Paeds · nephrology-urology-fluids-and-electrolytes
Lupus nephritis and systemic disease
Also known as Lupus nephritis · Lupus nephropathy · Childhood-onset lupus nephritis · SLE nephritis · ISN/RPS lupus nephritis classification
Fellowship guide to paediatric lupus nephritis: the six-class ISN/RPS 2003 classification (I minimal mesangial through VI advanced sclerosing), why Class IV diffuse proliferative dominates childhood disease, the immune-complex pathophysiology behind the full-house immunofluorescence pattern, and the induction-maintenance treatment paradigm built on mycophenolate mofetil or low-dose cyclophosphamide with glucocorticoids, treat-to-target proteinuria goals, and hydroxychloroquine for every patient.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
Overview & Definition
A 13-year-old girl who has been losing weight and complaining of swollen fingers presents with puffy eyelids, dark urine, and a blood pressure of 135 over 85. She has oral ulcers and a faint rash across her cheeks. Her urine shows blood and protein, her creatinine is mildly elevated, and her complement C3 and C4 are low. This is lupus nephritis, the renal manifestation of systemic lupus erythematosus, in which autoantibodies form immune complexes that deposit in the glomerulus and trigger inflammation. It is one of the most serious complications of childhood-onset lupus and the single strongest predictor of long-term morbidity and mortality in the disease. [9]
Renal involvement is both more frequent and more severe in children than in adults. Up to 50 to 80 percent of children with systemic lupus erythematosus develop nephritis, compared with roughly 30 to 40 percent of adults, and children are more likely to present with the aggressive proliferative Class III and IV patterns. Most childhood-onset lupus is diagnosed in adolescence, with a striking female predominance driven by oestrogen, and disease onset before puberty tends to be more severe. Because the kidney can be the first or only obviously affected organ, any child with unexplained haematuria, proteinuria, hypertension, or declining renal function needs lupus excluded. [9]
Classification
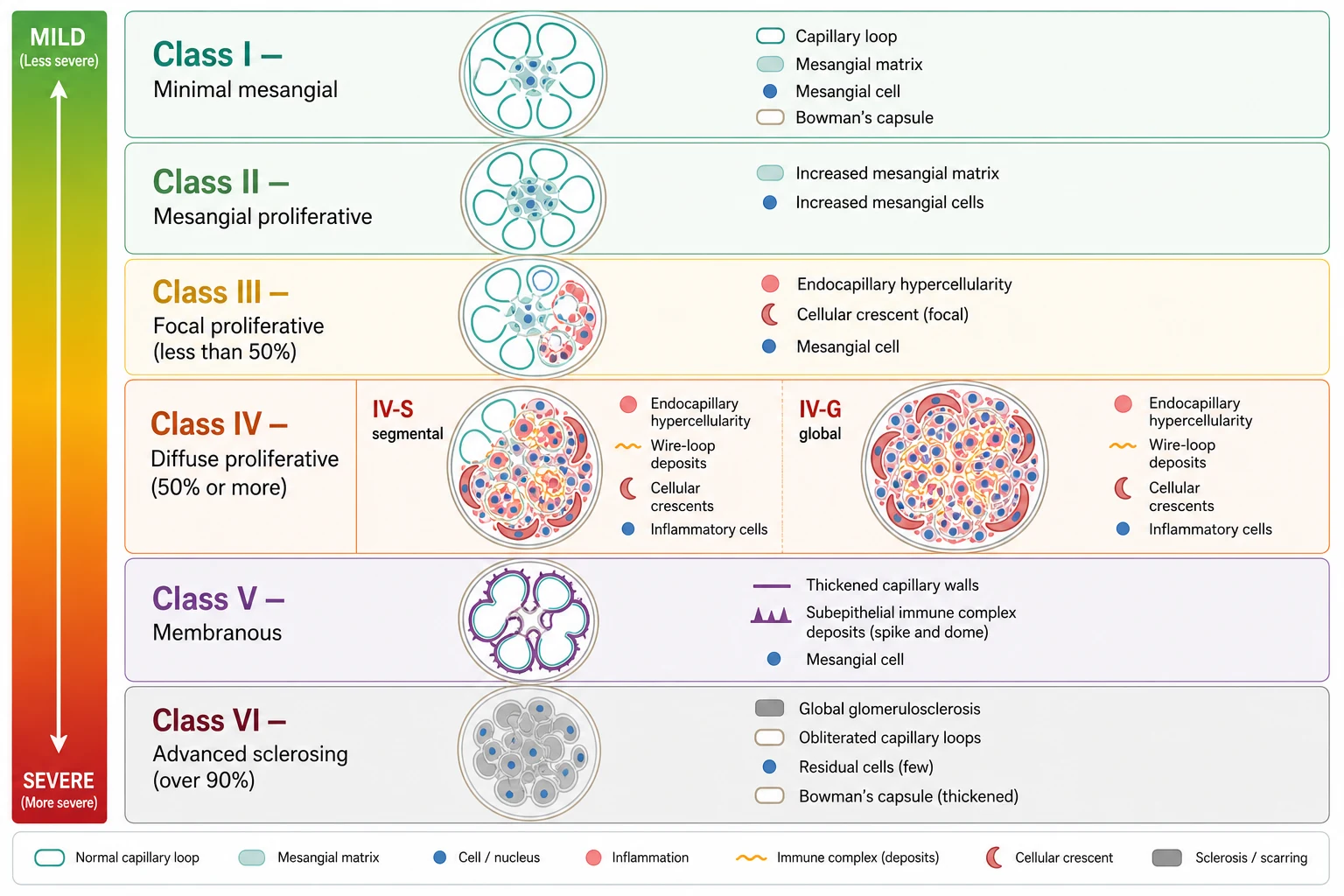
The classification that every candidate must know is the 2003 International Society of Nephrology and Renal Pathology Society system, usually abbreviated ISN/RPS. It assigns every lupus kidney biopsy to one of six classes based on the light microscopy pattern, the site of immune deposits, and the proportion of glomeruli involved. The classes run in order of increasing structural severity, and the practical divide is between mesangial-only disease (Classes I and II, generally mild), proliferative disease (Classes III and IV, generally severe and requiring immunosuppression), membranous disease (Class V, defined by heavy proteinuria), and end-stage scarring (Class VI). [1]
Class I is minimal mesangial lupus nephritis, in which the glomeruli look normal under light microscopy but mesangial immune deposits are visible on immunofluorescence. Class II is mesangial proliferative lupus nephritis, where mesangial hypercellularity of any degree accompanies the mesangial deposits. Both are clinically mild and usually present with microscopic haematuria and low-grade proteinuria. They do not require aggressive immunosuppression, and they never feature subendothelial deposits, necrosis, or crescents. If a child with apparent Class II disease develops nephrotic-range proteinuria or a rising creatinine, the biopsy should be questioned or repeated, because the clinical picture is too severe for the reported class. [1]
The proliferative classes are where the disease becomes dangerous. Class III is focal lupus nephritis, defined by active or inactive focal, segmental or global endocapillary or extracapillary glomerulonephritis involving fewer than 50 percent of glomeruli. Class IV is diffuse lupus nephritis, the most common and most severe pattern in children, defined by the same proliferative lesions but involving 50 percent or more of glomeruli. Class IV is further subdivided into IV-S, where at least half of the involved glomeruli have segmental lesions, and IV-G, where at least half have global lesions. Both Class III and IV show subendothelial immune deposits that are visible on light microscopy as wire-loop changes, and both can show necrosis and cellular crescents that indicate severe active inflammation. The 2018 revision of the classification simplified the reporting of Class IV and emphasised the NIH activity and chronicity indices, but the six-class framework and the proliferative-versus-non-proliferative divide remain the clinical and exam cornerstones. [1]
