Paeds · nephrology-urology-fluids-and-electrolytes
Inherited tubulopathies
Also known as Inherited tubulopathies · Bartter syndrome · Gitelman syndrome · Fanconi syndrome · Liddle syndrome · Salt-losing tubulopathy · Congenital chloride-losing diarrhoea mimic · Pseudoaldosteronism
Fellowship guide to the four archetypal inherited tubulopathies of childhood: Bartter syndrome (thick ascending limb, hypercalciuria, antenatal polyhydramnios), Gitelman syndrome (distal convoluted tubule, hypomagnesaemia and hypocalciuria), Fanconi syndrome (generalised proximal tubular failure with glycosuria, phosphaturia and proximal RTA), and Liddle syndrome (collecting duct ENaC gain-of-function with hypertension and suppressed renin and aldosterone). The blood pressure and the renin/aldosterone axis separate Liddle from the salt-losing trio, and the urine calcium and serum magnesium separate Bartter from Gitelman.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
A child who loses salt cannot hold their blood pressure, their potassium or their growth together for long, and the inherited tubulopathies are the prototypic salt-losing and potassium-shifting disorders of childhood. Each maps onto one segment of the nephron, and each segment has a characteristic transporter whose inherited failure produces a predictable biochemistry. Bartter is the thick ascending limb defect, the loop-diuretic equivalent, with hypercalciuria and nephrocalcinosis. Gitelman is the distal convoluted tubule defect, the thiazide equivalent, with hypocalciuria and hypomagnesaemia. Liddle is the collecting duct epithelial sodium channel gone into overdrive, and it is the one that raises the blood pressure. Fanconi is different in kind, a wholesale failure of the proximal tubule that loses glucose, phosphate, amino acids and bicarbonate and causes a metabolic acidosis rather than an alkalosis. [5] [6]
The bedside power of the topic comes from two branching questions. First, is the blood pressure high or normal and low, because a hypokalaemic alkalosis with hypertension points straight to Liddle while the same biochemistry with a normal or low blood pressure points to Bartter or Gitelman. Second, where do the renin and aldosterone sit, because Liddle suppresses both, primary hyperaldosteronism raises the aldosterone but not the renin, and Bartter and Gitelman raise both. Nail this two-step branch and you have answered the spine of every tubulopathy viva before you reach the genes. [7] [10]
Overview & Definition
An inherited tubulopathy is a monogenic disorder in which a single transporter or channel along the nephron fails, producing a predictable pattern of electrolyte and fluid loss. The defining biochemistry is determined by which segment broke. The salt-losing syndromes, Bartter and Gitelman, and the sodium-retaining syndrome, Liddle, all produce a hypokalaemic metabolic alkalosis. Fanconi syndrome stands apart, because a generalised proximal tubular failure wastes bicarbonate and produces a normal anion gap metabolic acidosis with glycosuria, phosphaturia and aminoaciduria. [5] [6]
The four syndromes are best held against the segment of nephron each one breaks. Bartter syndrome is the inherited failure of salt reabsorption in the thick ascending limb of the loop of Henle, and it mimics a loop diuretic given for life. Gitelman syndrome is the failure of the thiazide-sensitive sodium chloride cotransporter in the distal convoluted tubule, and it mimics a thiazide diuretic. Liddle syndrome is a gain-of-function of the epithelial sodium channel in the collecting duct, so the kidney retains sodium and loses potassium without any aldosterone driving it. Fanconi syndrome is a diffuse failure of proximal tubular reabsorption, so everything the proximal tubule normally reclaims, glucose, phosphate, amino acids and bicarbonate, is spilled into the urine. [3] [4]
What unifies the topic clinically is that all four present in childhood with electrolyte disturbance and growth failure, and that each has a treatment aimed at the segment that broke. The magnesium and potassium-sparing diuretic strategy suits Gitelman, the prostaglandin synthesis inhibitor strategy suits antenatal Bartter, the amiloride strategy suits Liddle, and the bicarbonate, phosphate and cause-specific strategy suits Fanconi. The examiner rewards the candidate who can move fluently from the bedside biochemistry to the segment, the gene and the treatment, and that fluency is what this page builds. [1] [2]
Classification
The classification rests on two axes, the segment of nephron that failed and the direction of the blood pressure and the renin and aldosterone axis. The figure lays the four syndromes onto their nephron sites with their genes, their biochemistry and their discriminator features, so the type can be inferred at the bedside from a chemistry panel, a blood pressure and a renin and aldosterone. [6] [8]
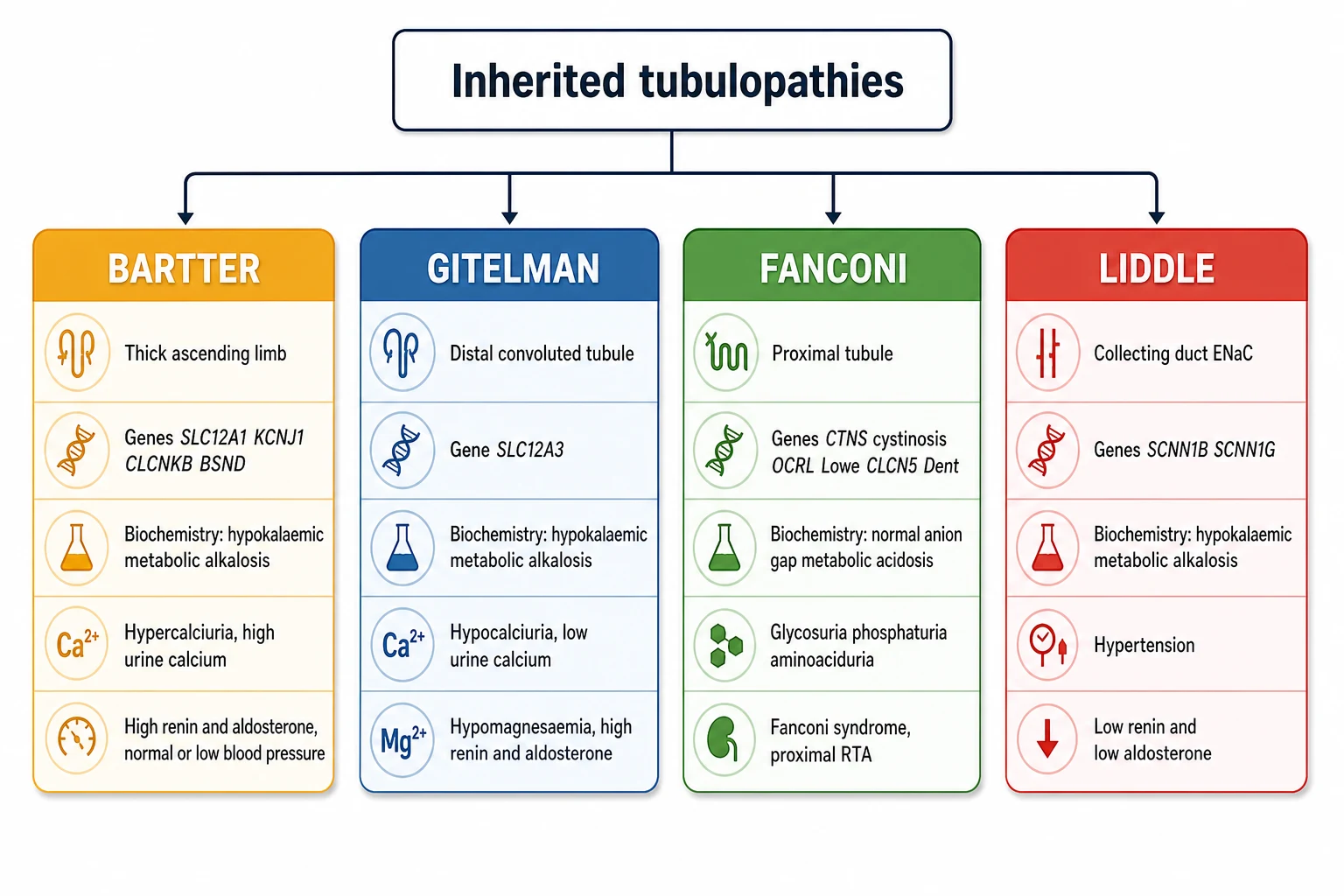
Bartter syndrome occupies the thick ascending limb. The defective transporters are the sodium-potassium-2-chloride cotransporter NKCC2 encoded by SLC12A1, the apical potassium channel ROMK encoded by KCNJ1, and the basolateral chloride channel ClC-Kb encoded by CLCNKB. A fourth subtype involves barttin, encoded by BSND, the shared subunit of ClC-Ka and ClC-Kb, and it carries sensorineural deafness because the same channel operates in the inner ear. Bartter is autosomal recessive, it presents antenatally or in early infancy, and its calling cards are hypercalciuria and nephrocalcinosis. [1] [5]
Gitelman syndrome occupies the distal convoluted tubule. The defective transporter is the thiazide-sensitive sodium chloride cotransporter NCC encoded by SLC12A3, and the inheritance is autosomal recessive. Gitelman presents later than Bartter, often in adolescence or adulthood, with muscle cramps, fatigue, tetany and palpitations. Its two discriminators from Bartter are hypocalciuria, a low urine calcium, and hypomagnesaemia, a low serum magnesium, and these two findings together are almost pathognomonic. [2] [3]
