriminating bedside clue. Ask how many days after the sore throat or gastroenteritis the urine turned dark. Ask about recent medications, a family history of renal disease or deafness (Alport), and, for suspected IgA vasculitis, the full tetrad — rash, abdominal pain, joint pain, and urine colour. In boys, ask about testicular pain or swelling. [6]
Examination measures blood pressure, because hypertension is both a presenting feature and a marker of severity. Look for oedema, which indicates significant proteinuria or nephrotic syndrome. For suspected IgA vasculitis, expose the lower limbs and buttocks to confirm palpable non-blanching purpura, examine the joints for effusion and tenderness, and assess the abdomen for tenderness, guarding, or a palpable mass that might signal intussusception. Examine the scrotum in boys. A urinalysis at the bedside confirms haematuria and semi-quantifies proteinuria. [7]
The synthesis is straightforward once the pieces are in place. A nephritic presentation confined to the kidney with normal C3 and synpharyngitic timing points to IgA nephropathy. The same nephritic picture within a tetrad of palpable purpura, arthritis, and abdominal pain in a young child points to IgA vasculitis nephritis. The red flag in either is sustained heavy proteinuria, hypertension, falling kidney function, or a biopsy with many crescents — these move the child from routine follow-up into the high-risk pathway. [8]
Investigations
Urinalysis is the first and most informative test. Glomerular haematuria shows dysmorphic red cells and red cell casts, and the presence of proteinuria on dipstick must be quantified with a spot urine protein-to-creatinine ratio. Proteinuria is the single most important prognostic and monitoring variable, so it is measured at every visit. [10]
Blood tests establish the renal function and the complement pattern. Serum creatinine and the estimated GFR quantify impairment, serum albumin falls in nephrotic presentations, and C3 and C4 are characteristically normal in both IgA nephropathy and IgA vasculitis — a normal C3 is what separates IgA disease from post-infectious glomerulonephritis, lupus, and membranoproliferative disease. Measure ASO and anti-DNase B when post-infectious disease is plausible, ANA and dsDNA to exclude lupus, and ANCA to exclude ANCA-associated vasculitis in atypical or crescentic cases. [8]
[8]The renal biopsy is the gold standard and is required whenever the diagnosis is in doubt or the clinical picture signals risk. Indications include proteinuria over 1 g per day, nephrotic syndrome, sustained renal impairment, and rapidly progressive disease. Light microscopy shows mesangial proliferation to variable degrees, immunofluorescence shows dominant or co-dominant mesangial IgA deposition (with IgG and C3), and electron microscopy shows electron-dense deposits in the mesangium. The biopsy yields the Oxford MEST-C score for IgA nephropathy and the ISKDC grade for IgA vasculitis nephritis, both of which direct therapy and prognosis. The International IgA Nephropathy Prediction Tool then combines the clinical features with MEST-C to estimate the five-year risk of losing half of kidney function, which is now central to deciding whether to start disease-specific therapy. [10]
Management — Resuscitation
Most children with IgA nephropathy or IgA vasculitis nephritis are haemodynamically stable and need no acute resuscitation. The exceptions are the medical emergencies within the spectrum. Rapidly progressive crescentic disease presents with a falling estimated GFR over days to weeks and requires urgent renal biopsy and immediate combination immunosuppression with high-dose corticosteroids and cyclophosphamide, mirroring the approach to other crescentic glomerulonephritides. Malignant hypertension requires controlled blood-pressure lowering, and nephrotic syndrome with significant oedema may need judicious diuretics and assessment for thromboembolism risk. [8]