[1]Class V is membranous lupus nephritis, defined by global or segmental subepithelial immune deposits or their morphological sequelae. It can occur alone or together with Class III or IV, and the combined pattern is recorded as, for example, Class V plus Class IV. Pure Class V typically presents with nephrotic-range proteinuria and carries a higher risk of thrombosis from the nephrotic state. Class VI is advanced sclerosing lupus nephritis, where over 90 percent of glomeruli are globally sclerosed without residual activity. This is irreversible scarring, the endpoint of longstanding untreated disease, and management shifts from immunosuppression to renal replacement therapy and transplantation. [1]
Epidemiology & Risk Factors
Systemic lupus erythematosus is relatively uncommon in children, with an estimated incidence of 0.3 to 0.9 per 100,000 children per year, but when it does occur in childhood the kidney is far more often involved. Between 50 and 80 percent of children with lupus develop nephritis, and in up to half of these the renal disease is evident at the time of the initial lupus diagnosis. The peak age of childhood-onset lupus is early adolescence, around 11 to 15 years, and the disease is rare before the age of five. [9]
Sex and ethnicity are powerful risk factors. The female-to-male ratio is roughly 5 to 1 before puberty but rises to 9 to 1 or higher after puberty, reflecting the permissive effect of oestrogen on the immune system. Children of African, Asian, Hispanic, and South Asian ancestry have both a higher incidence and a more severe phenotype, with a greater likelihood of proliferative Class IV disease and a higher risk of progression to end-stage kidney disease. This ethnic gradient, also seen in adults, reflects a combination of genetic susceptibility (including complement and interferon pathway variants) and socio-environmental factors. [9]
The single strongest predictor of a poor renal outcome is the histological class at biopsy. Class IV diffuse proliferative disease, especially with a high activity index (cellular crescents, fibrinoid necrosis), carries the highest risk of progression to end-stage kidney disease. Other adverse prognostic factors include African or Asian ancestry, delayed diagnosis and treatment, poor adherence to maintenance therapy, persistent proteinuria, hypertension, antiphospholipid antibodies, and a low complement that fails to normalise. The Childhood Arthritis and Rheumatology Research Alliance registry data show that variation in care and delays in achieving a complete renal response contribute to worse short-term kidney status in childhood-onset disease. [9]
Pathophysiology
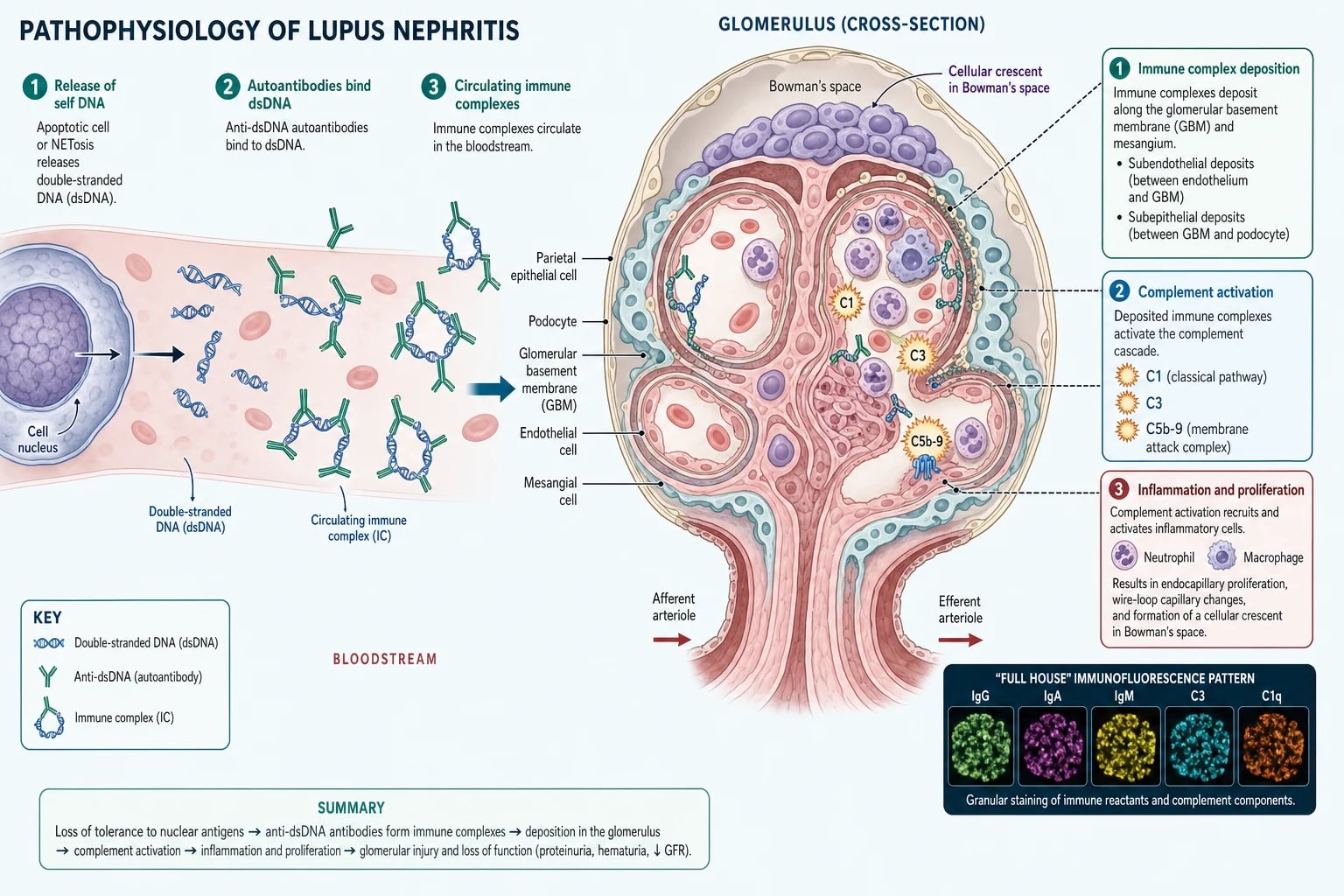
Lupus nephritis begins with a failure of immune tolerance to the body's own nuclear material. Apoptotic cells normally clear their nuclear debris cleanly, but in lupus this clearance is defective, and double-stranded DNA and other nucleosomes are released into the circulation. The immune system, having lost tolerance, generates high-affinity anti-dsDNA autoantibodies that bind these nuclear antigens to form circulating immune complexes. These complexes are not an innocent bystander: anti-dsDNA antibody titre correlates with disease activity and specifically with nephritis, and the complexes themselves are the agents that damage the kidney. [1]
The circulating immune complexes deposit in the glomerulus at three anatomical sites, and the site of deposition determines the class and severity. Mesangial deposits produce mild Class I and II disease. Subendothelial deposits, sitting between the endothelium and the glomerular basement membrane and therefore in contact with the bloodstream, trigger the most aggressive inflammation and produce the proliferative Class III and IV patterns with endocapillary proliferation, wire-loop lesions, necrosis, and crescents. Subepithelial deposits, sitting on the outer side of the basement membrane beneath the podocyte, produce the thickened capillary walls and spike-and-dome changes of membranous Class V disease. The same child can have deposits at more than one site, which is why combined patterns such as Class V plus Class IV occur. [1]
Once deposited, the immune complexes activate the classical complement pathway. C1q binds the antibodies, C3 convertase forms, and the cascade proceeds through C3 to the terminal C5b-9 membrane attack complex. Complement activation recruits neutrophils and macrophages, which release proteases and reactive oxygen species that injure the glomerular cells and basement membrane. The result is the active proliferative lesion: endocapillary hypercellularity that obliterates the capillary lumen, fibrinoid necrosis of the capillary wall, and cellular crescents formed by proliferating parietal epithelial cells in Bowman's space. These active lesions are potentially reversible with immunosuppression, which is why early induction therapy can rescue kidney function. Chronic lesions such as glomerular sclerosis, tubular atrophy, and interstitial fibrosis, by contrast, are irreversible and accumulate with each untreated flare. [1]