Liddle syndrome occupies the collecting duct. The epithelial sodium channel ENaC is overactive because a truncating mutation in the beta or gamma subunit, encoded by SCNN1B or SCNN1G, deletes the motif that normally targets the channel for degradation. ENaC stays in the membrane and reabsorbs sodium constitutively, driving potassium excretion and fluid retention. The inheritance is autosomal dominant, and the cardinal feature is early-onset hypertension with a hypokalaemic alkalosis and suppressed renin and aldosterone. [9] [10]
Fanconi syndrome is the generalised proximal tubulopathy. Rather than one transporter, the whole proximal cell fails to reabsorb, so glucose spills despite a normal blood glucose, phosphate spills causing hypophosphataemic rickets, amino acids spill, low molecular weight proteins spill, and bicarbonate spills causing a proximal renal tubular acidosis. The inherited causes in children are headed by cystinosis, encoded by CTNS, and include Lowe syndrome from OCRL, Dent disease from CLCN5 or OCRL, galactosaemia, hereditary fructose intolerance, tyrosinaemia type 1 and Fanconi-Bickel syndrome from SLC2A2. [4] [11]
Bartter syndrome
- Thick ascending limb (loop-diuretic equivalent)
- Genes SLC12A1, KCNJ1, CLCNKB, BSND, autosomal recessive
- Hypokalaemic metabolic alkalosis, high renin and aldosterone
- Hypercalciuria and nephrocalcinosis, normal or low blood pressure
- Antenatal polyhydramnios, prematurity, neonatal salt wasting
Gitelman syndrome
- Distal convoluted tubule (thiazide equivalent)
- Gene SLC12A3, autosomal recessive
- Hypokalaemic metabolic alkalosis, high renin and aldosterone
- Hypocalciuria and hypomagnesaemia, normal or low blood pressure
- Adolescent presentation with cramps, tetany and palpitations
Fanconi syndrome
- Generalised proximal tubular failure
- Cystinosis (CTNS), Lowe (OCRL), Dent (CLCN5), galactosaemia
- Normal anion gap metabolic acidosis, proximal RTA
- Glycosuria with normal glucose, phosphaturia, aminoaciduria
- Failure to thrive, rickets, polyuria in infancy
Liddle syndrome
- Collecting duct ENaC gain-of-function
- Genes SCNN1B, SCNN1G, autosomal dominant
- Hypokalaemic metabolic alkalosis, low renin and low aldosterone
- Hypertension, often early onset and severe
- Responds to amiloride, not to spironolactone or eplerenone
Epidemiology & Risk Factors
Gitelman syndrome is the commonest inherited salt-losing tubulopathy, with an estimated carrier frequency that yields a prevalence in the region of 1 in 40,000, while Bartter syndrome is rarer. Gitelman is often mild enough to escape diagnosis until adolescence or adulthood, when a chance electrolyte panel or an episode of tetany brings it to light. Bartter, especially the antenatal form, declares itself at birth with polyhydramnios and prematurity, so its incidence is better captured in neonatal series. Both are autosomal recessive, so consanguinity and a family history of salt wasting, hearing loss or unexplained infant deaths raise the pre-test probability. [2] [3]
The Bartter subtypes differ in severity and in their extrarenal features, and this is where the gene matters to the prognosis. Antenatal Bartter, from SLC12A1, KCNJ1 or BSND mutations, is the severe end, with polyhydramnios, prematurity, severe salt wasting, hypercalciuria and nephrocalcinosis. The BSND subtype and the CASR subtype add sensorineural deafness or hypocalcaemia respectively. Classic Bartter, from CLCNKB, is milder and can even resemble Gitelman with hypomagnesaemia and hypocalciuria, which is why the gene sometimes has to settle a clinical overlap. [1] [5]
Liddle syndrome is rare but important because it is a surgically and pharmacologically distinct cause of early hypertension that is easily mistaken for primary hyperaldosteronism. It is autosomal dominant, so an affected parent and a family history of early-onset hypertension or stroke should prompt screening of the electrolytes and the renin and aldosterone axis in children and young relatives. The trap is that the hypokalaemia may be mild or intermittent, so a normal potassium does not exclude Liddle in a hypertensive child with a family history. [9] [10]
Cystinosis is the single commonest inherited cause of Fanconi syndrome in children, with an incidence around 1 in 100,000 to 1 in 200,000 live births, and it is autosomal recessive from CTNS. It presents in infancy with failure to thrive, polyuria, vomiting, rickets and photophobia from corneal crystal deposition, and without cysteamine it progresses to end-stage kidney disease in the first decade. The other inherited Fanconi causes are individually rare, but the acquired causes, especially ifosfamide, tenofovir, valproate and outdated tetracyclines, are commoner in oncology and HIV cohorts and must be sought on the drug history. [12] [4]
Pathophysiology
Each tubulopathy is the loss of one transporter, and the biochemistry is the direct consequence of where that transporter sits. The thick ascending limb reabsorbs about a quarter of the filtered sodium chloride through the apical NKCC2 cotransporter, with potassium recycling through ROMK and chloride leaving through the basolateral ClC-Kb. This segment is also the site of calcium and magnesium reabsorption, driven by the lumen-positive potential that potassium recycling creates. When NKCC2, ROMK or ClC-Kb fails in Bartter syndrome, salt is lost, the medullary concentration gradient collapses, and calcium and magnesium reabsorption falls, which is why Bartter runs hypercalciuria and nephrocalcinosis. [1] [5]
The distal convoluted tubule reabsorbs a further 5 to 10 percent of filtered sodium chloride through the thiazide-sensitive NCC cotransporter, and it is the main site of magnesium reabsorption through the TRPM6 channel. When NCC fails in Gitelman syndrome, sodium and chloride are lost, and the mild chronic volume depletion drives secondary hyperaldosteronism that wastes potassium and hydrogen, producing the hypokalaemic alkalosis. Magnesium is lost because NCC-dependent transport sustains the electrical gradient for TRPM6, and calcium is retained, partly through increased proximal reabsorption, which is the mechanism behind the hypocalciuria. This is the single most examined mechanism in Gitelman, because hypomagnesaemia with hypocalciuria is the signature. [2] [8]
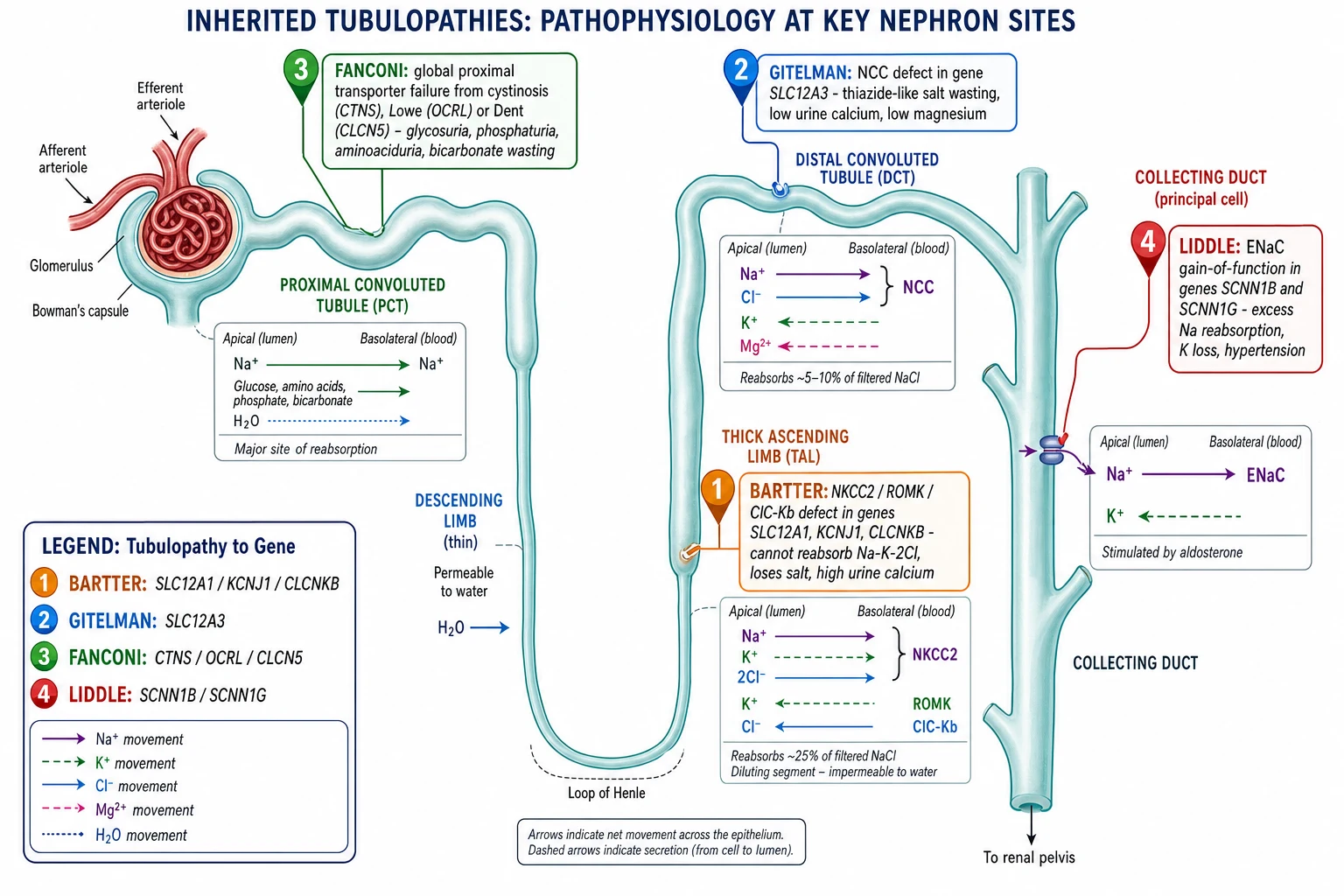
