Paeds · nephrology-urology-fluids-and-electrolytes
Oedema and nephrotic syndrome
Also known as Childhood nephrotic syndrome · Minimal change nephrotic syndrome · Steroid-sensitive nephrotic syndrome · Frequently relapsing nephrotic syndrome · Steroid-dependent nephrotic syndrome · Nephrotic-range proteinuria in children
Fellowship guide to oedema and nephrotic syndrome in children: the KDIGO 2021 definition built on oedema, nephrotic-range proteinuria and hypoalbuminaemia, the rule that minimal change disease dominates under ten years and responds to steroids in about ninety per cent, the initial prednisolone 60 mg/m2/day regimen capped at 60 mg/day and not prolonged beyond twelve weeks, the steroid-response classification that separates steroid-sensitive from steroid-resistant disease, and the three life-threatening complications — infection, thromboembolism and acute kidney injury — that kill children with nephrotic syndrome, not the proteinuria itself.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
The organising idea of this topic is that childhood nephrotic syndrome is a steroid-response disease with three dangerous complications. The KDIGO 2021 guideline and the IPNA recommendations frame the whole pathway around that idea: confirm with a first morning urine sample, treat with a defined steroid course, classify the response at four weeks, and remain alert for infection, thrombosis and hypovolaemia throughout. [1] [5] Almost every relapse is managed in the community with home urine testing, and the infrequently-relapsing steroid-sensitive child does well; the danger sits in the steroid-resistant child, in the frequently-relapsing or steroid-dependent child accumulating steroid toxicity, and in any child who becomes unwell during a relapse. [11]
This page covers the KDIGO definition, the response-based classification, the minimal-change pathophysiology, the initial and relapse steroid regimens, the steroid-sparing agents for frequently-relapsing and steroid-dependent disease, the work-up and management of steroid resistance, and the recognition and treatment of the complications. It links to the nephritic-syndrome and glomerulonephritis leaves for the haematuria-and-hypertension phenotype, which behaves nothing like minimal change disease. [1] [6]
Overview & Definition
Nephrotic syndrome is the clinical state produced when the glomerular filtration barrier loses its selectivity and leaks albumin faster than the liver can replace it. The KDIGO 2021 definition combines three findings: oedema, nephrotic-range proteinuria, and hypoalbuminaemia. [1] In children the proteinuria threshold is a first-morning urine protein-to-creatinine ratio of at least 200 mg/mmol (about 2 g/g), or 3+ or more on a dipstick reading, sustained rather than a single transient reading. The corresponding quantitative figure is roughly 40 mg/m2/hour of urinary protein, which is the historical reference standard. [10]
The serum albumin in nephrotic syndrome falls to 25 g/L or below, and most children also have hyperlipidaemia from the upregulated hepatic synthesis that accompanies albumin loss. The oedema is the sign that brings the child to attention: it is often first noticed around the eyes in the morning and is the reason parents seek help. The definition is deliberately a package, because oedema alone is non-specific and heavy proteinuria without a fall in albumin does not produce the full syndrome. [11]