FULL HOUSE
The hallmark of lupus nephritis on immunofluorescence is the full-house pattern, in which granular staining is present for all major immunoglobulin classes (IgG, IgA, IgM) and complement components (C3, C1q). This simultaneous deposition of multiple immunoglobulins and the early classical-pathway component C1q distinguishes lupus from other glomerulonephritides, where the deposition is more restricted (for example IgA-dominant in IgA nephropathy). Full-house immunofluorescence is not perfectly sensitive, but it is highly suggestive of lupus and is one of the histological findings that, together with the clinical picture, cements the diagnosis. [1]
In Australia and New Zealand, paediatric lupus nephritis is managed collaboratively between paediatric rheumatology and paediatric nephrology, with all children biopsied at diagnosis and treated according to EULAR and ACR-aligned protocols. Access to mycophenolate mofetil, calcineurin inhibitors, and biologic agents is available through tertiary paediatric centres. Indigenous Australian and Maori and Pacific children are over-represented among the more severe phenotypes, reflecting the global pattern of ethnic predisposition. [9]
Clinical Presentation
The presentation of lupus nephritis sits on a spectrum from silent renal involvement detected only on screening urinalysis to fulminant rapidly progressive glomerulonephritis. The most common scenario in adolescence is a girl with established or newly diagnosed systemic lupus who develops oedema, hypertension, and urinary abnormalities during a disease flare. The systemic features may precede or accompany the renal presentation: malar rash, oral or nasal ulcers, photosensitivity, alopecia, arthralgia or arthritis, fatigue, fever, and weight loss. A smaller but important group presents with the renal disease first, and the lupus is only diagnosed when the biopsy shows full-house immunofluorescence or the bloods return positive anti-dsDNA antibodies. [9]
The renal findings depend on the histological class. Proliferative Class III and IV disease produces an active urinary sediment with microscopic or macroscopic haematuria, proteinuria that can range from sub-nephrotic to nephrotic, red cell casts, and a variable rise in creatinine. Hypertension is common and reflects the inflammatory and ischaemic injury to the glomerulus. Pure membranous Class V disease presents differently, with heavy proteinuria in the nephrotic range, oedema, and little or no haematuria, because the subepithelial deposits injure the podocyte and the filtration barrier without the endocapillary inflammation that produces bleeding. A child with pure Class V may therefore look identical to a child with primary nephrotic syndrome until the lupus serology and biopsy clarify the cause. [9]
Rapidly progressive glomerulonephritis is the feared presentation, defined by a rapid loss of renal function over days to weeks, accompanied by active sediment and often oliguria. It usually corresponds to a crescentic Class IV pattern with a high activity index and requires urgent biopsy and immediate induction therapy. Extrarenal lupus activity often runs in parallel with the nephritis, and features such as neuropsychiatric lupus (seizures, psychosis, cognitive dysfunction), haematological cytopenias, and serositis may be present. The antiphospholipid syndrome, which can coexist with lupus, adds a thrombotic risk that is magnified by the nephrotic state in Class V disease. [9]
Differential Diagnosis
When a child presents with haematuria, proteinuria, and a falling complement, lupus nephritis is high on the list, but several other glomerulonephritides must be excluded. Post-infectious glomerulonephritis also causes haematuria, oedema, and a low C3, but it typically follows a streptococcal pharyngitis or skin infection by one to two weeks, the C4 is normal (isolated low C3), the anti-dsDNA is negative, and the disease is self-limiting. In lupus, by contrast, both C3 and C4 fall (classical pathway activation), the anti-dsDNA is positive, and the disease is chronic and progressive without treatment. [1]
IgA nephropathy presents with haematuria, often synchronous with an upper respiratory tract infection, but it lacks the multisystem features, the hypocomplementaemia, and the positive autoantibodies of lupus. The biopsy in IgA nephropathy shows mesangial IgA-dominant deposition rather than the full-house pattern. Membranous nephropathy, whether primary (anti-phospholipase A2 receptor positive) or secondary, mimics Class V lupus nephritis with nephrotic-range proteinuria, and the distinction rests on the lupus serology, the immunofluorescence pattern, and the search for secondary causes. [1]