In IgA vasculitis the acute issues are often extra-renal. Severe abdominal pain and gastrointestinal bleeding are treated with systemic corticosteroids, prednisolone 1 to 2 mg per kg per day up to a maximum of 60 to 80 mg, which shortens the duration of abdominal symptoms. Intussusception — classically ileocolic — presents with colicky pain, vomiting, and currant-jelly stool and requires urgent imaging (ultrasound with a target sign) and surgical or enema reduction. Steroids do not prevent IgA vasculitis nephritis when given for abdominal or joint disease, so they should not be prescribed for that purpose alone. [6]
Management — Definitive & Stepwise
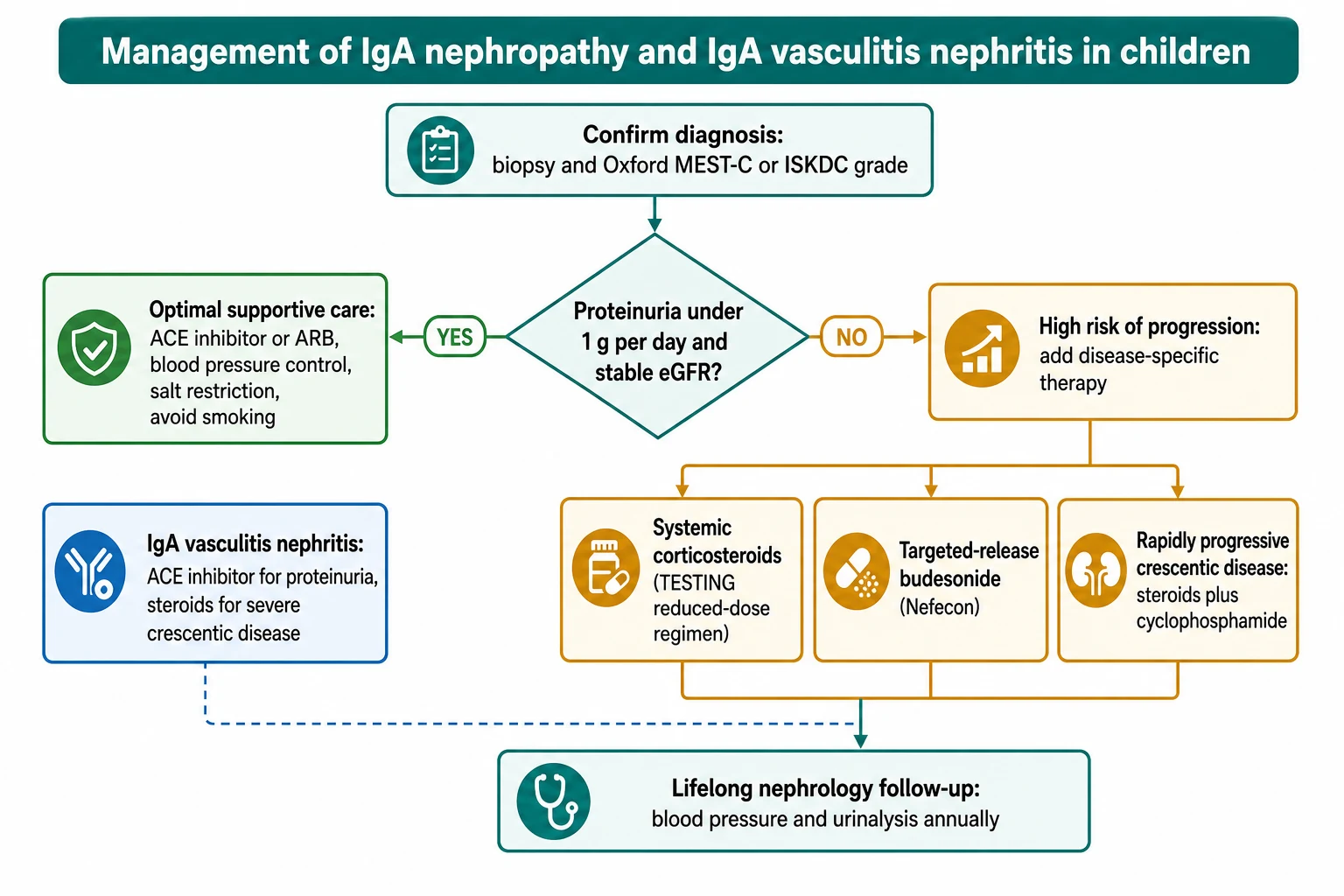
[8]The foundation for every patient, at every stage, is optimal supportive care, and it is the step most often under-prescribed. A renin-angiotensin system blocker — an ACE inhibitor or an angiotensin receptor blocker — is first-line because it lowers intraglomerular pressure, reduces proteinuria, and slows progression independent of its blood-pressure effect. The agent is titrated to drive proteinuria below 1 g per day while maintaining blood pressure at or below age-appropriate targets. Sodium restriction, weight management, exercise, and smoking avoidance in adolescents are reinforced at every visit, and the regimen is continued lifelong. [8]
Optimise supportive care first
Risk-stratify with the prediction tool
Add disease-specific therapy if high risk
Treat crescentic disease aggressively
Commit to lifelong surveillance
For the high-risk minority — those with persistent proteinuria over 0.75 to 1 g per day despite three to six months of optimised supportive care — disease-specific therapy is considered. Corticosteroids reduce the risk of losing half of kidney function. The TESTING trial established the benefit but also the harm: the full-dose regimen (prednisolone 0.8 to 1 mg per kg per day) carried a significant risk of serious infection, including two deaths from infection in the early part of the trial. The practical consequence is that a reduced-dose regimen — intravenous methylprednisolone pulses followed by lower-dose oral prednisolone — is now preferred, with tuberculin screening and infectious prophylaxis before starting. [9]
Systemic corticosteroids (IgA nephropathy)
Dose
Reduced-dose regimen: methylprednisolone 0.5 to 1 g intravenously daily for 3 days at months 0, 2 and 4, then oral prednisolone 0.4 to 0.6 mg per kg every other day. The historical full-dose TESTING regimen was prednisolone 0.8 to 1 mg per kg per day for 8 weeks then tapering, now avoided because of infection risk.
Targeted-release budesonide (Nefecon) is the steroid-sparing mucosal option. It delivers budesonide to the distal ileum — the Peyer patches where Gd-IgA1 is produced — and the NefIgArd trial showed that 16 mg per day reduces proteinuria while the patient takes it. It avoids much of the systemic steroid exposure but is not yet first-line in children, and its effect on long-term kidney failure outcomes continues to be evaluated. [5]
Targeted-release budesonide (Nefecon)
Dose
16 mg orally once daily for 9 months