The first practical rule is to confirm the proteinuria on a first morning urine sample, because orthostatic proteinuria — benign proteinuria present only when upright — is common in children and will give a falsely high reading on a random daytime sample. A first-morning protein-to-creatinine ratio below 50 mg/mmol makes nephrotic-range proteinuria unlikely. The combination of a first-morning ratio at or above 200 mg/mmol, a serum albumin of 25 g/L or below, and oedema is the package that commits the child to the nephrotic pathway. [10]
Classification
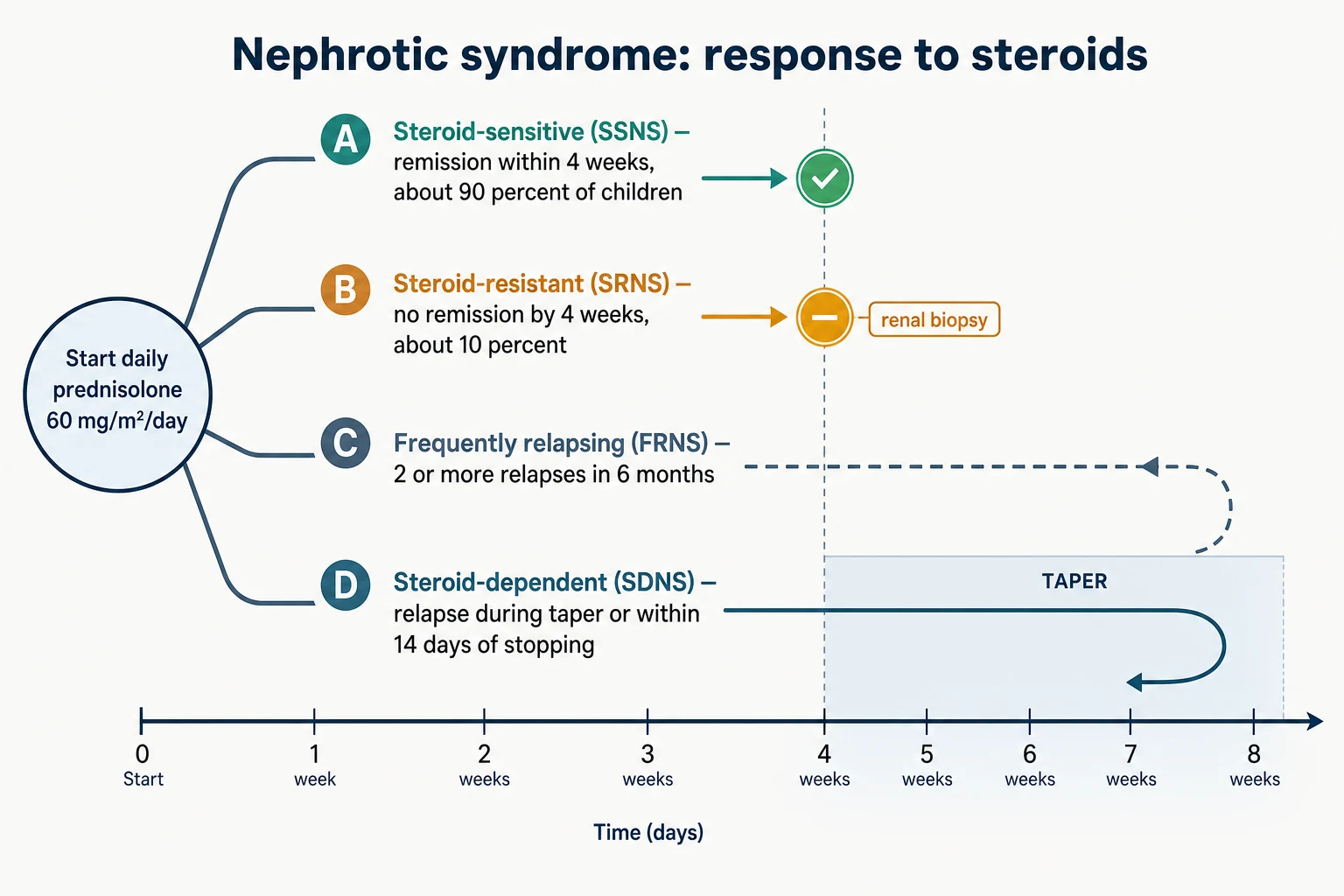
Classification in childhood nephrotic syndrome is response-based, not histology-based. The KDIGO 2021 guideline classifies children by what happens to the proteinuria after a defined course of daily steroids, because the response predicts the long-term course far better than the biopsy appearance does. [1] The histological label matters most for the steroid-resistant child, where a biopsy and genetic panel change management; for the steroid-sensitive child, biopsy is not part of routine first-line care. [5]
[1]A relapse is defined as proteinuria of 3+ or more (or a protein-to-creatinine ratio at or above 200 mg/mmol) for three consecutive days, and remission is trace or negative protein for three consecutive days. The frequently-relapsing label is applied once a child has had two or more relapses within six months or four or more within twelve months. Steroid dependence is the stricter label reserved for the child who relapses whenever the dose is lowered or within fourteen days of stopping, and it carries a higher chance of needing a steroid-sparing drug. These definitions are not academic: they trigger the move from a relapsing course managed with steroids alone to a course that needs a calcineurin inhibitor or rituximab. [1] [4]
Epidemiology & Risk Factors
Nephrotic syndrome is the commonest glomerular disease of childhood, with an incidence of one to three new cases per 100,000 children per year in Western populations and higher rates reported in South Asian and African populations. The peak age at first presentation is between two and six years, and boys are affected about twice as often as girls in the youngest age band, though the sex difference equalises by adolescence. [10]
The dominant histological pattern in young children is minimal change nephrotic syndrome, which accounts for around 85 per cent of cases under ten years. Focal segmental glomerulosclerosis becomes proportionally more common in adolescence and in steroid-resistant disease, and membranous nephropathy is uncommon in young children. The risk factors for a more difficult course are age under one year or over ten years at onset, initial steroid resistance, a higher number of relapses in the first year, and a family history of nephrotic syndrome, which raises the possibility of a monogenic cause. [6] [5]
Pathophysiology