[1]Vasculitis is the other critical exclusion. ANCA-associated vasculitis (granulomatosis with polyangiitis and microscopic polyangiitis) can produce a crescentic, rapidly progressive glomerulonephritis that overlaps with severe Class IV lupus nephritis, but the ANCA is positive and the anti-dsDNA is negative. Henoch-Schonlein purpura (IgA vasculitis) produces a purpuric rash on the lower limbs, abdominal pain, and arthritis alongside the nephritis, and the biopsy shows IgA deposition. In any child with suspected lupus nephritis, the combination of antinuclear antibody, anti-dsDNA, complement levels, and the renal biopsy with immunofluorescence resolves the diagnosis. A negative antinuclear antibody makes lupus nephritis very unlikely but not impossible, because about 2 to 5 percent of lupus patients are seronegative. [1]
Clinical & Bedside Assessment
Assessment has two goals: to confirm that the kidney is involved and to gauge the systemic disease activity that surrounds it. The history probes for the cardinal renal symptoms (foamy or tea-coloured urine, reduced output, facial or ankle swelling, headache from hypertension) and the systemic lupus features (malar or discoid rash, photosensitivity, oral and nasal ulcers, alopecia, arthralgia, Raynaud phenomenon, fatigue, fever, weight loss, chest pain from serositis, and any neurological symptoms suggesting neuropsychiatric lupus). A menstrual and gynaecological history matters, both because lupus targets adolescent girls and because cyclophosphamide and mycophenolate are teratogenic and demand reliable contraception. [9]
Examination looks for the stigmata of active lupus and the consequences of renal failure. Cutaneous findings include the malar (butterfly) rash that spares the nasolabial folds, discoid lesions, livedo reticularis suggesting antiphospholipid syndrome, and vasculitic rashes. Oral and nasal ulcers, alopecia, and synovitis support the diagnosis. Cardiovascular examination checks the blood pressure, which is often elevated, and looks for signs of fluid overload (periorbital and peripheral oedema, raised venous pressure, crackles, gallop). Abdominal examination may reveal hepatosplenomegaly or ascites, and a careful neurological examination screens for the cognitive, psychiatric, and focal deficits of neuropsychiatric lupus. [9]
The bedside assessment must include a urinalysis and blood pressure on every child with known or suspected lupus, because renal involvement can be clinically silent. Dipstick proteinuria and haematuria, confirmed by microscopy showing dysmorphic red cells and casts, are the earliest signs. The recognition that an adolescent girl with swollen joints and a facial rash also has blood and protein in her urine is the single bedside observation that triggers the pathway from suspicion to biopsy to treatment. A child with active urinary sediment and a rising creatinine needs same-day nephrology involvement. [9]
Investigations
The diagnosis of lupus nephritis rests on three legs: serology, urinalysis and renal function, and the renal biopsy with light microscopy, immunofluorescence, and electron microscopy. Serology confirms lupus and gauges activity. The antinuclear antibody is positive in over 95 percent of patients and is the screening test. Anti-dsDNA antibodies are highly specific for lupus, correlate with disease activity and nephritis, and are the antibody that forms the nephritogenic immune complexes. Complement C3 and C4 are low during active disease because the classical pathway is being consumed, and a rising anti-dsDNA with a falling complement is the serological signature of a flare. The antiphospholipid antibodies (lupus anticoagulant, anticardiolipin, and anti-beta-2-glycoprotein I) identify the thrombotic risk. [12]
Urine and renal function quantify the damage and monitor the response. The urine shows haematuria with dysmorphic red cells and red cell casts, proteinuria that should be quantified by the albumin-to-creatinine or protein-to-creatinine ratio, and the serum creatinine and estimated glomerular filtration rate define the degree of renal impairment. A spot urine protein-to-creatinine ratio above 0.5 indicates significant proteinuria, and a ratio above 3.0 defines nephrotic-range proteinuria. Novel biomarkers such as urinary neutrophil gelatinase-associated lipocalin (NGAL) can detect subclinical renal activity and predict flares before the proteinuria or creatinine changes, as demonstrated by Hinze and colleagues in childhood-onset lupus, though these remain adjunctive rather than routine. [12]