IgA vasculitis nephritis is managed on a parallel ladder. For mild isolated haematuria, supportive care and close monitoring suffice. ACE inhibition is used for proteinuria. Corticosteroids plus immunosuppression (cyclophosphamide, mycophenolate, or calcineurin inhibitors in selected cases) are reserved for severe crescentic or nephrotic disease, because the evidence that steroids prevent IgA vasculitis nephritis when given at rash onset is weak. The 2024 multicentre analysis by Barbour and colleagues confirmed that the histological and clinical factors predicting poor kidney outcomes in IgA vasculitis nephritis mirror those of IgA nephropathy — heavy proteinuria, impaired function at biopsy, and a high crescent count. [7]
Specific Subtypes & Scenarios
The isolated microscopic haematuria phenotype, often an asymptomatic adolescent boy found on screening, carries a relatively favourable prognosis in the short term but demands lifelong follow-up because progression can appear decades later. This group is managed with annual blood pressure and urinalysis and education about the importance of reporting infection-associated dark urine. [1]
The recurrent macroscopic haematuria phenotype, triggered by each mucosal infection, is common in younger patients. Episodes are managed supportively, and the trigger — recurrent tonsillitis, in particular — may warrant ear, nose and throat assessment, although tonsillectimony as a disease-modifying therapy has largely been abandoned outside Japan because the long-term benefit is unproven. [8]
The slowly progressive proteinuric phenotype is the group most often considered for disease-specific therapy. These patients have persistent proteinuria despite supportive care and are the population in whom the TESTING and NefIgArd evidence applies directly. The International IgA Nephropathy Prediction Tool is the decision aid that quantifies their risk and frames the conversation about steroids, budesonide, and the newer agents. [10]
The rapidly progressive crescentic phenotype, with crescents in over 50 percent of glomeruli and a falling estimated GFR, is the medical emergency within the spectrum and is treated with high-dose corticosteroids and cyclophosphamide, occasionally with plasma exchange. Recurrence of IgA nephropathy after renal transplantation is the most common form of recurrent glomerulonephritis in a graft; it usually appears slowly and rarely causes immediate graft loss, but it is a reason for ongoing vigilance and, increasingly, for considering budesonide or complement-targeted agents post-transplant. [8]
Complications & Pitfalls
The central long-term complication is progression to end-stage kidney disease. Approximately 20 to 40 percent of patients progress over 20 to 30 years, and the risk is concentrated in those with sustained proteinuria over 1 g per day, hypertension, impaired function at diagnosis, and adverse Oxford T and C scores. Nephrotic syndrome, malignant hypertension, and crescentic rapidly progressive disease are the acute and subacute complications. [1]
In IgA vasculitis the complications are often extra-renal and can be surgical. Intussusception, classically ileocolic, presents with colicky abdominal pain, vomiting, and currant-jelly stool; severe gastrointestinal bleeding can occur; and testicular pain and scrotal swelling can mimic torsion. Persistent renal impairment after an episode of nephritis is the renal concern. [6]
Other avoidable errors include using full-dose steroids without screening for tuberculosis and providing pneumococcal and influenza vaccination first, mistaking synpharyngitic timing for post-infectious disease and looking in vain for a low C3, and discharging a patient after an apparent recovery without arranging the lifelong surveillance that IgA disease demands. [8]
Prognosis & Disposition