The collecting duct is where aldosterone fine-tunes sodium and potassium, and it does so through the epithelial sodium channel ENaC on the apical membrane of the principal cell. Sodium enters through ENaC down its gradient, raising the lumen-negative potential that drives potassium secretion, and aldosterone increases the number and activity of ENaC channels. In Liddle syndrome a mutation in the beta or gamma subunit deletes the PY motif that ubiquitin tags use to remove ENaC from the membrane, so ENaC accumulates and stays open regardless of aldosterone. Sodium is reabsorbed relentlessly, the extracellular volume expands, potassium and hydrogen are secreted, and the result is hypertension with a hypokalaemic alkalosis and renin and aldosterone both suppressed by the volume expansion. [9] [10]
Fanconi syndrome is mechanistically different because the whole proximal cell fails rather than one channel. The proximal tubule reabsorbs the bulk of the filtered load, glucose and amino acids and phosphate and bicarbonate and low molecular weight proteins, through a battery of transporters powered by the basolateral sodium-potassium ATPase. In cystinosis the accumulation of lysosomal cystine impairs cellular energy metabolism and the apical endocytic machinery, so the transporters are not recycled and the cell leaks. The result is global proximal wasting, glycosuria with a normal blood glucose, phosphaturia with hypophosphataemic rickets, aminoaciduria, low molecular weight proteinuria and a proximal renal tubular acidosis from bicarbonate wasting. [4] [12]
The shared theme across the salt-losing syndromes is secondary hyperaldosteronism. Bartter and Gitelman both lose salt, so they contract the extracellular volume, and the renin-angiotensin-aldosterone axis responds by raising renin and aldosterone. The high aldosterone drives the distal secretion of potassium and hydrogen, which is why both syndromes show a hypokalaemic alkalosis despite being salt-losing states. Liddle produces the same biochemistry by a different route, direct ENaC overactivity, and crucially without raising aldosterone, because the volume expansion suppresses it. This single difference, raised versus suppressed renin and aldosterone, is what separates Liddle from the salt-losing trio at the bedside. [7] [8]
Clinical Presentation
Bartter syndrome presents in one of two patterns. The antenatal form declares itself with polyhydramnios, premature birth and a salt-wasting neonate who fails to thrive, drinks and urinates excessively, vomits, and may show fever from the profound prostaglandin E2 excess. Nephrocalcinosis is found on the early ultrasound, and the biochemistry is a hypokalaemic hypochloraemic metabolic alkalosis with hypercalciuria. The classic form, usually from CLCNKB, presents later in childhood with failure to thrive, polyuria, salt craving and a milder alkalosis, and it can overlap with Gitelman. [1] [3]
Gitelman syndrome presents later and more subtly, typically in adolescence or young adulthood. The patient complains of fatigue, muscle cramps, thirst and salt craving, and may come to attention through tetany, Chvostek or Trousseau signs, carpopedal spasm, or palpitations. Growth can be affected in childhood, and affected adults are often slightly short. The symptoms track the hypokalaemia and the hypomagnesaemia, and a prolonged QT interval on the electrocardiogram flags the small but real risk of ventricular arrhythmia. Unlike Bartter, there is no polyhydramnios and no nephrocalcinosis. [2] [3]
Fanconi syndrome presents in infancy with the consequences of global proximal wasting. The infant fails to thrive despite an adequate intake, vomits, drinks and urinates excessively, and develops rickets from the phosphate wasting, with bowing of the legs, widened wrists and frontal bossing. The urine contains glucose despite a normal blood glucose. In cystinosis there is added photophobia from corneal crystal deposition, and a fair complexion, and without treatment the child progresses to renal failure. The presentation is therefore a small, unwell, polyuric infant with rickets and glycosuria, and the Fanconi screen confirms it. [4] [12]
Liddle syndrome presents with hypertension, and the hypertension is what brings it to attention, often early in life and sometimes severe. The patient may be asymptomatic apart from the high blood pressure found on a routine check, or may present with headache. The hypokalaemia can cause muscle weakness, cramps or polyuria, but it can also be absent at the time of presentation, which is why Liddle is missed. A family history of early-onset hypertension or early stroke in a relative is the clue that the hypertension is inherited rather than essential. [9] [10]
Atypical presentations deserve attention because they are how each syndrome is missed. An adolescent with recurrent tetany or seizures and a low magnesium has Gitelman syndrome until excluded. A neonate with polyhydramnios and unexplained salt wasting has antenatal Bartter until excluded. An infant with failure to thrive, glycosuria and rickets has Fanconi syndrome from cystinosis until excluded. And any child or young adult with hypertension and a family history of early stroke has Liddle until the renin, aldosterone and genetics are back. The key is to send the electrolytes, the renin and aldosterone, the urine calcium and the serum magnesium early. [7] [6]
Differential Diagnosis
The first differential question is not which tubulopathy but whether the biochemistry is an alkalosis or an acidosis, because that single step separates Fanconi from the other three. A metabolic acidosis with glycosuria, phosphaturia and aminoaciduria points to Fanconi syndrome and away from the salt-losing trio. A hypokalaemic metabolic alkalosis points to Bartter, Gitelman or Liddle, and the next question is the blood pressure. [8] [6]
Within the hypokalaemic alkaloses, the blood pressure and the renin and aldosterone do the sorting. Hypertension with suppressed renin and aldosterone is Liddle syndrome. Hypertension with high aldosterone and suppressed renin is primary hyperaldosteronism, an adrenal adenoma or hyperplasia, which is rare in children but must be considered. A normal or low blood pressure with high renin and high aldosterone is a salt-losing state, and within that group the urine calcium and the serum magnesium split Bartter from Gitelman. [10] [8]