The renal biopsy is the definitive investigation and is indicated in every child with suspected lupus nephritis, because the histological class determines the treatment. The biopsy provides the ISN/RPS class, the activity index (a measure of reversible inflammation scored from cellular crescents, fibrinoid necrosis, and other active lesions), and the chronicity index (a measure of irreversible damage scored from glomerular sclerosis, tubular atrophy, and interstitial fibrosis). The activity-versus-chronicity distinction is clinically vital: high activity with low chronicity predicts a good response to immunosuppression, while a high chronicity index predicts irreversible damage and a poorer outcome. Immunofluorescence shows the full-house pattern, and electron microscopy confirms the site of the immune deposits. A biopsy is also indicated when a child on treatment deteriorates, to distinguish a flare (new active lesions) from cumulative chronic damage or drug toxicity. [1]
Management — Resuscitation
Resuscitation targets the immediate threats to the kidney and the child before the definitive immunosuppression begins. Severe hypertension from the nephritic process needs prompt control to prevent hypertensive encephalopathy and further renal injury, using calcium channel blockers such as amlodipine (0.1 to 0.2 mg per kg per dose once daily, maximum 10 mg per day) or, in a hypertensive emergency, an intravenous infusion such as labetalol (0.25 to 1 mg per kg per hour) or nicardipine (0.5 to 1 microgram per kg per minute). Fluid overload from oliguric renal failure is treated with salt restriction and loop diuretics such as furosemide (1 to 2 mg per kg per dose), and dialysis is reserved for refractory hyperkalaemia, severe acidosis, or fluid overload that does not respond to diuretics. [9]
The nephrotic state of Class V disease carries a thrombotic risk that must be addressed. Severe hypoalbuminaemia (albumin below 25 g per litre) warrants prophylactic anticoagulation, because renal vein thrombosis and pulmonary embolism are recognised complications of nephrotic-range proteinuria, especially when antiphospholipid antibodies are present. Concurrent electrolyte and acid-base disturbances (hyponatraemia from fluid retention, hyperkalaemia from renal failure, hypocalcaemia, and metabolic acidosis) are corrected with the standard paediatric approaches, while the team prepares for induction therapy. [9]
Before starting any immunosuppression, the child needs screening for latent infections that the drugs will reactivate. Tuberculosis screening (chest radiograph, interferon-gamma release assay, or tuberculin skin test), hepatitis B and C, and human immunodeficiency virus serology are sent. Live vaccines (notably measles, mumps, and rubella, and varicella) should be given before immunosuppression begins or deferred until the immunosuppression is reduced. The pneumococcal, influenza, hepatitis B, and human papillomavirus vaccines are safe and recommended, and bone health is protected with calcium, vitamin D, and consideration of a bisphosphonate for children on prolonged glucocorticoids. [7]
Management — Definitive & Stepwise
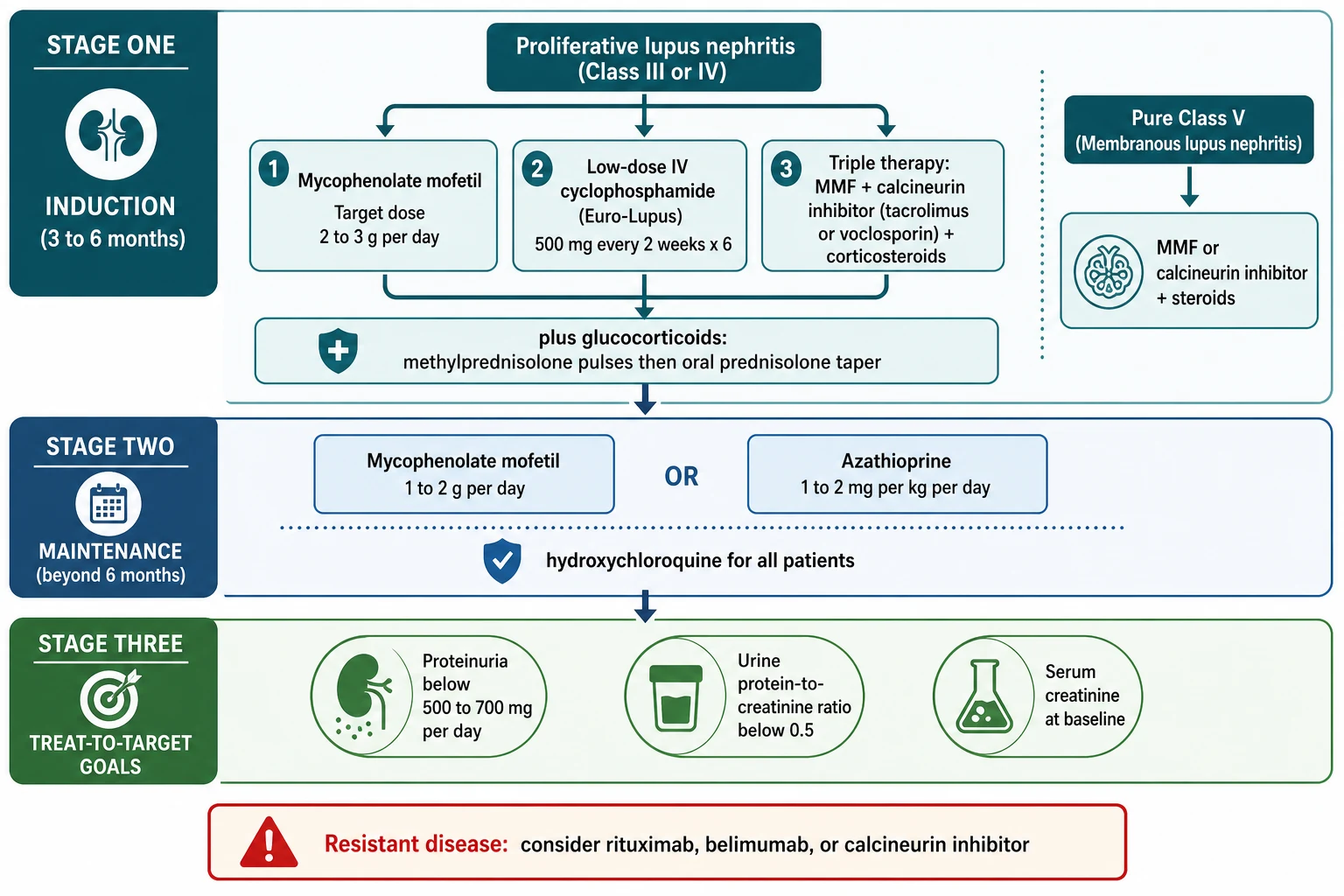
[7]Definitive management is built on an induction-maintenance paradigm that applies to proliferative Class III and IV disease, the patterns that dominate childhood lupus nephritis. The goal of induction, lasting 3 to 6 months, is to suppress the active inflammation and prevent irreversible glomerular damage. The goal of maintenance, lasting years, is to consolidate the remission and prevent flares. The current evidence-based induction regimen, endorsed by both the EULAR and the American College of Rheumatology guidelines, is mycophenolate mofetil plus glucocorticoids, or low-dose intravenous cyclophosphamide plus glucocorticoids, with a calcineurin-inhibitor-based triple-therapy option that is particularly effective in Asian populations. [7]
Mycophenolate mofetil has become the preferred induction agent for most children because it matches cyclophosphamide for efficacy while avoiding gonadal toxicity. The pivotal trials established this equivalence: Ginzler and colleagues showed that mycophenolate was at least as effective as intravenous cyclophosphamide for induction, and the Aspreva Lupus Management Study (ALMS) by Appel and colleagues confirmed that mycophenolate mofetil was non-inferior to cyclophosphamide for induction of remission. The target dose is 2 to 3 g per day in adults and older children, achieved by gradual up-titration to manage gastrointestinal side effects, and Sundel and colleagues confirmed mycophenolate efficacy specifically in adolescent patients. Mycophenolate is teratogenic and demands reliable contraception in adolescent girls. [2]
Mycophenolate mofetil
Dose
Target 2 to 3 g per day in two divided doses for induction; 1 to 2 g per day for maintenance. Start at a lower dose and up-titrate over 2 to 4 weeks to minimise gastrointestinal adverse effects.