IgA nephropathy is a chronic, slowly progressive disease. Children generally fare better than adults, but they are not exempt from progression, and because they carry the disease for many more decades, the cumulative risk is substantial. Approximately 20 to 40 percent progress to end-stage kidney disease over 20 to 30 years. The predictors are consistent across studies: sustained proteinuria over 1 g per day, hypertension, reduced estimated GFR at diagnosis, and the Oxford T and C scores. The International IgA Nephropathy Prediction Tool, validated in children one and two years after biopsy, turns these into an individual risk estimate that should be shared with the family. [10]
Severity
Low-risk IgA nephropathy
Isolated microscopic haematuria or only minor proteinuria, normal blood pressure and eGFR, favourable Oxford score. Prognosis excellent in the short term; needs lifelong surveillance.
Severity
Moderate-risk IgA nephropathy
Persistent moderate proteinuria, controlled blood pressure, stable eGFR. Manage with optimised supportive care; consider disease-specific therapy if proteinuria rises.
Severity
High-risk or crescentic IgA disease
Heavy persistent proteinuria despite supportive care, hypertension, falling eGFR, or crescents on biopsy. Requires disease-specific therapy and specialist centre involvement.
IgA vasculitis nephritis has a broadly favourable prognosis in children, with complete recovery in most. The minority with heavy proteinuria, nephrotic syndrome, or crescents in over 50 percent of glomeruli have a worse long-term renal outlook, and these are the patients who need careful follow-up and sometimes aggressive therapy. The 2024 multicentre analysis confirmed that the same histological and clinical factors predict outcome in IgA vasculitis nephritis as in IgA nephropathy. [7]
Disposition is outpatient nephrology follow-up for the majority. Admission is reserved for rapidly progressive disease, nephrotic syndrome requiring biopsy, severe hypertension, and the surgical complications of IgA vasculitis. At discharge from any acute episode, the single most important safety-net is the commitment to lifelong annual blood pressure and urinalysis, because the renal consequences of IgA disease can emerge years or decades after an apparently benign presentation. [8]
Special Populations
Children and adolescents are the core of paediatric practice. IgA nephropathy is uncommon before age five and rises through adolescence, while IgA vasculitis peaks at age four to six years, so the age of the child in front of you strongly biases the likely diagnosis. Drug doses must be weight-based throughout childhood, and the growth and bone effects of corticosteroids weigh more heavily in a child than in an adult. [9]
Pregnancy in a young woman with IgA nephropathy is a high-stakes scenario. Pre-existing hypertension or proteinuria substantially increases the risks of pre-eclampsia, fetal loss, and a permanent decline in kidney function, so pre-conception counselling, optimisation of blood pressure and proteinuria with a pregnancy-safe regimen, and risk stratification are essential. Angiotensin blockers must be stopped before conception because of fetal renal toxicity. [8]
Recurrence of IgA nephropathy after renal transplantation is the most common form of recurrent glomerulonephritis in a graft. It usually appears slowly and rarely causes immediate graft loss, but it influences the choice of living-related versus deceased donor and increasingly informs the decision to use budesonide or complement-targeted agents post-transplant. A related donor must be counselled about the small risk of sharing the predisposition. [8]
Indigenous and East Asian populations show a higher prevalence of IgA nephropathy and sometimes a more aggressive course, and socioeconomic disadvantage can delay diagnosis by limiting access to urinalysis, biopsy, and specialist therapy. Equity of access to nephrology and to the newer targeted therapies is an explicit consideration in the KDIGO 2025 guideline. [8]