Minimal change nephrotic syndrome is a podocyte disease driven by immune dysregulation rather than by inflammation or immune-complex deposition. The leading model is that an aberrant T-cell response releases a circulating permeability factor that acts on the podocyte, causing its foot processes to efface and flattening the slit diaphragm. The barrier loses its size-and-charge selectivity, and albumin — small and negatively charged, normally held back by the polyanionic barrier — leaks freely into the urine. [2] [10]
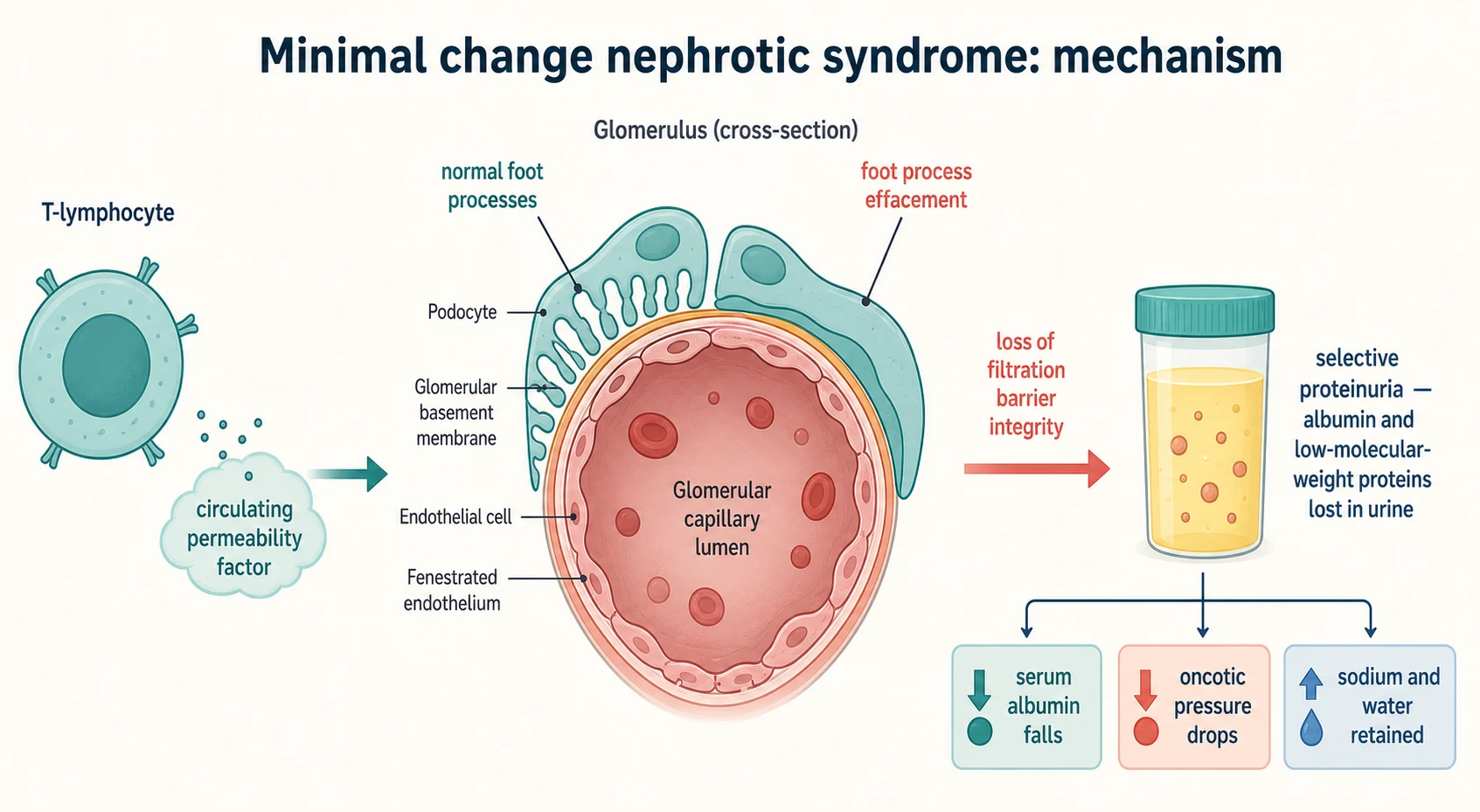
The oedema that follows is the product of two mechanisms acting together. The loss of albumin lowers the plasma oncotic pressure, shifting fluid into the interstitium and contracting the intravascular volume; this is the underfill pathway. At the same time the diseased kidney retains sodium directly through increased activity of the epithelial sodium channel, which is the overfill or primary-sodium-retention pathway. The balance between the two varies between children and even within a single relapse, and it is the reason a nephrotic child can be oedematous yet intravascularly depleted — the dangerous state that diuretics worsen. [2]
The selectivity of the protein loss matters clinically. In minimal change disease the leak is selective, dominated by albumin and smaller proteins rather than by large immunoglobulins, which is why the urine is frothy and the serum IgG is low while complement is normal. In focal segmental glomerulosclerosis and in the more damaged glomeruli of steroid-resistant disease the leak is non-selective. The accompanying urinary loss of binding proteins explains several of the complications: loss of immunoglobulin and alternative-complement factor B raises infection risk, loss of antithrombin drives thrombosis, loss of thyroxine-binding globulin can lower free thyroid hormone, and loss of cholecalciferol-binding protein contributes to bone disease. [8] [10]
Clinical Presentation
The typical child presents between two and six years with oedema that has built up over days to a week. Parents first notice puffiness around the eyes in the morning that may be dismissed as allergy, followed by swelling of the ankles and legs during the day, and sometimes abdominal distension from ascites or scrotal or labial oedema. The urine is often described as frothy. A preceding upper respiratory tract infection is reported in many children in the days before onset, but it is a trigger rather than the cause. [10]
The examination must establish the extent and distribution of the oedema and, crucially, the intravascular volume status. Look for periorbital and peripheral oedema, ascites, pleural effusions, and scrotal or labial oedema. Then assess for hypovolaemia: tachycardia, a narrow pulse pressure, prolonged capillary refill, cool peripheries, oliguria and abdominal pain are the signs that the oedema is interstitial while the circulating volume is depleted. The absence of haematuria, hypertension and renal failure is the pattern that fits minimal change disease; their presence demands consideration of a nephritic or steroid-resistant process and prompts biopsy. [7]