The urine calcium is the discriminator between Bartter and Gitelman, and it is worth committing exactly. Bartter syndrome runs hypercalciuria, a high urine calcium, because the thick ascending limb is the main site of calcium reabsorption and it has failed. Gitelman syndrome runs hypocalciuria, a low urine calcium, because the distal convoluted tubule defect paradoxically increases proximal calcium reabsorption. Add the serum magnesium, which is normal or mildly low in Bartter but characteristically low in Gitelman, and the two are separated at the bedside. The one exception is classic Bartter from CLCNKB, which can mimic Gitelman with hypomagnesaemia and hypocalciuria and may need a gene test to settle. [2] [3]
BUCKET GLoW
Several mimics must not be missed. Chronic diuretic abuse, particularly loop diuretics, produces a Bartter-like alkalosis with hypercalciuria, and thiazide abuse produces a Gitelman-like alkalosis with hypocalciuria, so a drug and a urine diuretic screen belong in every unexplained alkalosis. Vomiting produces a hypochloraemic alkalosis, but the urine chloride is low rather than high, distinguishing it from the renal salt-wasting causes. Congenital chloride-losing diarrhoea produces a hypochloraemic alkalosis from infancy, but the diarrhoea is the clue. Apparent mineralocorticoid excess and the licorice-induced form of it produce a Liddle-like picture, and ectopic ACTH causing cortisol to saturate the mineralocorticoid receptor is another low-renin hypertension mimic. [8] [10]
Clinical & Bedside Assessment
Begin with growth, because every untreated tubulopathy stunts growth, and growth failure is often the presenting complaint. Plot weight, height and body mass index, and ask whether the child has fallen away from their centiles. In Bartter and Fanconi the failure to thrive is severe and early, while in Gitelman it is milder and may only declare as short stature in adolescence. A child whose growth recovers on treatment is being treated adequately, and one who does not is under-treated or has another diagnosis. [7] [4]
Assess the volume status carefully, because the salt-losing syndromes deplete the extracellular fluid. The Bartter neonate is often wasted, hypotonic and dehydrated, with sunken eyes and dry mucous membranes, and signs of postural hypotension in an older child with Gitelman suggest significant volume depletion. Examine for the tetany of hypomagnesaemia and hypocalcaemia, with Chvostek and Trousseau signs, and for muscle weakness from hypokalaemia. Take the blood pressure properly and repeatedly, because an elevated pressure in a child with a hypokalaemic alkalosis redirects the diagnosis to Liddle and away from the salt-losing trio. [2] [9]
Examine for the extrarenal clues to a specific cause. Photophobia and a fair complexion point to cystinosis, the cataracts and hypotonia of Lowe syndrome, the hepatosplenomegaly of the storage disorders, and the bowing of the legs and widened wrists of hypophosphataemic rickets in Fanconi. Sensorineural deafness points to the BSND subtype of Bartter. In Liddle, look for the end-organ effects of chronic hypertension, and take a careful family history of early-onset hypertension or stroke, because the autosomal dominant inheritance is a major clue. [1] [12]
The history identifies the precipitants and the inherited pattern. Ask about consanguinity, family history of salt wasting, deafness, kidney stones, failure to thrive or early hypertension and stroke. In the cystinosis suspect, ask about photophobia and excessive thirst from infancy. Take a careful drug history, because diuretic abuse mimics Bartter and Gitelman, and ifosfamide, tenofovir, valproate and outdated tetracyclines cause acquired Fanconi. Ask about diarrhoea, which would redirect the alkalosis toward a gastrointestinal cause with a low urine chloride. [8] [6]
Investigations
The first panel is drawn from one cannula and answers the family-level question at once. Send serum electrolytes with chloride and bicarbonate, urea and creatinine, magnesium, calcium and phosphate, glucose, and a venous blood gas for the pH. A urine sample gives the pH and the spot urine calcium, sodium, potassium, chloride, creatinine and, if available, magnesium. The anion gap and the bicarbonate establish whether this is an alkalosis or an acidosis, the potassium names the direction, and the urine calcium and the serum magnesium begin to split Bartter from Gitelman. [7] [8]
The renin and aldosterone levels are the next step and the most powerful discriminator, because they separate Liddle from primary hyperaldosteronism and from the salt-losing trio. In Liddle both renin and aldosterone are suppressed. In primary hyperaldosteronism aldosterone is high and renin is suppressed. In Bartter and Gitelman both renin and aldosterone are high. A random renin and aldosterone, interpreted with the posture and the salt intake, usually suffices, and the combination with the blood pressure names the family before any gene is sequenced. [10] [2]
The urine calcium and the serum magnesium settle Bartter against Gitelman. Bartter runs hypercalciuria, expressed as a high urine calcium-to-creatinine ratio, and a normal or only mildly low serum magnesium. Gitelman runs hypocalciuria, a low urine calcium-to-creatinine ratio, and a characteristically low serum magnesium. In the overlap case, the clinic often proceeds to genetic testing, because the CLCNKB classic Bartter subtype can mimic Gitelman biochemically. The fractional excretion of chloride is high in the salt-losing states, confirming renal salt wasting rather than extrarenal loss. [3] [5]
For Fanconi syndrome the investigations extend to the Fanconi screen and its cause. The urine shows glycosuria with a normal blood glucose, phosphaturia with a low serum phosphate, aminoaciduria, and low molecular weight proteinuria with a normal albumin. The serum shows a metabolic acidosis with a low bicarbonate and a low potassium. To find the cause, measure leukocyte cystine for cystinosis, examine the slit lamp for corneal crystals, screen the galactose and fructose pathways where the history fits, and send a Fanconi gene panel covering CTNS, OCRL, CLCN5 and SLC2A2. [4] [11]
