Paeds · nephrology-urology-fluids-and-electrolytes
Polycystic kidney disease and inherited nephropathies
Also known as ARPKD · ADPKD · Alport syndrome · Nephronophthisis · Inherited cystic kidney disease · Ciliopathy
Fellowship guide to polycystic kidney disease and inherited nephropathies in children: the neonatal-onset ARPKD driven by PKHD1 and fibrocystin with bilateral enlarged echogenic kidneys and congenital hepatic fibrosis, the later-presenting ADPKD from PKD1 or PKD2 encoding polycystin with height-adjusted total kidney volume guiding tolvaptan therapy, Alport syndrome with its type IV collagen defect causing progressive haematuric nephropathy with sensorineural hearing loss and the ACE-inhibitor-at-diagnosis strategy, and nephronophthisis as the autosomal recessive ciliopathy causing tubulointerstitial fibrosis with disproportionate anaemia and ESKD in childhood.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
Overview & Definition
A newborn is found on antenatal ultrasound to have bilateral massively enlarged echogenic kidneys. He is born with respiratory distress from pulmonary hypoplasia, and within days his blood pressure climbs and his creatinine rises. This is autosomal recessive polycystic kidney disease, one of a family of inherited conditions in which a single gene defect in a kidney structural protein slowly destroys renal architecture. The inherited nephropathies share three things: a genetic cause, a progressive course toward kidney failure, and the need for lifelong surveillance that begins in childhood and spans generations. [1]
These diseases are best understood by what is broken. In the polycystic kidney diseases, the defective protein sits on the primary cilium of the tubular epithelial cell, the antenna-like sensory organelle that lines every nephron, and its failure turns orderly tubules into disorganised cysts. In Alport syndrome, the defective protein is type IV collagen, the meshwork that forms the glomerular basement membrane, and its failure makes the filter fragile and leaky. In nephronophthisis, the defective protein is a ciliary or centrosomal protein that triggers tubular cell death and interstitial fibrosis. Each gene, each protein, each mechanism produces a recognisable clinical picture, and the exam reward lies in matching the gene to the phenotype. [3]
Classification
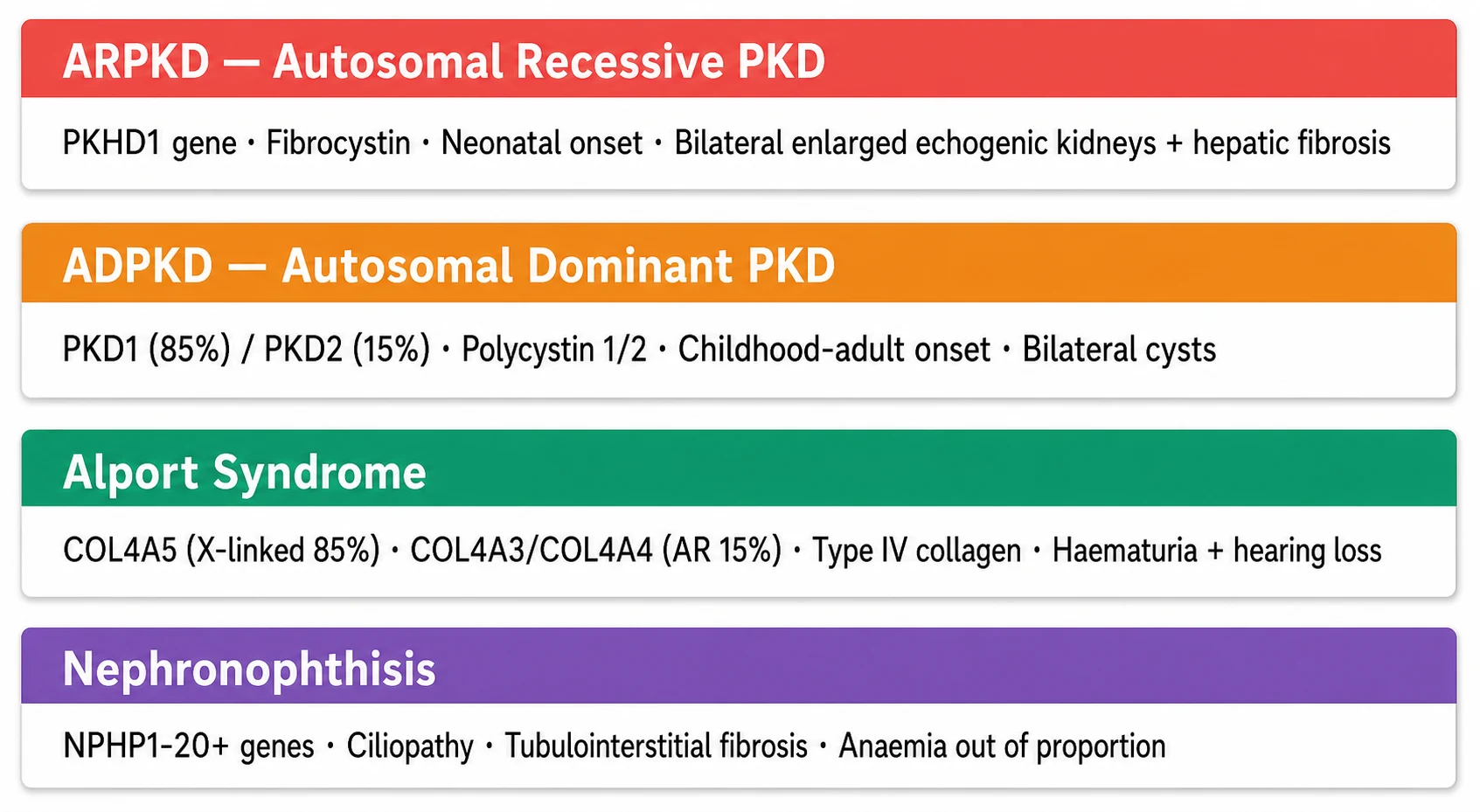
The classification follows the defective protein and its structural role. The ciliopathies are diseases of the primary cilium. Autosomal recessive polycystic kidney disease results from mutations in PKHD1, which encodes fibrocystin or polyductin, a protein found on the primary cilium of collecting duct and biliary epithelium, and the disease characteristically presents in the neonatal period with bilateral enlarged echogenic kidneys accompanied by congenital hepatic fibrosis. Autosomal dominant polycystic kidney disease results from mutations in PKD1, which encodes polycystin 1 and accounts for about 85 percent of cases, or PKD2, which encodes polycystin 2 and accounts for about 15 percent, and it tends to present later, often with cysts detected in childhood or adolescence. [3]