Low-dose intravenous cyclophosphamide, the Euro-Lupus regimen, is the alternative induction agent and remains valuable, especially in severe disease with crescents or in resource settings where mycophenolate access is limited. It gives 500 mg intravenously every 2 weeks for 6 doses, a markedly lower cumulative exposure than the older high-dose NIH regimen (0.5 to 1 g per square metre monthly for 6 months). The dose reduction preserves efficacy while cutting toxicity. The major concern with cyclophosphamide is gonadal toxicity, which is dose-dependent and age-dependent and threatens future fertility, particularly in adolescent girls approaching reproductive age. This is why mycophenolate is preferred first-line in children, and why gonadotropin-releasing hormone agonist protection is considered for girls receiving cyclophosphamide. [4]
The multi-target or triple-therapy approach combines mycophenolate mofetil with a calcineurin inhibitor (tacrolimus or cyclosporin) and glucocorticoids, and has shown superior remission rates to intravenous cyclophosphamide in trials, particularly in East Asian populations. Calcineurin inhibitors stabilise the podocyte cytoskeleton and reduce proteinuria independently of their immunosuppressive effect. Tacrolimus is dosed at 0.05 to 0.1 mg per kg per day in two divided doses, with trough-level monitoring and surveillance for nephrotoxicity, hypertension, hyperkalaemia, and glucose intolerance. Voclosporin, a novel calcineurin inhibitor that does not require therapeutic drug monitoring, demonstrated efficacy in the AURORA 1 trial by Rovin and colleagues and is approved for adult lupus nephritis, with paediatric experience emerging. All induction regimens are accompanied by glucocorticoids: intravenous methylprednisolone pulses (500 mg to 1 g, or 30 mg per kg, daily for 3 days) followed by oral prednisolone (0.5 to 1 mg per kg per day, maximum 60 mg per day) tapered to the lowest effective dose. [5]
Confirm class and activity
Induction for 3 to 6 months
Add hydroxychloroquine
Maintenance beyond 6 months
Treat to target
Maintenance therapy follows induction and is essential to prevent relapse. The two evidence-based options are mycophenolate mofetil (1 to 2 g per day) and azathioprine (1 to 2 mg per kg per day). The landmark trial by Contreras and colleagues demonstrated that mycophenolate and azathioprine were superior to long-term intravenous cyclophosphamide for maintenance, with fewer flares and less toxicity. Hydroxychloroquine (5 mg per kg per day in children) is prescribed for every patient with lupus nephritis regardless of class, because it reduces flares, improves renal survival, and lowers the thrombotic and metabolic risks of glucocorticoids. The dose is weight-based to minimise retinal toxicity, and annual ophthalmological screening is required. [4]
The treat-to-target framework defines success in measurable renal terms. A complete renal response requires the urine protein-to-creatinine ratio to fall below 0.5 (or proteinuria below 500 mg per day), the creatinine to return to baseline, and the active sediment to resolve. A partial renal response is a 50 percent or greater reduction in proteinuria with stable or improving renal function. Achieving a complete response within the first year predicts long-term renal survival, and persistent proteinuria is the strongest predictor of progression to end-stage kidney disease. This is why every clinic visit quantifies the protein-to-creatinine ratio, the creatinine, the complement, and the anti-dsDNA titre, and adjusts therapy toward the target. [7]
Refractory disease, defined as failure to achieve a partial response after 6 months of standard induction, demands escalation. The options are a switch between mycophenolate and cyclophosphamide, the addition of a calcineurin inhibitor (multi-target therapy), or a biologic agent. Rituximab, a chimeric anti-CD20 monoclonal antibody that depletes B cells, is the most commonly used biologic. The LUNAR trial by Rovin and colleagues did not meet its primary endpoint as an add-on to mycophenolate and steroids for proliferative disease, but rituximab is widely used off-label in refractory or relapsing lupus nephritis and is effective in clinical practice for patients who have failed standard therapy. Belimumab, a monoclonal antibody that neutralises B-cell activating factor, is approved as an add-on for lupus nephritis and reduces the risk of renal flares. Plasma exchange and intravenous immunoglobulin have limited evidence and are reserved for specific scenarios such as concomitant catastrophic antiphospholipid syndrome. [6]
Specific Subtypes & Scenarios
Class IV diffuse proliferative lupus nephritis is the dominant paediatric subtype and the one that drives most of the morbidity and the exam questions. It involves 50 percent or more of glomeruli in active or chronic proliferative lesions, shows subendothelial deposits and wire-loop changes, and frequently features cellular crescents and fibrinoid necrosis that mark severe active inflammation. It is the pattern most likely to present with an active sediment, hypertension, and a rising creatinine, and it carries the highest risk of progression to end-stage kidney disease if not treated promptly. Standard induction with mycophenolate or low-dose cyclophosphamide plus glucocorticoids, followed by maintenance, is the treatment, and the activity-versus-chronicity index on the biopsy refines the prognosis. [1]
Pure membranous Class V lupus nephritis presents a different clinical problem. The subepithelial deposits injure the podocyte and produce nephrotic-range proteinuria with oedema but little haematuria and preserved renal function in the early stages. The treatment of pure Class V is mycophenolate mofetil or a calcineurin inhibitor plus glucocorticoids, with a particular focus on managing the nephrotic state: proteinuria control, oedema with diuretics and salt restriction, lipid management, and thromboprophylaxis for severe hypoalbuminaemia. Class V combined with Class III or IV is treated as the proliferative component dictates, with induction for the active proliferative lesions. [7]
Combined Class V plus IV is a common and challenging scenario in which both subepithelial and subendothelial deposits coexist. The child presents with both heavy proteinuria (from the membranous component) and an active sediment with a rising creatinine (from the proliferative component). Treatment follows the proliferative arm, because the subendothelial inflammation is the immediate threat to renal function, but the proteinuria often takes longer to resolve. Childhood-onset lupus nephritis, as a whole, is more aggressive than adult-onset disease: the Childhood Arthritis and Rheumatology Research Alliance registry documented substantial variation in care and outcomes, with a significant proportion of children not achieving a complete renal response in the short term. [9]
ALMS induction trial
Key finding
The Aspreva Lupus Management Study was a randomised trial comparing mycophenolate mofetil with intravenous cyclophosphamide for induction of proliferative lupus nephritis. Mycophenolate was non-inferior to cyclophosphamide for achieving remission, with comparable efficacy and a favourable safety profile, supporting mycophenolate as first-line induction.
Practice change
Mycophenolate mofetil is the preferred first-line induction agent for proliferative lupus nephritis in children, matching cyclophosphamide for efficacy while avoiding gonadal and bladder toxicity.