Genetic testing confirms the diagnosis, settles the overlap, and enables cascade testing and counselling. A next-generation sequencing panel covering the Bartter genes SLC12A1, KCNJ1, CLCNKB, BSND and CASR, the Gitelman gene SLC12A3, the Liddle genes SCNN1B and SCNN1G, and the Fanconi genes CTNS, OCRL and CLCN5 is now the standard. Genetic confirmation matters most when the biochemistry overlaps, when the family is being counselled about recurrence, and when a specific therapy like cysteamine for cystinosis hangs on the diagnosis. [1] [6]
Management — Resuscitation
The salt-losing syndromes can present in crisis, and the resuscitation priorities are volume, potassium and magnesium. A Bartter neonate with severe salt wasting and dehydration needs intravenous fluid and salt replacement, with the potassium corrected and the magnesium maintained, because the biochemistry drives the symptoms. The correction should be cautious and monitored, because over-rapid shifts can precipitate arrhythmia, and the long-term therapy is the goal of the resuscitation. [1] [5]
Gitelman syndrome rarely presents as an acute emergency, but a severe hypokalaemia or hypomagnesaemia can cause tetany, seizures or a ventricular arrhythmia, and these need intravenous magnesium and potassium. Intravenous magnesium is given as magnesium sulfate, with cardiac monitoring, and the oral therapy is re-established as soon as tolerated. A patient who presents with palpitations or a prolonged QT interval needs continuous cardiac monitoring until the potassium and magnesium are stabilised. [2] [8]
Liddle syndrome can present with a hypertensive emergency, and the resuscitation is blood pressure control. Amiloride is the definitive agent because it blocks the epithelial sodium channel directly, and it lowers the blood pressure and corrects the potassium. In a severe hypertensive crisis the standard paediatric intravenous antihypertensives are used as a bridge, but the long-term answer is oral amiloride, and spironolactone or eplerenone should not be expected to work because aldosterone is already suppressed. [9] [10]
Fanconi syndrome presents acutely with dehydration from polyuria, acidosis, and the metabolic consequences of phosphate wasting, and the resuscitation is fluid, bicarbonate and potassium. An intravenous bicarbonate infusion corrects the acidosis partially and carefully, potassium is replaced, and the phosphate and vitamin D are begun once the child is stable. The underlying cause is treated in parallel, with cysteamine for cystinosis and removal of an offending drug, because no amount of electrolyte replacement replaces treating the cause. [4] [12]
Magnesium sulfate (acute symptomatic hypomagnesaemia in Gitelman)
Dose
Intravenous magnesium sulfate 25 to 50 mg per kg per dose, equivalent to 0.1 to 0.2 mmol per kg of elemental magnesium, over several hours with cardiac monitoring
Management — Definitive & Stepwise
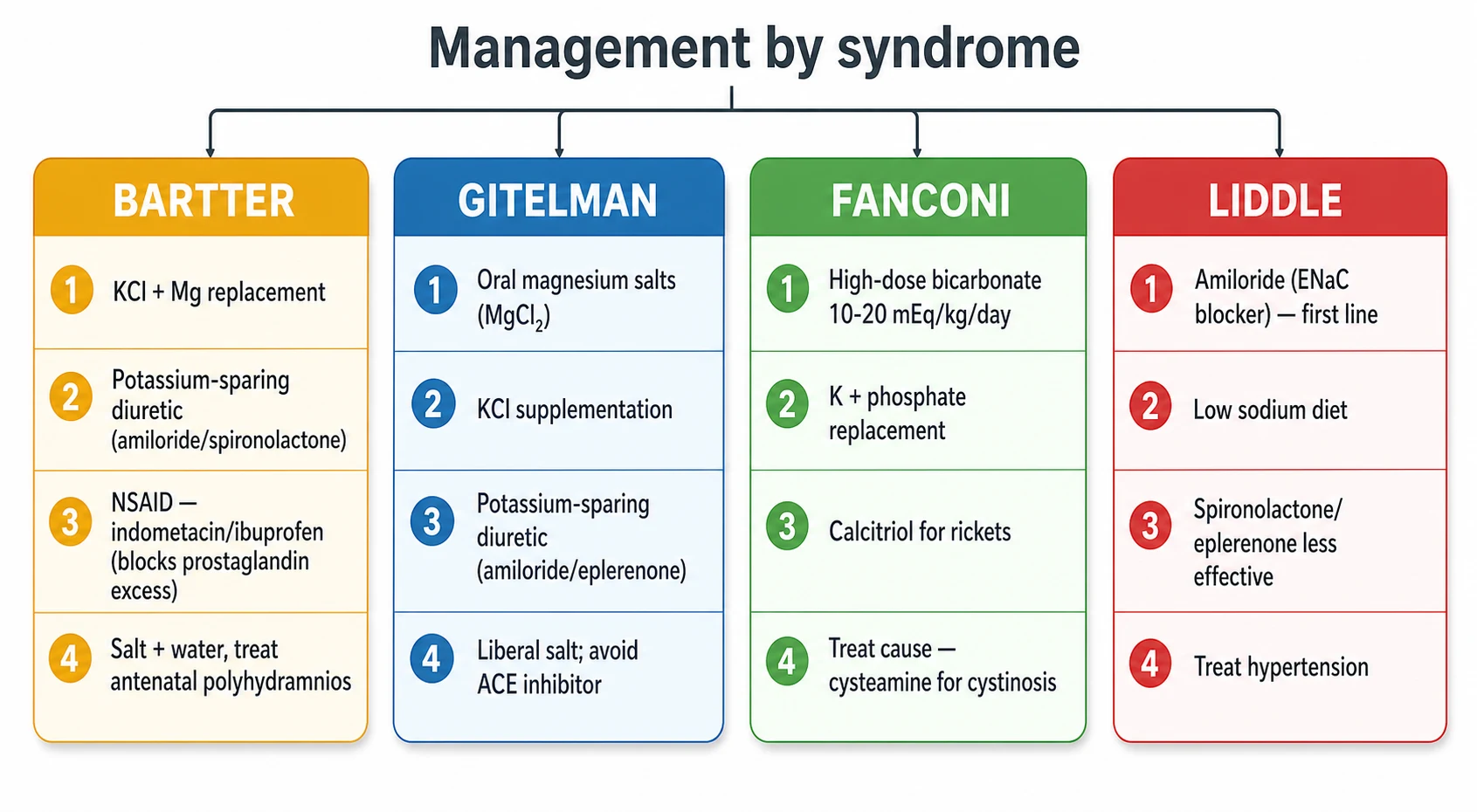
The definitive therapy of each syndrome is dictated by the segment that broke, and getting the agent right is the difference between a child who thrives and one who does not. Bartter syndrome is treated with potassium chloride and magnesium replacement, potassium-sparing diuretics such as amiloride or spironolactone to reduce potassium wasting, and a prostaglandin synthesis inhibitor, because the salt-losing state drives a marked prostaglandin E2 excess that worsens the polyuria and the salt wasting. Indometacin or ibuprofen at 1 to 3 mg per kg per day is the standard, and it is most valuable in the antenatal and neonatal forms. [1] [5]
Gitelman syndrome is treated with oral magnesium and potassium replacement and a potassium-sparing diuretic, because the hypomagnesaemia drives much of the symptom burden and the potassium wasting. Oral magnesium is given as the chloride, lactate or aspartate salt, in divided doses through the day to reduce diarrhoea, and the dose is titrated to the serum magnesium and the symptoms. Amiloride or eplerenone reduces the potassium and magnesium wasting. Angiotensin-converting enzyme inhibitors and angiotensin receptor blockers should be avoided, because they can precipitate significant hypotension in the salt-losing state. A liberal salt intake supports the intravascular volume. [2] [3]
Fanconi syndrome is treated by replacing what the proximal tubule wastes and by treating the cause. Bicarbonate is given at the high proximal-RTA dose of 10 to 20 mEq per kg per day in divided doses, because the kidney keeps wasting bicarbonate above the lowered threshold. Potassium is added, because the increased distal bicarbonate delivery drives potassium wasting. Phosphate is replaced for the rickets, with calcitriol to support the bone mineralisation. The cause-specific therapy is central, with cysteamine for cystinosis, dietary exclusion for galactosaemia and hereditary fructose intolerance, and withdrawal of an offending drug such as ifosfamide or tenofovir. [4] [12]
Liddle syndrome is treated with amiloride, which blocks the epithelial sodium channel directly and corrects both the hypertension and the potassium loss. Triamterene is an alternative. Spironolactone and eplerenone are ineffective, because they act through the mineralocorticoid receptor and aldosterone is already suppressed in Liddle. A low-sodium diet supports the blood pressure control, and lifelong amiloride is the rule, because the genetic defect does not remit. The blood pressure and potassium response to amiloride is itself diagnostic, and a patient whose hypertension and hypokalaemia resolve on amiloride has Liddle until the genetics prove otherwise. [9] [10]
Bartter syndrome — the salt and prostaglandin ladder
Confirm hypokalaemic alkalosis with hypercalciuria and high renin and aldosterone
Start potassium chloride and magnesium replacement, weight-based and revised as the child grows
Add a potassium-sparing diuretic, amiloride or spironolactone, to reduce potassium wasting
Add a prostaglandin synthesis inhibitor, indometacin or ibuprofen 1 to 3 mg per kg per day, for the antenatal and neonatal forms
Monitor growth, electrolytes and renal ultrasound for nephrocalcinosis; arrange genetic confirmation and hearing assessment
Gitelman syndrome — the magnesium ladder
Confirm hypokalaemic alkalosis with hypocalciuria, hypomagnesaemia and high renin and aldosterone
Start oral magnesium as chloride, lactate or aspartate in divided doses, titrated to symptoms and serum magnesium
Add potassium chloride and a potassium-sparing diuretic, amiloride or eplerenone