[3]The basement membrane diseases are Alport syndrome and its spectrum. Type IV collagen forms the meshwork of the glomerular basement membrane, and its alpha-3, alpha-4 and alpha-5 chains form a specialised network that is critical for filter integrity. Mutations in COL4A5, located on the X chromosome, cause the X-linked form that accounts for about 85 percent of Alport syndrome, with affected males following a severe course and carrier females variably affected. Mutations in COL4A3 or COL4A4 on autosomes cause the autosomal recessive form in about 15 percent and the milder autosomal dominant form more rarely. Nephronophthisis sits at the interface: its causative genes, now numbering over 20, encode proteins of the centrosome and primary cilium, classifying it as a ciliopathy, but its pathology is tubulointerstitial rather than cystic in the classic sense, producing shrunken kidneys with corticomedullary scarring and medullary microcysts. [8]
Epidemiology & Risk Factors
The epidemiology of these conditions reflects their genetic architecture. Autosomal recessive polycystic kidney disease has an incidence of about 1 in 20,000 live births and is one of the most common inherited causes of kidney failure presenting in infancy. Carrier frequency is estimated at 1 in 70 individuals of Northern European descent. Autosomal dominant polycystic kidney disease is far more common, with a prevalence of about 1 in 1000 to 1 in 2500, and is the most common monogenic cause of kidney failure worldwide, though most of its burden falls in adulthood because cysts accumulate over decades. [1]
Alport syndrome affects about 1 in 5000 to 1 in 10,000 individuals, and the X-linked form from COL4A5 accounts for the majority. The risk of progression is determined by the genotype: males with X-linked Alport syndrome almost universally progress to kidney failure, typically by the second or third decade, while females with X-linked Alport syndrome have variable outcomes, with about 15 to 30 percent developing kidney failure by age 60. In autosomal dominant polycystic kidney disease, PKD1 mutations produce a more severe course than PKD2, with kidney failure occurring on average 20 years earlier, and very early-onset presentation, defined as symptoms before age 15, carries the heaviest burden. [6]
Nephronophthisis is the most common genetic cause of kidney failure in the first three decades of life, and about 15 to 20 percent of affected children have additional extrarenal features involving the retina, central nervous system, liver or skeleton, reflecting the shared ciliary biology of these organ systems. The syndromic forms include Senior-Loken syndrome with retinitis pigmentosa, Joubert syndrome with cerebellar vermis hypoplasia, and Mainzer-Saldino syndrome with skeletal dysplasia. A positive family history, consanguinity and a known family mutation are the strongest risk factors across all four conditions, and they drive the genetic counselling that is inseparable from the diagnosis. [8]
Pathophysiology
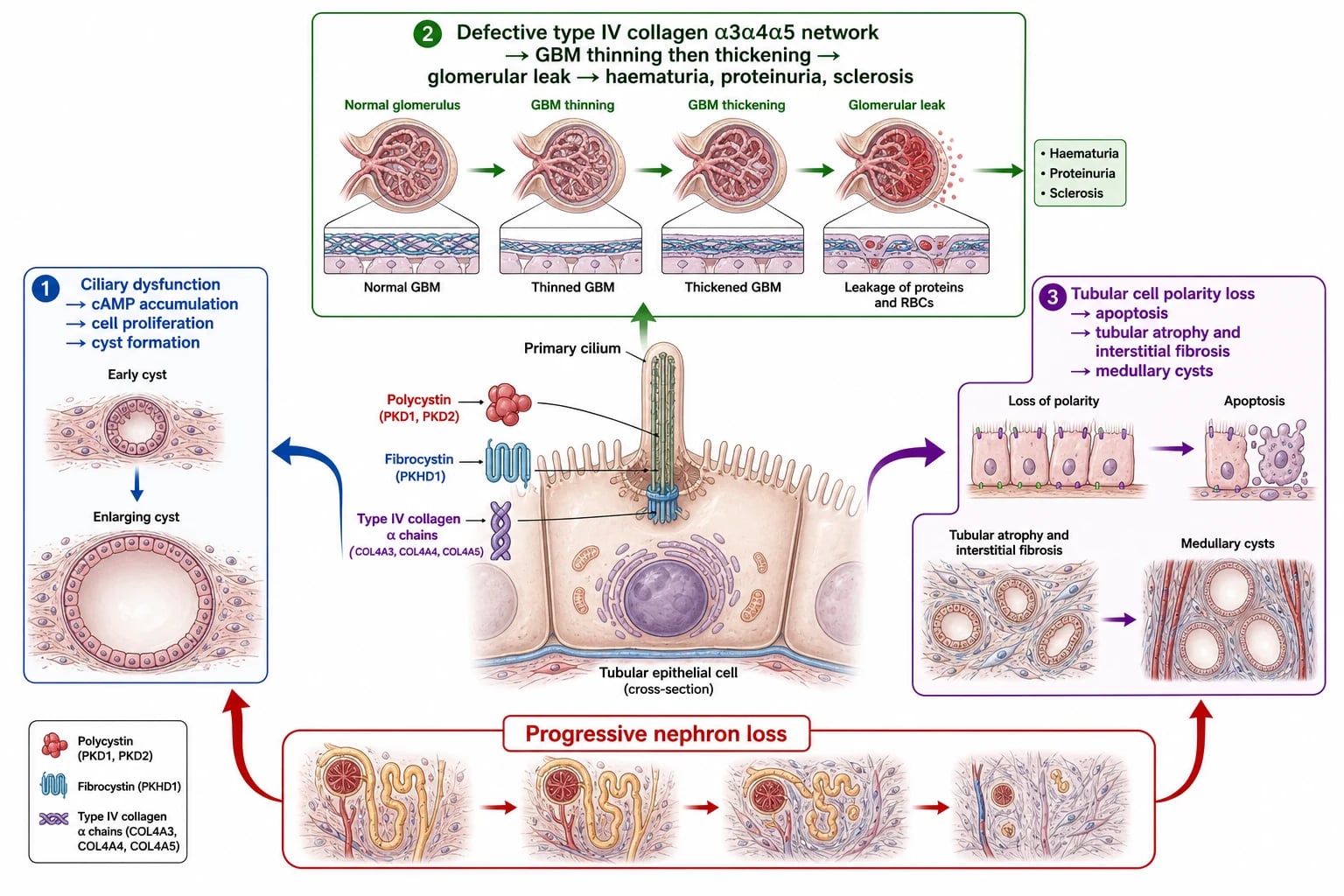
The polycystic kidney diseases are diseases of the primary cilium. Polycystin 1 and polycystin 2, encoded by PKD1 and PKD2, form a complex on the cilium of tubular epithelial cells that senses luminal flow and calcium influx, and when this complex is defective, the cell loses its ability to regulate proliferation, planar polarity and fluid secretion. Cyclic AMP accumulates, driven by vasopressin V2 receptor signalling, and it activates pathways that promote cell proliferation and chloride-driven fluid secretion into the enlarging cyst cavity. Cysts detach from their parent tubule, grow by accumulating fluid, and compress and destroy the surrounding normal nephrons. Fibrocystin, encoded by PKHD1, plays a parallel role in the collecting ducts and biliary tree, which explains why autosomal recessive polycystic kidney disease simultaneously affects the kidneys and the liver. [3]
CILIA