Evidence, Guidelines & Regional Differences
The KDIGO 2025 Clinical Practice Guideline for the Management of IgA Nephropathy and IgA Vasculitis is the current international reference, and its publication as a standalone IgA guideline — rather than a chapter within the general glomerular disease document — reflects how rapidly the field has moved. It codifies optimal supportive care as the foundation, formalises the place of corticosteroids and targeted-release budesonide, and anticipates the complement- and B-cell-targeted agents that are now entering practice. [8]
TESTING — corticosteroids in IgA nephropathy
Key finding
Corticosteroids reduced the risk of a 50 percent decline in estimated GFR or end-stage kidney disease compared with supportive care alone, but the full-dose regimen (prednisolone 0.8 to 1 mg per kg per day) caused excess serious infections, including fatal infection, prompting early modification to a reduced-dose regimen.
Practice change
Steroids work but are toxic at full dose; a reduced-dose regimen with infectious screening and prophylaxis is the modern standard.
NefIgArd part A — targeted-release budesonide
Key finding
Targeted-release budesonide 16 mg per day for 9 months significantly reduced proteinuria compared with placebo in patients already on optimised supportive care, confirming a steroid-sparing effect from mucosal IgA modulation.
Practice change
Budesonide is a valid steroid-sparing option for proteinuric IgA nephropathy at high risk of progression, with a favourable systemic safety profile.
The Oxford MEST-C classification (2009, updated 2016) and the International IgA Nephropathy Prediction Tool provide the evidence base for risk stratification and are now the shared language between pathologist and clinician. The 2016 update formalised crescents as the C score after the Haas multicentre study, and the 2024 paediatric validation of the prediction tool extended its use to children one and two years after biopsy. [3]
In Australia and New Zealand, the approach to IgA nephropathy and IgA vasculitis nephritis follows the KDIGO 2025 guideline through the paediatric nephrology services in each state, with access to renal biopsy, the newer targeted therapies through specialist centres, and long-term registries that track progression. IgA vasculitis management in general paediatrics emphasises supportive care and steroid use confined to severe gastrointestinal disease, in line with international consensus. [8]
Controversies persist. The role of tonsillectomy has largely been abandoned outside Japan because long-term benefit is unproven. The choice between corticosteroids, targeted-release budesonide, and the newer complement- and B-cell-targeted agents (sibeprenlimab, atacicept, atrasentan, iptacopan) is evolving rapidly, and access to these expensive biologics varies by region. Whether mild childhood IgA vasculitis nephritis is over-treated with immunosuppression remains a live debate. Regional differences exist in biopsy thresholds, steroid regimens, and funding of newer therapies. [7]
Exam Pearls
IgA nephropathy is the most common primary glomerulonephritis worldwide. Its classic presentation is synpharyngitic macroscopic haematuria — dark urine within one to two days of an upper respiratory infection — and the normal C3 and short latency distinguish it from post-infectious glomerulonephritis, which waits one to two weeks and has a low C3. The Oxford MEST-C score classifies the biopsy: M, E, S, T, and C, with T (tubular atrophy or interstitial fibrosis) the strongest predictor of progression and C (crescents) added in the 2016 update. [2]