Blood pressure in minimal change disease is usually normal, though a slightly elevated reading can occur with steroid therapy or with fluid overload. Growth is usually normal at first presentation, but growth failure and stunted height emerge in the frequently-relapsing child on long-term steroids. A child under one year with nephrotic syndrome is in a different category: congenital or infantile nephrotic syndrome is usually genetic and does not respond to steroids, and the pathway diverges early. [9]
Differential Diagnosis
The first differential is between nephrotic and nephritic presentations, because they demand different management. A nephritic picture — haematuria, hypertension, renal impairment and a lower-grade proteinuria — points to post-infectious glomerulonephritis, IgA nephropathy or IgA vasculitis nephritis, none of which is managed with high-dose steroids in the first instance. The nephrotic child, by contrast, has dominant oedema and heavy proteinuria with little or no haematuria and a normal blood pressure. [1]
Nephrotic versus nephritic presentation
The second differential, once nephrotic syndrome is confirmed, is the cause of the protein leak. In a child under ten years presenting with the classic picture, minimal change disease is assumed and a biopsy is not performed before treatment. A biopsy at first presentation is reserved for the atypical child — age under one or over ten years, overt haematuria, hypertension, hypocomplementaemia, or renal failure — in whom the diagnosis is unlikely to be minimal change disease and the treatment may differ. Secondary causes, including systemic lupus erythematosus and hepatitis B, should be excluded where the picture suggests them. [6]
Finally, oedema itself has a differential that must be excluded before nephrotic syndrome is diagnosed. Heart failure, liver disease, protein-losing enteropathy, severe malnutrition, allergic angioedema and salt poisoning can all swell a child. The first morning urine protein-to-creatinine ratio and the serum albumin separate nephrotic oedema from the rest: a normal ratio and a normal albumin direct the investigation elsewhere. [11]
Clinical & Bedside Assessment
The bedside assessment hinges on measuring the proteinuria reliably and on judging the fluid status. The urine dipstick at the bedside gives a rapid read, but the quantitative anchor is the first-morning protein-to-creatinine ratio, which corrects for urine concentration and is the value used to confirm the diagnosis and to define remission and relapse. A ratio below 50 mg/mmol is normal, 50 to 200 mg/mmol is intermediate, and 200 mg/mmol or above is nephrotic-range. [10]
Assessing the nephrotic child on the ward
Confirm nephrotic-range proteinuria on a first morning protein-to-creatinine ratio and check the serum albumin and renal function.
Examine the distribution of oedema: periorbital, peripheral, ascites, pleural effusion, scrotal or labial.
Judge intravascular volume: heart rate, blood pressure, capillary refill, peripheral temperature, urine output and abdominal pain.
Screen for complications: temperature for infection, calf tenderness or chest signs for thrombosis, urine output for acute kidney injury.
Check weight daily and plot on a growth chart; teach the family home urine dipstick testing.