Encourage a liberal salt intake and avoid angiotensin-converting enzyme inhibitors and angiotensin receptor blockers
Monitor serum magnesium and potassium, the electrocardiogram for a prolonged QT, and growth
Liddle syndrome — the amiloride ladder
Confirm hypokalaemic alkalosis with hypertension and suppressed renin and aldosterone
Start amiloride, titrated to the blood pressure and the potassium
Prescribe a low-sodium diet and review the family for early-onset hypertension
Do not use spironolactone or eplerenone as first line, because aldosterone is already suppressed
Confirm genetically with SCNN1B or SCNN1G and offer cascade testing to affected relatives
Fanconi syndrome — the replace and treat the cause ladder
Confirm glycosuria with normal glucose, phosphaturia, aminoaciduria and a normal anion gap acidosis
Start high-dose bicarbonate at 10 to 20 mEq per kg per day in divided doses, with potassium
Replace phosphate and add calcitriol for the rickets
Treat the cause: cysteamine for cystinosis, dietary exclusion for galactosaemia and hereditary fructose intolerance, drug withdrawal for ifosfamide or tenofovir
Monitor growth, serum bicarbonate, phosphate and bone mineralisation, and renal function
Specific Subtypes & Scenarios
Antenatal Bartter syndrome is the severe end of the Bartter spectrum and deserves its own handling. It presents with polyhydramnios from fetal polyuria, prematurity, and a salt-wasting neonate with hypercalciuria, nephrocalcinosis, fever and failure to thrive. The polyhydramnios may be treated antenatally with indometacin to the mother, and the neonate needs aggressive salt and water replacement, potassium, magnesium and a prostaglandin synthesis inhibitor. The prognosis has improved with modern neonatal care, but growth and renal function remain long-term concerns, and lifelong therapy is the rule. [1] [5]
Cystinosis is the archetype of inherited Fanconi syndrome and the one the exam most often targets. It is an autosomal recessive lysosomal storage disorder from CTNS, with an incidence around 1 in 100,000 to 1 in 200,000 live births. Cystine accumulates in every cell, but the proximal tubule fails first, producing a generalised Fanconi syndrome. Without treatment it progresses to end-stage kidney disease in the first decade. Cysteamine depletes the lysosomal cystine and dramatically slows the disease, which is why early diagnosis by leukocyte cystine and slit-lamp examination matters. The Fanconi itself needs high-dose bicarbonate, potassium, phosphate and calcitriol. [12] [4]
Dent disease and Lowe syndrome are the X-linked proximal tubulopathies and overlap with Fanconi. Dent disease, from CLCN5 or OCRL, presents in boys with low molecular weight proteinuria, hypercalciuria, nephrocalcinosis and progressive renal failure, often without a full Fanconi picture. Lowe syndrome, the oculocerebrorenal syndrome from OCRL, adds congenital cataracts, hypotonia and intellectual disability to the Fanconi. Both are X-linked, so the family history and the male sex are clues, and the cataracts of Lowe are present from infancy. [11] [4]
Liddle syndrome in childhood is the prototypic monogenic hypertension, and it is worth knowing as a cause of early-onset, often severe, hypokalaemic hypertension. The genetics are autosomal dominant in SCNN1B or SCNN1G, the renin and aldosterone are suppressed, and the blood pressure responds to amiloride. It is easily mistaken for primary hyperaldosteronism, but the aldosterone is low rather than high, and the response to amiloride with the suppressed aldosterone is the bedside signature. Family screening identifies affected relatives whose hypertension may have been labelled essential. [9] [10]
Acquired Fanconi syndrome is common in oncology and HIV cohorts and is reversible when the cause is removed. Ifosfamide, tenofovir, didanosine, valproate, outdated tetracyclines and several antiretrovirals produce a proximal tubulopathy by poisoning the proximal tubule, and heavy metals and paraquat are rarer causes. The clinical picture is the same Fanconi biochemistry, and the management is withdrawal of the offending drug with electrolyte and bicarbonate support in the interim. Monitoring for recovery of tubular function after cessation is part of the follow-up. [4] [11]
Complications & Pitfalls
The cardinal pitfall is confusing Liddle with Bartter or Gitelman, because the treatment is different and the prognosis is different. A hypokalaemic metabolic alkalosis with hypertension is Liddle, and treating it with spironolactone or eplerenone fails, because aldosterone is already suppressed. The blood pressure and the renin and aldosterone must be checked before committing to a pathway, and the blood pressure response to amiloride is the bedside confirmation. This single error, attributing Liddle to a salt-losing state, is the most damaging in the topic. [9] [8]
The second pitfall is missing cystinosis in an infant with Fanconi syndrome. Cystinosis is the commonest inherited cause of Fanconi in children, and without cysteamine it progresses to end-stage kidney disease in the first decade. Every infant with failure to thrive, polyuria, glycosuria and a normal anion gap acidosis needs a leukocyte cystine and a slit-lamp examination, because early cysteamine changes the trajectory of the disease. Missing the diagnosis, or delaying it, costs the child a kidney and the stature. [12] [4]
The third pitfall is undertreating Gitelman syndrome, particularly the hypomagnesaemia. The oral magnesium dose is large and often limited by diarrhoea, and the symptoms, fatigue, cramps, tetany and palpitations, track the magnesium as much as the potassium. A patient whose symptoms persist on potassium alone is usually magnesium-deficient, and the magnesium needs uptitration, sometimes with the chloride or aspartate salt for tolerance. The prolonged QT and the small arrhythmia risk justify aggressive magnesium replacement. [2] [3]
The fourth pitfall is over-reliance on the urine calcium without the serum magnesium, or vice versa, to split Bartter from Gitelman. The two are meant to be read together, and the classic CLCNKB Bartter can mimic Gitelman with hypomagnesaemia and hypocalciuria, so a single discordant value should prompt a gene test rather than a confident label. The fifth pitfall is missing the drug causes of an alkalosis, because diuretic abuse mimics Bartter and Gitelman perfectly, and a urine diuretic screen belongs in every unexplained alkalosis. [8] [5]