Lupus nephritis in pregnancy, relevant to older adolescents and young women in transition, requires careful planning. Active nephritis, especially proliferative disease, increases the risk of pre-eclampsia, fetal loss, and preterm delivery, and mycophenolate and cyclophosphamide are teratogenic and contraindicated in pregnancy. Disease should be in stable remission before conception, and maintenance is switched to pregnancy-compatible agents such as azathioprine, hydroxychloroquine, and low-dose glucocorticoids. The antiphospholipid syndrome adds a thrombotic and recurrent-miscarriage risk that is managed with aspirin and heparin. [7]
Complications & Pitfalls
The complications of lupus nephritis are the consequences of the disease itself, the immunosuppression used to treat it, and the glucocorticoids. Disease-driven complications include progression to end-stage kidney disease, the endpoint that induction and maintenance therapy are designed to prevent; hypertension that persists and damages the kidney further; and the thrombotic events of the nephrotic state and antiphospholipid syndrome. The strongest predictor of progression to end-stage kidney disease is persistent proteinuria, which is why the treat-to-target proteinuria goal exists. [9]
The immunosuppressive drugs carry serious risks that must be anticipated and monitored. Mycophenolate and azathioprine cause leukopenia, anaemia, and thrombocytopenia, demanding regular full blood counts, and they increase the susceptibility to infection. Cyclophosphamide adds the risks of haemorrhagic cystitis (mitigated by mesna and aggressive hydration), secondary malignancy, and irreversible gonadal toxicity, which is the dose-dependent and age-dependent threat to fertility that makes mycophenolate the preferred first-line agent in children. Calcineurin inhibitors cause nephrotoxicity, hypertension, hyperkalaemia, and glucose intolerance, and require trough-level monitoring. Biologics increase the risk of infusion reactions and infection. Every child on long-term immunosuppression needs vaccination with pneumococcal, influenza, hepatitis B, and human papillomavirus vaccines (avoiding live vaccines) and vigilance for opportunistic infection. [7]
Glucocorticoid toxicity is a major contributor to long-term morbidity and must be actively managed. Growth suppression, weight gain, hypertension, glucose intolerance and steroid-induced diabetes, osteoporosis and avascular necrosis, cataracts and glaucoma, and mood disturbance and psychosis all accompany prolonged glucocorticoid therapy. The strategy is to use pulses for rapid control of severe inflammation and then taper the oral dose as quickly as the disease allows, using steroid-sparing agents (mycophenolate, calcineurin inhibitors, biologics) to maintain remission on the lowest possible glucocorticoid dose. Bone protection with calcium, vitamin D, and bisphosphonate consideration, ophthalmological surveillance, and blood pressure and glucose monitoring are part of every long-term plan. [7]
The most dangerous clinical pitfall is delay. Delaying the biopsy in a child with active urinary sediment and falling complement delays the diagnosis of proliferative Class IV disease and allows irreversible glomerular scarring to accumulate. Under-treating severe disease, by using inadequate induction or stopping maintenance prematurely, invites relapse and cumulative damage. Over-treating mild Class II disease with aggressive immunosuppression exposes the child to needless toxicity for a pattern that usually needs only hydroxychloroquine. Failing to achieve the proteinuria target, dismissing persistent proteinuria as acceptable, and neglecting the thromboprophylaxis of severe nephrotic Class V are all avoidable errors. [1]
Prognosis & Disposition
The prognosis of lupus nephritis in children has improved dramatically with the induction-maintenance paradigm and mycophenolate-based therapy, but it remains a serious disease with a significant minority progressing to end-stage kidney disease. The strongest predictor of long-term renal survival is the achievement of a complete renal response, defined as proteinuria below 500 mg per day and creatinine at baseline, within the first year. Persistent proteinuria, a high chronicity index, African or Asian ancestry, delayed treatment, and poor adherence all predict progression to end-stage kidney disease. Roughly 10 to 20 percent of children with severe proliferative disease progress to end-stage kidney disease over the long term despite optimal therapy. [9]
Severity
Class I or II lupus nephritis
Mesangial-only disease with mild proteinuria and normal renal function. Hydroxychloroquine and monitoring. Excellent renal prognosis.
Severity
Class III or V lupus nephritis
Focal proliferative or membranous disease needing induction and maintenance. Good prognosis with adherence, with risk of progression if undertreated.
Severity
Class IV lupus nephritis
Diffuse proliferative disease with crescents, high activity index, and rising creatinine. High risk of end-stage kidney disease. Needs urgent induction and lifelong surveillance.
Children who progress to end-stage kidney disease are managed with dialysis and renal transplantation, and lupus nephritis can recur in the transplant graft, though recurrence rates are lower than in diseases such as atypical haemolytic uraemic syndrome. Transplantation is generally deferred until the lupus has been in clinical and serological remission for at least 6 to 12 months to minimise recurrence, and the outcomes in children are comparable to transplantation for other causes of end-stage kidney disease. Long-term mortality in childhood-onset lupus nephritis is driven by cardiovascular disease (accelerated atherosclerosis from chronic inflammation and glucocorticoids), infection (from immunosuppression), and renal failure. [9]
Disposition depends on the severity and the treatment phase. Acute severe presentations (rapidly progressive glomerulonephritis, hypertensive emergency, severe nephrotic state) are admitted for induction, blood pressure control, and complication management, often in a high-dependency or intensive care setting. Stabilised children on maintenance therapy are managed as outpatients with regular monitoring of the protein-to-creatinine ratio, creatinine, complement, anti-dsDNA, full blood count, and drug levels. Every child needs a structured transition to adult nephrology and rheumatology services in late adolescence, because loss to follow-up in the transition period is a major cause of relapse and progression. [9]
Special Populations
Adolescent girls are the largest paediatric group with lupus nephritis, and their management is shaped by two reproductive considerations. First, mycophenolate, cyclophosphamide, and the newer biologics are teratogenic or have unknown fetal effects, so reliable contraception and pregnancy planning are non-negotiable, and treatment must be switched to pregnancy-compatible agents before conception is attempted. Second, cyclophosphamide threatens future ovarian reserve, which is the rationale for mycophenolate first-line and for gonadotropin-releasing hormone agonist protection when cyclophosphamide is used. Counselling the adolescent and her family about these issues, with sensitivity and confidentiality, is a core skill. [7]
Children of African, Asian, Hispanic, and South Asian ancestry have more severe disease. The proliferative Class IV pattern, the high activity index, and the risk of progression to end-stage kidney disease are all over-represented in these groups, reflecting a combination of genetic susceptibility and socio-environmental factors. This ethnic gradient, also documented in the adult ALMS subgroup analyses, means that these children warrant a lower threshold for biopsy, close monitoring, and aggressive induction. [9]