The daily ward routine pairs a weight and a urine dipstick with the fluid-status examination. A rising weight with falling urine output and rising urea signals fluid retention and impending acute kidney injury, while a falling weight with tachycardia and cool peripheries signals effective diuresis that is threatening the intravascular volume. The family is taught to test the first morning urine at home with dipsticks once the child is established on treatment, because home monitoring detects relapse early and shortens each relapse course. [11]
Examination must also look for the stigmata of long-term steroid toxicity in the frequently-relapsing child: growth failure, cushingoid features, hypertension, striae, cataracts and reduced bone density. These are the costs of repeated steroid courses, and they are the reason steroid-sparing therapy is introduced once the frequently-relapsing or steroid-dependent thresholds are crossed. [4]
Investigations
The first-episode investigation panel is deliberately limited in the classic young child, because minimal change disease is assumed and treatment should not wait for a biopsy. The baseline panel confirms the syndrome, screens for complications and infection risk, and excludes the atypical features that would prompt a biopsy. [1]
[1] [5]A renal biopsy is performed when the presentation is atypical or when the child proves steroid-resistant after four weeks of daily prednisolone. In minimal change disease the light microscopy is normal and the diagnosis rests on electron microscopy, which shows the diffuse effacement of the podocyte foot processes. Focal segmental glomerulosclerosis shows segmental sclerosis in only some glomeruli, which is why a single biopsy can miss it, and membranous nephropathy shows subepithelial immune deposits. [6]
Genetic testing has become part of the work-up of steroid-resistant disease because a monogenic cause is found in a substantial fraction of cases, and identifying it spares the child ineffective immunosuppression. The IPNA recommendations advise a genetic panel after steroid resistance is confirmed, alongside the biopsy, because a pathogenic variant predicts a low likelihood of response to calcineurin inhibitors and rituximab and alters the counselling about recurrence after transplant. [5]
Management — Resuscitation
The resuscitation scenarios in nephrotic syndrome are not the proteinuria but the complications: hypovolaemia, severe infection and thromboembolism. A child who presents with shock from intravascular depletion must be resuscitated with colloid rather than with crystalloid, because the problem is a low oncotic pressure and the oedema will only worsen with saline. [7]
Spontaneous bacterial peritonitis is treated empirically with a third-generation cephalosporin such as cefotaxime or ceftriaxone intravenously once cultures are drawn, and the regimen is broadened if a resistant organism is identified. The child is nursed with attention to fluid balance, and the nephrotic state is treated in parallel with steroids if it is a first presentation. Children with nephrotic syndrome should receive pneumococcal vaccination, and some units add oral penicillin V prophylaxis during a relapse. [11]
A suspected thromboembolism is a medical emergency. Deep vein thrombosis, pulmonary embolism, renal vein thrombosis and cerebral sinus venous thrombosis all occur, and the index of suspicion must be high for any new focal symptom. Imaging is urgent — Doppler ultrasound for limb thrombosis, computed tomography pulmonary angiography for pulmonary embolism, and magnetic resonance venography for cerebral sinus thrombosis — and anticoagulation with low-molecular-weight heparin is started once the diagnosis is confirmed or strongly suspected. [8] [12]
Management — Definitive & Stepwise
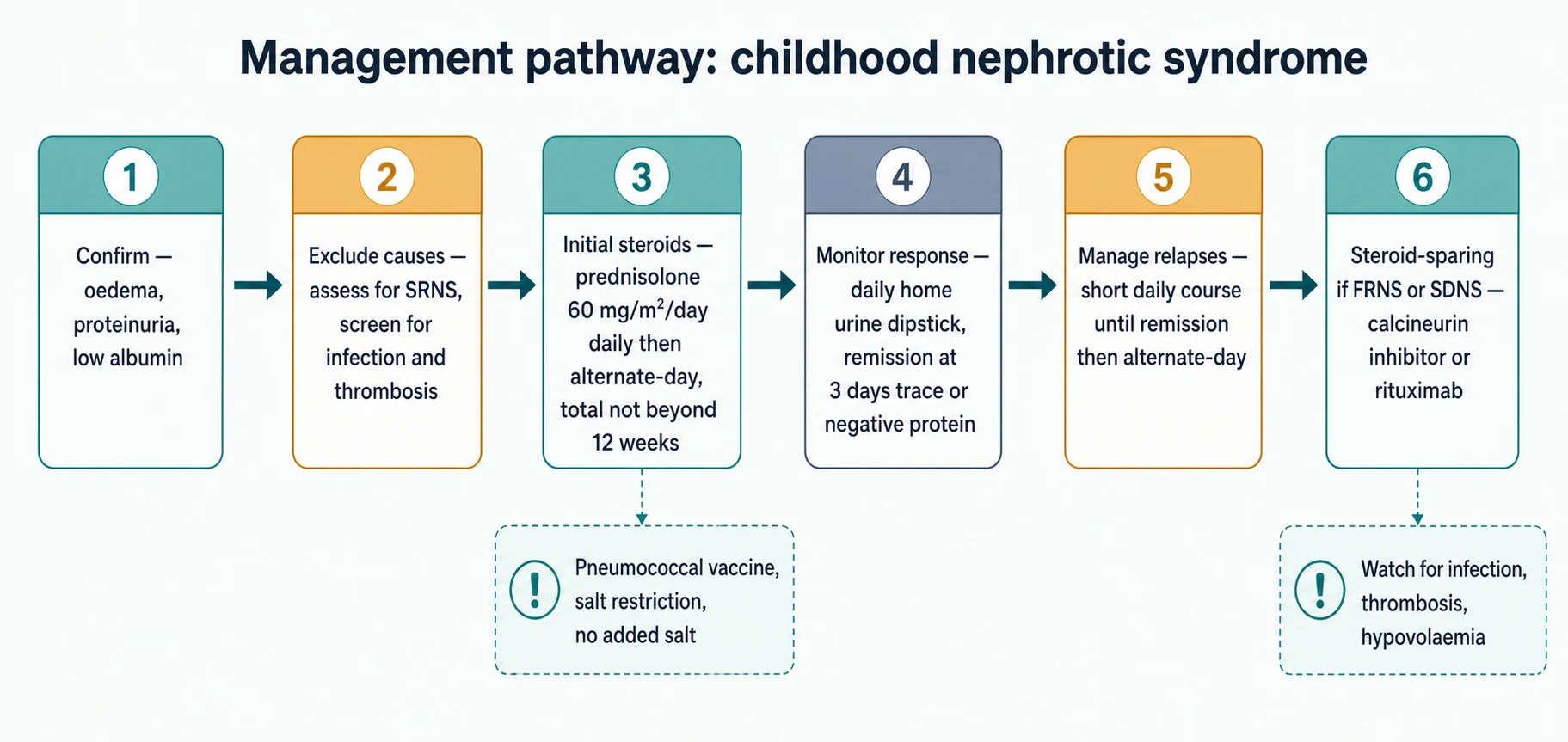
The definitive treatment of the first episode is a defined course of oral prednisolone, and the structure of that course is what the KDIGO 2021 guideline fixes. The aim is to induce remission quickly, convert to alternate-day dosing to limit toxicity, and keep the total duration within twelve weeks, because the evidence shows that prolonging the course does not reduce relapses. [1] [3]
Prednisolone (first-line, first episode and relapse)
Dose
60 mg/m2/day orally once daily (maximum 60 mg/day) until remission for 3 days, then 40 mg/m2 on alternate days (maximum 50 mg/dose) for 4 weeks; initial course total duration kept within 12 weeks