Alport syndrome follows a different mechanism entirely. Type IV collagen alpha-3, alpha-4 and alpha-5 chains assemble into a specialised network in the glomerular basement membrane, the lens capsule and the cochlea. When one chain is missing, the network cannot form, and the basement membrane undergoes progressive thinning, splitting and thickening with multilamellation. The filter becomes fragile, allowing red blood cells and protein to leak through, producing the characteristic persistent haematuria. Over years the repetitive injury drives glomerular sclerosis and tubulointerstitial fibrosis, and the same collagen defect in the cochlear basement membrane produces the high-frequency sensorineural hearing loss that appears in late childhood or adolescence. [6]
Nephronophthisis converges on a third mechanism: defective ciliary and centrosomal proteins cause tubular epithelial cells to undergo apoptosis, producing tubular atrophy with thickened tubular basement membranes, interstitial fibrosis and eventual medullary microcyst formation. Unlike the polycystic diseases, the kidneys are typically normal-sized or small, and the anaemia is disproportionately severe for the degree of renal impairment, possibly because the tubulointerstitial destruction compromises erythropoietin production early. This triad of shrunken echogenic kidneys, disproportionate anaemia and a slowly progressive course toward kidney failure is the signature that distinguishes nephronophthisis from the cystic diseases. [8]
Clinical Presentation
Autosomal recessive polycystic kidney disease is the condition that presents earliest and most dramatically. The classical picture is bilateral enlarged echogenic kidneys detected on antenatal ultrasound, often with oligohydramnios from fetal urinary failure, which produces pulmonary hypoplasia and the respiratory distress, limb contractures and characteristic facies of Potter sequence. In the neonatal period the massively enlarged kidneys may cause a distended abdomen and may impede ventilation, and early complications include severe hypertension, electrolyte disturbance and respiratory failure. A milder presentation in older infants and children includes hypertension, polyuria and polydipsia from impaired concentrating ability, failure to thrive and progressive kidney dysfunction, often accompanied by features of portal hypertension from congenital hepatic fibrosis such as splenomegaly and variceal bleeding. [1]
Autosomal dominant polycystic kidney disease in children is often detected incidentally when cysts are found on ultrasound performed for a family history or an unrelated complaint. Most children are asymptomatic, but some present with haematuria from cyst rupture, flank pain from cyst haemorrhage or infection, hypertension, or urinary tract infection. Very early-onset disease, with symptoms or significant cyst burden before age 15, portends a more aggressive course and is the group in which tolvaptan therapy is most likely to be considered. The clinical burden accumulates across decades, with most patients reaching kidney failure in middle adulthood, but the seeds are sown in childhood, which is why surveillance begins early. [5]
Alport syndrome classically presents in a young boy with persistent microscopic haematuria, sometimes with episodic gross haematuria triggered by upper respiratory infection. The family history may reveal a male relative with deafness and kidney failure, and the mother may herself have haematuria if the inheritance is X-linked. Sensorineural hearing loss appears in late childhood or adolescence, is never congenital, and affects the high frequencies first. Ocular findings include anterior lenticonus, which causes progressive myopia, and macular flecks. Females with X-linked Alport syndrome may present with isolated haematuria and can develop proteinuria, hypertension and progressive kidney disease, sometimes reaching kidney failure in later life. [6]