The rare prepubertal child with lupus nephritis (onset before 10 years, and rarely before 5 years) tends to have more aggressive disease with a higher frequency of proliferative patterns and organ involvement, and deserves early aggressive therapy. Children with antiphospholipid syndrome complicating their lupus need lifelong anticoagulation after a thrombotic event, and the combination of nephrotic-range proteinuria and antiphospholipid antibodies is particularly thrombogenic. Young people transitioning to adult care need a structured handover of the biopsy findings, the treatment history, the cumulative cyclophosphamide dose, and the reproductive and cardiovascular risk profile, because these define the adult management plan. [9]
Evidence, Guidelines & Regional Differences
Two guideline frameworks dominate current practice. The 2019 joint EULAR and European Renal Association-European Dialysis and Transplant Association recommendations, updated by Fanouriakis and colleagues, endorse mycophenolate mofetil or low-dose intravenous cyclophosphamide with glucocorticoids as first-line induction for proliferative disease, and mycophenolate or azathioprine for maintenance, with hydroxychloroquine for all patients and a treat-to-target goal of complete renal response. The 2024 American College of Rheumatology guideline reinforces these principles, emphasises early biopsy and induction, and addresses the role of calcineurin inhibitors and biologic agents in refractory disease. Both align on the central message: induce with mycophenolate or low-dose cyclophosphamide plus steroids, maintain for years, and treat to a proteinuria target. [8]
The evidence base for induction is built on three pivotal trials. Ginzler and colleagues demonstrated mycophenolate efficacy against cyclophosphamide, and the ALMS trial confirmed non-inferiority. The Contreras maintenance trial established mycophenolate and azathioprine as superior to long-term cyclophosphamide. The AURORA 1 trial established voclosporin as an effective add-on for adult lupus nephritis, and the LUNAR trial, though negative for its primary endpoint, informed the off-label use of rituximab in refractory disease. The Childhood Arthritis and Rheumatology Research Alliance consensus treatment plans, led by Mina and colleagues, have standardised paediatric induction and enable collaborative outcome research. [10]
Regional differences in lupus nephritis management are shaped by ethnicity and drug access. The multi-target (triple-therapy) approach combining mycophenolate with a calcineurin inhibitor is favoured in East Asia, where trials showed superior remission rates, and it is increasingly adopted elsewhere. Voclosporin access is expanding but remains concentrated in high-income health systems. In resource-limited settings, cyclophosphamide remains more accessible than mycophenolate or biologics, and the approach leans on the low-dose Euro-Lupus regimen. The International Pediatric Nephrology Association supports the adaptation of these guidelines to local capacity, while preserving the core principles of early biopsy, adequate induction, sustained maintenance, and treat-to-target monitoring. [9]
Exam Pearls
The ISN/RPS 2003 classification assigns six classes, and the exam-critical distinction is proliferative versus non-proliferative. Class I (minimal mesangial) and Class II (mesangial proliferative) are mild, mesangial-only, and need little immunosuppression. Class III (focal proliferative, under 50 percent of glomeruli) and Class IV (diffuse proliferative, 50 percent or more) are the severe patterns that demand induction therapy, and Class IV is the most common and most severe paediatric pattern, subdivided into IV-S segmental and IV-G global. Class V (membranous) is defined by subepithelial deposits and nephrotic-range proteinuria. Class VI (advanced sclerosing, over 90 percent global sclerosis) is irreversible. [1]
The pathophysiology chain is high-yield: loss of tolerance to nuclear antigens, anti-dsDNA antibody formation, circulating immune complex deposition in the glomerulus (mesangial, subendothelial, or subepithelial), classical complement pathway activation with low C3 and C4, and the full-house immunofluorescence pattern (granular IgG, IgA, IgM, C3, and C1q). Subendothelial deposits produce wire-loop lesions and the proliferative pattern, while subepithelial deposits produce the membranous pattern. A rising anti-dsDNA with a falling complement signals a flare. [1]
The induction-maintenance paradigm is the treatment backbone. Induction for 3 to 6 months uses mycophenolate mofetil (target 2 to 3 g per day) or low-dose Euro-Lupus cyclophosphamide (500 mg every 2 weeks for 6 doses) plus glucocorticoid pulses and a prednisolone taper. Maintenance for years uses mycophenolate (1 to 2 g per day) or azathioprine (1 to 2 mg per kg per day). Hydroxychloroquine (5 mg per kg per day) is given to every patient. Mycophenolate is preferred over cyclophosphamide first-line in children because cyclophosphamide is gonadotoxic. Treat to target: complete renal response is proteinuria below 500 mg per day with creatinine at baseline, and achieving this in the first year predicts long-term renal survival. [7]
References
- [1]Weening JJ, D'Agati VD, Schwartz MM, et al The classification of glomerulonephritis in systemic lupus erythematosus revisited. J Am Soc Nephrol, 2004.PMID 14747370
- [2]Ginzler EM, Dooley MA, Aranow C, et al Mycophenolate mofetil or intravenous cyclophosphamide for lupus nephritis. N Engl J Med, 2005.PMID 16306519
- [3]Appel GB, Contreras G, Dooley MA, et al Mycophenolate mofetil versus cyclophosphamide for induction treatment of lupus nephritis. J Am Soc Nephrol, 2009.PMID 19369404
- [4]Contreras G, Pardo V, Leclercq B, et al Sequential therapies for proliferative lupus nephritis. N Engl J Med, 2004.PMID 14999109
- [5]Rovin BH, Teng YKO, Ginzler EM, et al Efficacy and safety of voclosporin versus placebo for lupus nephritis (AURORA 1): a double-blind, randomised, multicentre, placebo-controlled, phase 3 trial. Lancet, 2021.PMID 33971155
- [6]Rovin BH, Furie R, Latinis K, et al Efficacy and safety of rituximab in patients with active proliferative lupus nephritis: the Lupus Nephritis Assessment with Rituximab study. Arthritis Rheum, 2012.PMID 22231479
- [7]Fanouriakis A, Kostopoulou M, Cheema K, et al 2019 Update of the Joint European League Against Rheumatism and European Renal Association-European Dialysis and Transplant Association (EULAR/ERA-EDTA) recommendations for the management of lupus nephritis. Ann Rheum Dis, 2020.PMID 32220834
- [8]Sammaritano LR, Askanase A, Bermas BL, et al 2024 American College of Rheumatology (ACR) Guideline for the Screening, Treatment, and Management of Lupus Nephritis. Arthritis Rheumatol, 2025.PMID 40331662
- [9]Smitherman EA, Chahine RA, Beukelman T, et al Childhood-Onset Lupus Nephritis in the Childhood Arthritis and Rheumatology Research Alliance Registry: Short-Term Kidney Status and Variation in Care. Arthritis Care Res (Hoboken), 2023.PMID 36775844
- [10]Mina R, von Scheven E, Ardoin SP, et al Consensus treatment plans for induction therapy of newly diagnosed proliferative lupus nephritis in juvenile systemic lupus erythematosus. Arthritis Care Res (Hoboken), 2012.PMID 22162255
- [11]Sundel R, Solomons N, Lisk L Efficacy of mycophenolate mofetil in adolescent patients with lupus nephritis: evidence from a two-phase, prospective randomized trial. Lupus, 2012.PMID 22922564
- [12]Hinze CH, Suzuki M, Klein-Gitelman M, et al Neutrophil gelatinase-associated lipocalin is a predictor of the course of global and renal childhood-onset systemic lupus erythematosus disease activity. Arthritis Rheum, 2009.PMID 19714584