The initial course gives daily prednisolone 60 mg/m2/day, capped at 60 mg/day, until the urine is trace or negative for three consecutive days, at which point the dose is switched to 40 mg/m2 on alternate days. The total duration of the first course is kept within twelve weeks. The Teeninga trial established that extending the initial prednisolone course beyond this does not lower the relapse rate, which is why shorter, toxicity-sparing courses are now standard. [3]
A relapse is treated with daily prednisolone 60 mg/m2/day until the urine is trace or negative for three days, followed by 40 mg/m2 on alternate days for four weeks. Families are taught to test the first morning urine at home and to start contact with the team at the first sign of 3+ protein, so that most relapses are short and managed without admission. Salt restriction — no added salt, avoidance of processed foods — runs throughout, because sodium retention is central to the oedema. [1] [11]
Specific Subtypes & Scenarios
Frequently-relapsing and steroid-dependent nephrotic syndrome are the common subtypes that force the move beyond steroids. Once a child meets the frequently-relapsing or steroid-dependent definition, a steroid-sparing agent is added to reduce cumulative steroid exposure and the complications that come with it. The KDIGO 2021 guideline names the calcineurin inhibitors and rituximab as the agents with the strongest evidence. [1]
Steroid-sparing agents for FRNS and SDNS
The Iijima randomised trial established rituximab as effective for the child with complicated, frequently-relapsing or steroid-dependent disease, showing that a single course reduced the risk of relapse at one year compared with placebo. Rituximab is given as 375 mg/m2 per dose intravenously, and its effect is monitored through the B-cell count and the immunoglobulin level. It is not a first-line agent for the uncomplicated child, but it has transformed the management of the child accumulating steroid and calcineurin-inhibitor toxicity. [4]
Steroid-resistant nephrotic syndrome is the subtype with the gravest prognosis, because it carries a real risk of progression to end-stage kidney disease. The definition is failure to achieve remission after four weeks of daily prednisolone 60 mg/m2/day, and the management that follows is entirely different from the steroid-sensitive pathway. A renal biopsy defines the histology, a genetic panel seeks a monogenic cause, and a calcineurin inhibitor — usually tacrolimus or ciclosporin — is introduced as the first-line immunosuppressant, with around half of non-genetic cases achieving partial or complete remission. [5] [6]
Congenital and infantile nephrotic syndrome, presenting in the first three months or first year of life, is usually genetic — classically the NPHS1 Finnish type — and does not respond to steroids. Management is supportive, centred on albumin infusions, nutrition and infection control, with bilateral nephrectomy and transplantation as the definitive treatment for the severe cases. The ERKNet-ESPN consensus guides this rare but important pathway. [9]
Complications & Pitfalls
The complications of nephrotic syndrome are the events that harm and kill children, and they are infection, thromboembolism and acute kidney injury. None of them is caused by the proteinuria directly; all of them arise from the physiological consequences of the leak. Infection risk is driven by the urinary loss of immunoglobulin and alternative-complement factor B, and the classic serious infection is pneumococcal peritonitis, though gram-negative and other organisms occur. [11]
Thromboembolism occurs in roughly three per cent of children with nephrotic syndrome, and the risk rises with the severity and duration of the relapse, with a serum albumin below 20 g/L, with a fibrinogen above 6 g/L, with haemoconcentration, and with immobility or central venous lines. The mechanism is the urinary loss of antithrombin alongside increased hepatic synthesis of fibrinogen and platelet activation. Deep vein thrombosis, pulmonary embolism, renal vein thrombosis and cerebral sinus thrombosis are all recognised, and a Chinese cohort study confirmed pulmonary embolism as a real and under-recognised risk. [8] [12]