Nephronophthisis presents differently again, with a child who develops polyuria, polydipsia, secondary enuresis and a slowly progressive decline in kidney function, often accompanied by an anaemia that seems disproportionate to the modest creatinine rise. Growth failure, sodium wasting and a tendency to dehydration are common, and the child may present with a crisis of acute kidney injury precipitated by an intercurrent illness. The syndromic forms declare themselves through their extrarenal features: a child with retinitis pigmentosa and night blindness may have Senior-Loken syndrome, one with hypotonia, ataxia and abnormal eye movements may have Joubert syndrome, and the combination of skeletal abnormalities and renal failure suggests Mainzer-Saldino syndrome. [8]
Differential Diagnosis
The first diagnostic task is to distinguish the inherited nephropathies from acquired and other structural causes. A child with bilateral cystic kidneys may have multicystic dysplastic kidneys, which are part of the CAKUT spectrum, and these are distinguished by their non-uniform cysts and disorganised parenchyma rather than the symmetric, radially arranged cysts of ARPKD. Simple cortical cysts, though rare in young children, can occur, and a family history of adult-onset cysts in a child with a few cysts raises the possibility of early ADPKD. Tuberous sclerosis complex, with its cutaneous manifestations, seizures and renal angiomyolipomas, must be considered in any child with multiple renal cysts. [9]
[9]The child with persistent haematuria demands a different differential. Benign familial haematuria from COL4A3 or COL4A4 heterozygosity, now recognised as the most common cause of persistent microscopic haematuria, must be distinguished from Alport syndrome, because thin basement membrane nephropathy carries a much milder course though it can progress in some families. IgA nephropathy presents with episodic gross haematuria following infection, as does Alport, but the family history of hearing loss and the progression pattern help separate them, and renal biopsy with electron microscopy showing the lamellated, thickened basement membrane is definitive. Post-infectious glomerulonephritis presents with acute nephritic syndrome, complement consumption and a preceding streptococcal infection, and it resolves rather than progresses. [6]
The child with progressive kidney failure and disproportionate anaemia raises nephronophthisis, but the differential includes any cause of chronic tubulointerstitial disease, including reflux nephropathy with scarring, obstructive uropathy, and autosomal dominant tubulointerstitial kidney disease from UMOD or REN mutations. The renal ultrasound is the first discriminator: nephronophthisis shows small, echogenic kidneys with loss of corticomedullary differentiation and possible medullary cysts, while reflux nephropathy shows focal cortical scarring and calyceal blunting. Genetic testing ultimately resolves the diagnosis in most cases, and a renal biopsy showing the characteristic tubular basement membrane thickening, tubular atrophy and interstitial fibrosis with absence of significant glomerular disease supports nephronophthisis. [8]
Clinical & Bedside Assessment