Prognosis & Disposition
Prognosis depends on the syndrome, the underlying gene, and the timeliness and adequacy of therapy. Gitelman syndrome is compatible with a normal lifespan, though the symptom burden from fatigue, cramps and tetany can be considerable, and a small arrhythmia risk persists. Bartter syndrome, especially the antenatal form, carries a harder prognosis in infancy, with neonatal mortality from salt wasting and long-term concerns about growth and renal function, but modern neonatal care and prostaglandin synthesis inhibitors have improved the outlook substantially. [3] [1]
Liddle syndrome has an excellent prognosis once amiloride is established, because the blood pressure and the potassium respond well and the long-term risk is the cardiovascular damage from uncontrolled hypertension. The prognosis is therefore governed by how early the diagnosis is made and the family is screened, because an affected relative whose hypertension has been labelled essential is at risk of early stroke. Family screening and early amiloride change the trajectory. [9] [10]
Fanconi syndrome from cystinosis progresses to end-stage kidney disease without cysteamine, typically in the first decade, but with cysteamine the progression is dramatically slowed and renal survival into adulthood is achievable. The other inherited Fanconi causes have variable prognoses, and the acquired causes are reversible when the offending drug is withdrawn. Growth and bone are the key paediatric markers, and a child whose growth recovers on therapy is being treated adequately. [12] [4]
Disposition follows the severity. The Bartter neonate, the patient with severe hypokalaemia or a prolonged QT, the hypertensive emergency in Liddle, and the acidotic Fanconi infant need admission, with high-dependency or intensive care for the unstable. The stable patient, once the biochemistry is corrected and the oral therapy is established, goes to the ward for titration and then home with outpatient nephrology follow-up. Most management is outpatient, with electrolyte titration, growth and blood pressure monitoring, and the surveillance specific to the cause. [7] [6]
The monitoring plan covers growth, biochemistry, bone, blood pressure and the cause-specific risks. Serial electrolytes, magnesium, bicarbonate and the urine calcium-to-creatinine ratio confirm adequate therapy. Height and weight plotted over time confirm growth recovery. The electrocardiogram monitors the QT in Gitelman. The renal ultrasound monitors nephrocalcinosis in Bartter. The blood pressure monitors Liddle. Leukocyte cystine and slit-lamp examination monitor cystinosis, and the bone mineralisation and phosphate monitor Fanconi. Adherence is the single biggest determinant of outcome, and it is the thing most often lost in adolescence. [1] [2]
Special Populations
Neonates dominate the severe end of the tubulopathy spectrum, and they need special handling. Antenatal Bartter presents with polyhydramnios and prematurity, the salt-wasting neonate needs aggressive fluid, salt, potassium and magnesium replacement with a prostaglandin synthesis inhibitor, and the dose must be weight-based and revised frequently as the infant grows. The polyuria and salt wasting produce dehydration that intercurrent illness worsens, so the family must be taught sick-day management early. Cystinosis presents in the first year, and early cysteamine is the determinant of long-term renal survival. [1] [12]
Adolescents with the salt-losing syndromes face the twin challenges of symptom control and transition to adult care. Gitelman symptoms can be disabling in adolescence, with fatigue limiting school and sport, and the magnesium and potassium need careful titration through growth. Adherence is the leading determinant of outcome, and it is most often lost in adolescence, so a structured transition programme with peer support and honest conversations about the lifelong nature of the therapy is essential. The young person who arrives in adult care with a stable regimen and an understanding of their disease is a transition done well. [2] [7]
The hypertensive child with suspected Liddle needs family screening, because the autosomal dominant inheritance means affected relatives may carry a label of essential hypertension. Testing the parents and siblings, and offering cascade testing to the wider family, identifies relatives whose blood pressure and cardiovascular risk can be reduced with amiloride. The genetic counselling for the autosomal recessive Bartter and Gitelman families addresses the recurrence risk, and the cystinosis families need counselling about the 25 percent recurrence and the role of preimplantation genetic diagnosis. [9] [3]
The child with complex chronic disease on multiple drugs is at risk of acquired Fanconi, particularly from ifosfamide in oncology and tenofovir in HIV. Electrolyte, bicarbonate and phosphate monitoring during and after these treatments is essential, and drug cessation is the definitive step where the disease allows it. A medication review is part of every assessment of an unexplained proximal tubulopathy, because the acquired causes are common and reversible, and missing them leaves the child on unnecessary replacement therapy. [4] [11]
Evidence, Guidelines & Regional Differences
The Bartter and Gitelman syndromes are anchored by two international consensus documents. The 2021 European Rare Kidney Disease Reference Network (ERKNet) and European Society for Paediatric Nephrology consensus on Bartter syndrome, led by Konrad and colleagues, sets out the gene-based classification, the diagnostic criteria and the management. The 2017 Kidney Disease Improving Global Outcomes (KDIGO) controversies conference on Gitelman syndrome, led by Blanchard and colleagues, sets out the diagnostic approach, the symptomatic management and the gaps in the evidence. Together these two documents define the current standard of care. [1] [2]