Acute kidney injury complicates a substantial minority of nephrotic admissions, and its mechanisms are hypovolaemia from the underfill state, acute tubular necrosis from prolonged hypoperfusion, interstitial oedema of the kidney, and, rarely, intrarenal thrombosis. The Rheault study of children hospitalised with nephrotic syndrome quantified the burden of acute kidney injury in this setting and emphasised that it is usually reversible with attention to volume status. The pitfall is treating the oedema with diuretics alone in a child whose intravascular volume is depleted, which deepens the hypoperfusion and worsens the injury. [7]
The endocrine and metabolic complications accumulate over time: hypocalcaemia and bone disease from cholecalciferol loss, hypothyroidism from thyroxine-binding globulin loss, iron-deficiency anaemia from transferrin loss, and the growth failure and reduced bone density of long-term steroid therapy. These are managed with calcium and vitamin D supplementation, thyroid replacement where indicated, and attention to nutrition and bone health throughout the relapsing course. [10]
Prognosis & Disposition
The prognosis of childhood nephrotic syndrome is determined almost entirely by the steroid response. Steroid-sensitive disease carries an excellent long-term prognosis: the majority of children stop relapsing by late childhood or adolescence, retain normal kidney function, and grow into adults without renal disease. The frequently-relapsing and steroid-dependent child does well in the long term provided steroid toxicity is contained with steroid-sparing agents, though the burden of treatment is real. [10]
Steroid-resistant disease carries the heavy prognosis of its underlying histology and genetics. Children who respond partially or completely to a calcineurin inhibitor retain kidney function for longer, but a substantial minority progress to end-stage kidney disease over years, and the risk is highest in those with a pathogenic genetic variant and in those with focal segmental glomerulosclerosis. The transition to dialysis and transplant planning begins early in this group, and counselling about the risk of recurrence of focal segmental glomerulosclerosis in the transplant graft is part of the preparation. [5] [6]
The disposition is shared care between the general paediatrician and a paediatric nephrologist. A first episode is usually managed as a partnership, with the general team leading the day-to-day steroid course and home urine testing, and nephrology involved for atypical features, steroid resistance, and the move to steroid-sparing therapy. Infrequently-relapsing steroid-sensitive disease can be managed largely in primary and general paediatric care. The transition to adult care in late adolescence is a period of risk for the relapsing patient, and a structured handover covering the response history, the steroid-sparing agents used, the vaccination status and the complication history is essential. [11]
Special Populations
Children from South Asian, African and certain Indigenous populations carry a higher incidence of nephrotic syndrome and, in some groups, a higher proportion of the more difficult histologies such as focal segmental glomerulosclerosis and of genetic variants. Aboriginal, Torres Strait Islander and Maori and Pacific children in Australia and New Zealand have higher rates of renal disease overall, and the threshold for screening, for nephrology involvement, and for culturally safe education about home urine testing and medication adherence is lower. [10]
Children from migrant, refugee and socioeconomically disadvantaged backgrounds face barriers to the sustained home urine testing and reliable medication supply that nephrotic syndrome management demands. Language-appropriate education about dipstick technique, about the steroid course, and about the danger signs of peritonitis and thrombosis is the practical priority, and the provision of dipsticks, a written relapse plan and a direct contact route to the team can prevent admissions. [11]
Children with complex chronic conditions and those who are technology-dependent, including transplant recipients and children on dialysis, may develop nephrotic-range proteinuria from recurrent or de novo glomerular disease, and the management is integrated with their primary renal care. The principles do not change — confirm the proteinuria, define the response, treat the complications — but the threshold for nephrology-led care is the default from the outset. [6]
Evidence, Guidelines & Regional Differences