The bedside assessment of a child with a suspected inherited nephropathy answers three questions: which disease, how far has it progressed, and what complications are present. The history is where the diagnosis is most often made. A detailed three-generation family history probes for kidney failure, dialysis, transplantation, premature death, hearing loss and consanguinity. The pregnancy and birth history establish whether there was oligohydramnios, antenatal renal abnormalities, neonatal respiratory distress or hypertension. In the older child, the symptom history asks about polyuria, polydipsia, enuresis, haematuria, abdominal pain, headaches, failure to thrive and, for Alport syndrome specifically, hearing difficulties and visual changes. [6]
Examination is systematic and looks for both the renal and the extrarenal features. Blood pressure is measured with an appropriate cuff and plotted on centile charts, because hypertension is a common and early complication in all cystic kidney diseases. Abdominal examination may reveal ballotable enlarged kidneys in ARPKD or ADPKD, or hepatosplenomegaly suggesting portal hypertension from congenital hepatic fibrosis. Growth parameters are plotted meticulously because growth failure accompanies progressive kidney disease. Otoscopic and tuning-fork examination screens for hearing loss, though formal pure-tone audiometry is required for confirmation. Eye examination looks for the anterior lenticonus, macular flecks and corneal endothelial vesicles of Alport syndrome. In suspected nephronophthisis-related ciliopathies, a careful developmental and neurological examination assesses for cerebellar signs, hypotonia, and ocular motor abnormalities suggesting Joubert syndrome. [6]
Family screening is not just a diagnostic step but a clinical intervention in its own right. Parents and siblings of a child with an inherited nephropathy are offered clinical evaluation, renal ultrasound, urinalysis for haematuria or proteinuria, and targeted genetic testing once the family mutation is known. Identifying an affected but asymptomatic sibling allows early initiation of surveillance and nephroprotection, and identifying unaffected carriers informs reproductive counselling. The psychosocial impact of genetic testing in children and families is substantial, and genetic counselling by a trained professional must accompany testing, particularly for autosomal recessive conditions where carrier status has implications for future pregnancies. [11]
Investigations
The investigation strategy confirms the inherited nephropathy, defines its genetic basis, and surveys the complications that require treatment. Renal ultrasound is the first-line imaging modality and is often diagnostic in the cystic diseases. In ARPKD it shows bilateral enlarged echogenic kidneys with loss of corticomedullary differentiation and a characteristic radial array of small cysts, while in ADPKD it shows bilateral cysts of varying sizes scattered through the cortex and medulla. In nephronophthisis the kidneys are small and echogenic with indistinct corticomedullary junctions, and medullary cysts may be visible. The international imaging consensus provides standardised diagnostic criteria for cystic kidney diseases in children, including the age-dependent number of cysts required to diagnose ADPKD in a child at risk. [9]
Genetic testing has become central to the diagnosis and management of inherited nephropathies. Next-generation sequencing gene panels covering PKHD1, PKD1, PKD2, COL4A3, COL4A4, COL4A5 and the NPHP genes are now the standard approach, and they identify pathogenic variants in the majority of affected children. In Alport syndrome, genetic testing is recommended for all suspected cases because it confirms the diagnosis, determines the inheritance pattern which profoundly affects prognosis and counselling, and identifies at-risk relatives. The classification of COL4A3, COL4A4 and COL4A5 variants follows ACMG criteria refined for Alport syndrome, and the 2024 ERKNet guideline provides a structured approach to genetic testing, surveillance and treatment. [7]