The pathophysiology and the bedside discriminators are well summarised in the salt-losing tubulopathy reviews of Kleta and colleagues and the Bartter and Gitelman review of Fulchiero and colleagues, and the clinical approach of Kermond and colleagues brings the paediatric perspective. The metabolic alkalosis core curriculum of Do and colleagues lays out the renin, aldosterone, blood pressure and urine chloride algorithm that splits Liddle from primary hyperaldosteronism and from the salt-losing trio, and the Liddle review of Tetti and colleagues and the low-renin hypertension review of Athimulam and colleagues cover the ENaC disorders. [5] [8]
ANZ and UK paediatric nephrology practice follows the ERKNet and KDIGO consensus documents for the Bartter and Gitelman syndromes, with local unit differences in the formulation of oral magnesium and potassium and in the choice of prostaglandin synthesis inhibitor for antenatal Bartter. Oral magnesium chloride is the preferred salt where available, and amiloride is the standard first-line agent for Liddle. Genetic testing through a tubulopathy gene panel is now routine, and cysteamine for cystinosis is initiated and monitored through the metabolic and nephrology services. Always confirm the local paediatric nephrology protocol before prescribing, and document the guideline you are following. [1] [7]
The international consensus treats Bartter with salt, potassium, magnesium and a prostaglandin synthesis inhibitor, Gitelman with oral magnesium, potassium and a potassium-sparing diuretic, Liddle with amiloride, and Fanconi with high-dose bicarbonate, phosphate, calcitriol and cause-specific therapy such as cysteamine for cystinosis. Regional differences exist in the oral magnesium formulation, the indometacin dosing convention in neonatal Bartter, and the surveillance intervals for nephrocalcinosis and the prolonged QT, all of which should be checked against the local protocol. [2] [4]
The evidence is weakest in three places. The long-term renal and growth outcome of antenatal Bartter despite apparently adequate therapy remains drawn from observational cohorts, and the optimal duration and dose of the prostaglandin synthesis inhibitors in the neonate is extrapolated. The symptom burden and the arrhythmia risk in Gitelman are increasingly recognised but the magnesium target that best controls symptoms and risk is inferred rather than proven. And the optimal family screening strategy for Liddle, balancing the yield of cascade testing against the burden of treating a label, varies between units. These are the corners where specialist nephrology discussion is most valuable. [5] [9]
Exam Pearls
Commit the blood pressure branch first, because it names the family before any gene test. A hypokalaemic metabolic alkalosis with hypertension is Liddle until proven otherwise, while the same biochemistry with a normal or low blood pressure is Bartter or Gitelman. Confirm with renin and aldosterone, which Liddle suppresses, primary hyperaldosteronism splits with high aldosterone and low renin, and Bartter and Gitelman raise both. This two-step branch, blood pressure then renin and aldosterone, answers the spine of every tubulopathy viva. [9] [10]
The urine calcium and the serum magnesium split Bartter from Gitelman, and the rule is worth memorising exactly. Bartter runs hypercalciuria, because the thick ascending limb is the main site of calcium reabsorption and it has failed, and the serum magnesium is normal or only mildly low. Gitelman runs hypocalciuria and a characteristically low serum magnesium. The mnemonic is that Bartter is the loop-diuretic equivalent, with high urine calcium, and Gitelman is the thiazide equivalent, with low urine calcium and low magnesium. The classic CLCNKB Bartter can overlap and may need a gene test to settle. [2] [3]
Cystinosis is the commonest inherited cause of Fanconi syndrome in children, and it is examiner gold. It is autosomal recessive from CTNS, it presents in infancy with failure to thrive, polyuria, rickets and photophobia from corneal crystals, and without cysteamine it progresses to end-stage kidney disease in the first decade. Every infant with a Fanconi urine needs a leukocyte cystine and a slit-lamp examination, and cysteamine changes the trajectory of the disease. Dent disease and Lowe syndrome are the X-linked proximal tubulopathies to know alongside it. [12] [4]
The treatment is dictated by the segment that broke, and examiners test this directly. Bartter needs a prostaglandin synthesis inhibitor, indometacin or ibuprofen, because the salt-losing state drives a prostaglandin E2 excess. Gitelman needs oral magnesium, because the hypomagnesaemia drives the symptoms. Fanconi needs high-dose bicarbonate at 10 to 20 mEq per kg per day, with phosphate and calcitriol, and cause-specific therapy. Liddle needs amiloride, which blocks the epithelial sodium channel directly, and spironolactone and eplerenone are ineffective because aldosterone is already suppressed. [1] [8]
References
- [1]Konrad M, et al Diagnosis and management of Bartter syndrome: executive summary of the consensus and recommendations from the European Rare Kidney Disease Reference Network Working Group for Tubular Disorders. Kidney Int, 2021.PMID 33509356
- [2]Blanchard A, et al Gitelman syndrome: consensus and guidance from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference. Kidney Int, 2017.PMID 28003083
- [3]Fulchiero R, et al Bartter Syndrome and Gitelman Syndrome. Pediatr Clin North Am, 2019.PMID 30454738
- [4]Foreman JW Fanconi Syndrome. Pediatr Clin North Am, 2019.PMID 30454741
- [5]Kleta R, et al Salt-Losing Tubulopathies in Children: What's New, What's Controversial? J Am Soc Nephrol, 2018.PMID 29237739
- [6]Iancu D, et al Inherited Renal Tubulopathies-Challenges and Controversies. Genes (Basel), 2020.PMID 32150856
- [7]Kermond R, et al A clinical approach to tubulopathies in children and young adults. Pediatr Nephrol, 2023.PMID 35585366
- [8]Do C, et al Metabolic Alkalosis Pathogenesis, Diagnosis, and Treatment: Core Curriculum 2022. Am J Kidney Dis, 2022.PMID 35525634
- [9]Tetti M, et al Liddle Syndrome: Review of the Literature and Description of a New Case. Int J Mol Sci, 2018.PMID 29534496
- [10]Athimulam S, et al Low-Renin Hypertension. Endocrinol Metab Clin North Am, 2019.PMID 31655771
- [11]Albuquerque ALB, et al Inherited Fanconi syndrome. World J Pediatr, 2023.PMID 36729281
- [12]Elmonem MA, et al Cystinosis: a review. Orphanet J Rare Dis, 2016.PMID 27102039