The two frameworks that shape practice are the KDIGO 2021 guideline for the management of glomerular diseases and the IPNA clinical practice recommendations for steroid-resistant nephrotic syndrome. KDIGO 2021 sets the definition, the steroid regimen, the response-based classification, and the place of calcineurin inhibitors and rituximab; the IPNA recommendations add the specific pathway for the steroid-resistant child, including the genetic panel and the biopsy strategy. The KDIGO Controversies Conference on glomerular diseases underpins the pathophysiological framing. [1] [5] [2]
The principal controversy remains the duration of the initial steroid course. The historical long courses of the 1980s gave way to shorter regimens after the Teeninga trial showed that extending prednisolone beyond twelve weeks did not reduce relapses, and current practice favours shorter, toxicity-sparing courses within the KDIGO window. The place of rituximab continues to evolve as longer follow-up accumulates on its safety and on the durability of remission, and the role of newer agents such as ofatumumab and sparsentan in the paediatric steroid-resistant population is still being defined. [3] [4]
A live area of evidence is the genetic work-up of steroid-resistant disease. As the gene panels grow, a monogenic cause is identified in an increasing fraction of cases, and this changes management by redirecting treatment away from ineffective immunosuppression and by informing transplant counselling. The integration of genetics into the IPNA pathway reflects this shift, and the proportion of children in whom a genetic diagnosis is made is expected to rise as testing becomes more accessible. [5]
Exam Pearls
Three rules that mark a fellowship answer
The high-yield minutiae an examiner rewards: the first morning sample is non-negotiable; minimal change disease dominates under ten years and clears with steroids in about ninety per cent; the initial prednisolone dose is 60 mg/m2/day capped at 60 mg/day for a total course within twelve weeks; steroid resistance is failure at four weeks and changes everything; pneumococcal peritonitis is the classic infection; antithrombin loss drives the thrombosis; hypovolaemia needs 20 per cent albumin with furosemide; and rituximab at 375 mg/m2 per dose is the evidence-based option for complicated frequently-relapsing or steroid-dependent disease. [1] [4]
References
- [1]Rovin BH; Adler SG; Barratt J; et al Executive summary of the KDIGO 2021 Guideline for the Management of Glomerular Diseases. Kidney Int, 2021.PMID 34556300
- [2]Rovin BH; Caster DJ; Cattran DC; et al Management and treatment of glomerular diseases (part 2): conclusions from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference. Kidney Int, 2019.PMID 30665569
- [3]Teeninga N; Kist-van Holthe JE; van Rijswijk N; et al Extending prednisolone treatment does not reduce relapses in childhood nephrotic syndrome. J Am Soc Nephrol, 2013.PMID 23274956
- [4]Iijima K; Sako M; Nozu K; et al Rituximab for childhood-onset, complicated, frequently relapsing nephrotic syndrome or steroid-dependent nephrotic syndrome: a multicentre, double-blind, randomised, placebo-controlled trial. Lancet, 2014.PMID 24965823
- [5]Trautmann A; Vivarelli M; Samuel S; et al IPNA clinical practice recommendations for the diagnosis and management of children with steroid-resistant nephrotic syndrome. Pediatr Nephrol, 2020.PMID 32382828
- [6]Tullus K; Webb H; Bagga A; et al Management of steroid-resistant nephrotic syndrome in children and adolescents. Lancet Child Adolesc Health, 2018.PMID 30342869
- [7]Rheault MN; Zhang L; Selewski DT; et al AKI in Children Hospitalized with Nephrotic Syndrome. Clin J Am Soc Nephrol, 2015.PMID 26450933
- [8]Kerlin BA; Haworth K; Smoyer WE; et al Venous thromboembolism in pediatric nephrotic syndrome. Pediatr Nephrol, 2014.PMID 23812352
- [9]Boyer O; Schaefer F; Haffner D; et al Management of congenital nephrotic syndrome: consensus recommendations of the ERKNet-ESPN Working Group. Nat Rev Nephrol, 2021.PMID 33514942
- [10]Bagga A; Mantan M Nephrotic syndrome in children. Indian J Med Res, 2005.PMID 16106086
- [11]Gipson DS; Massengill SF; Yao L; et al Management of childhood onset nephrotic syndrome. Pediatrics, 2009.PMID 19651590
- [12]Zhu H; Qi J; Schoepf J; et al Prevalence and Associated Risk Factors of Pulmonary Embolism in Children and Young Adults With Nephrotic Syndrome: A Chinese Large Cohort Study. J Thorac Imaging, 2021.PMID 34269751