IgA vasculitis is the most common childhood vasculitis, peaking at age four to six years, and is classified by the EULAR, PRINTO and PRES Ankara 2008 criteria: mandatory palpable purpura or petechiae plus at least one of diffuse abdominal pain, arthritis or arthralgia, renal involvement, or IgA deposition on biopsy. Renal involvement appears in 20 to 50 percent of children, almost always within four weeks of the rash, and the kidney lesion is histologically identical to IgA nephropathy. [6]
The shared pathogenesis is the multi-hit hypothesis: galactose-deficient IgA1 is produced, antiglycan autoantibodies form against it, circulating immune complexes are generated, and these deposit in the mesangium to activate complement (C3 and the membrane attack complex C5b to 9) and injure the glomerulus. Treatment begins with an ACE inhibitor or ARB for everyone, adds reduced-dose corticosteroids or targeted-release budesonide for the high-risk proteinuric minority, and reserves steroids plus cyclophosphamide for rapidly progressive crescentic disease. All patients need lifelong nephrology follow-up. Recurrence after renal transplant is the most common recurrent glomerulonephritis in a graft. [8]
References
- [1]Working Group of the International IgA Nephropathy Network and the Renal Pathology Society, Cattran DC, Coppo R, Cook HT, Feehally J, Roberts IS The Oxford classification of IgA nephropathy: rationale, clinicopathological correlations, and classification. Kidney Int, 2009.PMID 19571791
- [2]Trimarchi H, Barratt J, Cattran DC, Cook HT, Coppo R, Haas M, et al Oxford Classification of IgA nephropathy 2016: an update from the IgA Nephropathy Classification Working Group. Kidney Int, 2017.PMID 28341274
- [3]Haas M, Verhave JC, Liu ZH, Alpers CE, Barratt J, Becker JU, et al A Multicenter Study of the Predictive Value of Crescents in IgA Nephropathy. J Am Soc Nephrol, 2017.PMID 27612994
- [4]Lv J, Xu D, Perkovic V, Ma X, Johnson DW, Woodward M, et al Corticosteroid therapy in IgA nephropathy. J Am Soc Nephrol, 2012.PMID 22539830
- [5]Barratt J, Lafayette R, Kristensen J, Stone A, Cattran D, Floege J, et al Results from part A of the multi-center, double-blind, randomized, placebo-controlled NefIgArd trial, which evaluated targeted-release formulation of budesonide for the treatment of primary immunoglobulin A nephropathy. Kidney Int, 2023.PMID 36270561
- [6]Ozen S, Pistorio A, Iusan SM, Bakkaloglu A, Herlin T, Brik R, et al EULAR/PRINTO/PRES criteria for Henoch-Schonlein purpura, childhood polyarteritis nodosa, childhood Wegener granulomatosis and childhood Takayasu arteritis: Ankara 2008. Part II: Final classification criteria. Ann Rheum Dis, 2010.PMID 20413568
- [7]Barbour SJ, Coppo R, Er L, Pillebout E, Russo ML, Alpers CE, et al Histologic and Clinical Factors Associated with Kidney Outcomes in IgA Vasculitis Nephritis. Clin J Am Soc Nephrol, 2024.PMID 38261310
- [8]Floege J, Barratt J, Cook HT, Noronha IL, Reich HN, Suzuki Y, et al Executive summary of the KDIGO 2025 Clinical Practice Guideline for the Management of Immunoglobulin A Nephropathy (IgAN) and Immunoglobulin A Vasculitis (IgAV). Kidney Int, 2025.PMID 40975525
- [9]Zhang Y, Hu YT, Lv JC, Zhang H Corticosteroids in the treatment of IgA nephropathy: lessons from the TESTING trial. Pediatr Nephrol, 2023.PMID 36881171
- [10]Barbour SJ, Coppo R, Er L, Russo ML, Liu ZH, Ding J, et al Application of the updated International IgA Nephropathy Prediction Tool in children one or two years post-biopsy. Kidney Int, 2024.PMID 39094695
- [11]Suzuki H, Moldoveanu Z, Julian BA, Wyatt RJ, Novak J Autoantibodies Specific for Galactose-Deficient IgA1 in IgA Vasculitis With Nephritis. Kidney Int Rep, 2019.PMID 31844808