The complication survey runs in parallel. Urinalysis and protein quantification assess for haematuria and proteinuria, with first-morning albumin-to-creatinine ratio tracking progression in Alport and ADPKD. Blood pressure is measured and, where indicated, confirmed with ambulatory monitoring to capture nocturnal hypertension. Estimated GFR is calculated using the bedside Schwartz equation for children. In ARPKD, liver function tests, abdominal ultrasound with Doppler for portal hypertension, and platelet count to screen for hypersplenism survey the hepatic fibrosis. Audiometry screens for hearing loss in Alport syndrome, performed from age five and repeated annually, and ophthalmology review assesses for anterior lenticonus and macular changes. In suspected nephronophthisis-related ciliopathies, ophthalmological assessment including electroretinography screens for retinal dystrophy, and brain magnetic resonance imaging assesses for the molar tooth sign of Joubert syndrome. [8]
Management — Resuscitation
The resuscitation phase applies to the child who presents acutely unwell with an inherited nephropathy, most often the neonate with ARPKD. Respiratory distress from pulmonary hypoplasia may require mechanical ventilation, and the massively enlarged kidneys may splint the diaphragm and necessitate ventilatory strategies that accommodate the restricted lung capacity. Severe hypertension is common and dangerous in neonatal ARPKD, and it is treated with intravenous antihypertensives titrated carefully, because these infants are exquisitely sensitive to volume and blood pressure changes. Fluid and electrolyte management must account for the polyuria and salt wasting that accompany impaired concentrating ability, and sodium supplementation may be required. [1]
Fluid management demands precision in all cystic kidney diseases. Children with ARPKD and dysplastic kidneys are often polyuric and salt-wasting, and they can present dehydrated with a rising creatinine that mimics acute-on-chronic failure. Cautious rehydration with isotonic saline improves these children, while sodium supplementation maintains intravascular volume. Conversely, a child who has progressed to oliguric kidney failure must be fluid-restricted to insensible losses plus urine output, because excess accumulates rapidly and causes pulmonary and cerebral oedema. Daily weights and strict input-output charting are the most reliable guides, and symptomatic uraemia, severe acidosis or uncontrollable fluid and biochemical disturbance is an indication for urgent renal replacement therapy. [1]
Acute complications of Alport syndrome and nephronophthisis that bring a child to resuscitation are typically those of their progressive kidney disease: acute kidney injury precipitated by intercurrent illness or nephrotoxins, severe anaemia from relative erythropoietin deficiency, or hypertensive emergency. The approach follows standard principles of managing decompensated chronic kidney disease, addressing the trigger, supporting the airway and circulation, correcting electrolyte and acid-base disturbance, and arranging dialysis if medical therapy fails. Gastrointestinal bleeding from portal hypertension in a child with ARPKD and congenital hepatic fibrosis is managed with endoscopic intervention, octreotide and, in refractory cases, transjugular intrahepatic portosystemic shunt, with the hepatology team closely involved. [1]
Management — Definitive & Stepwise
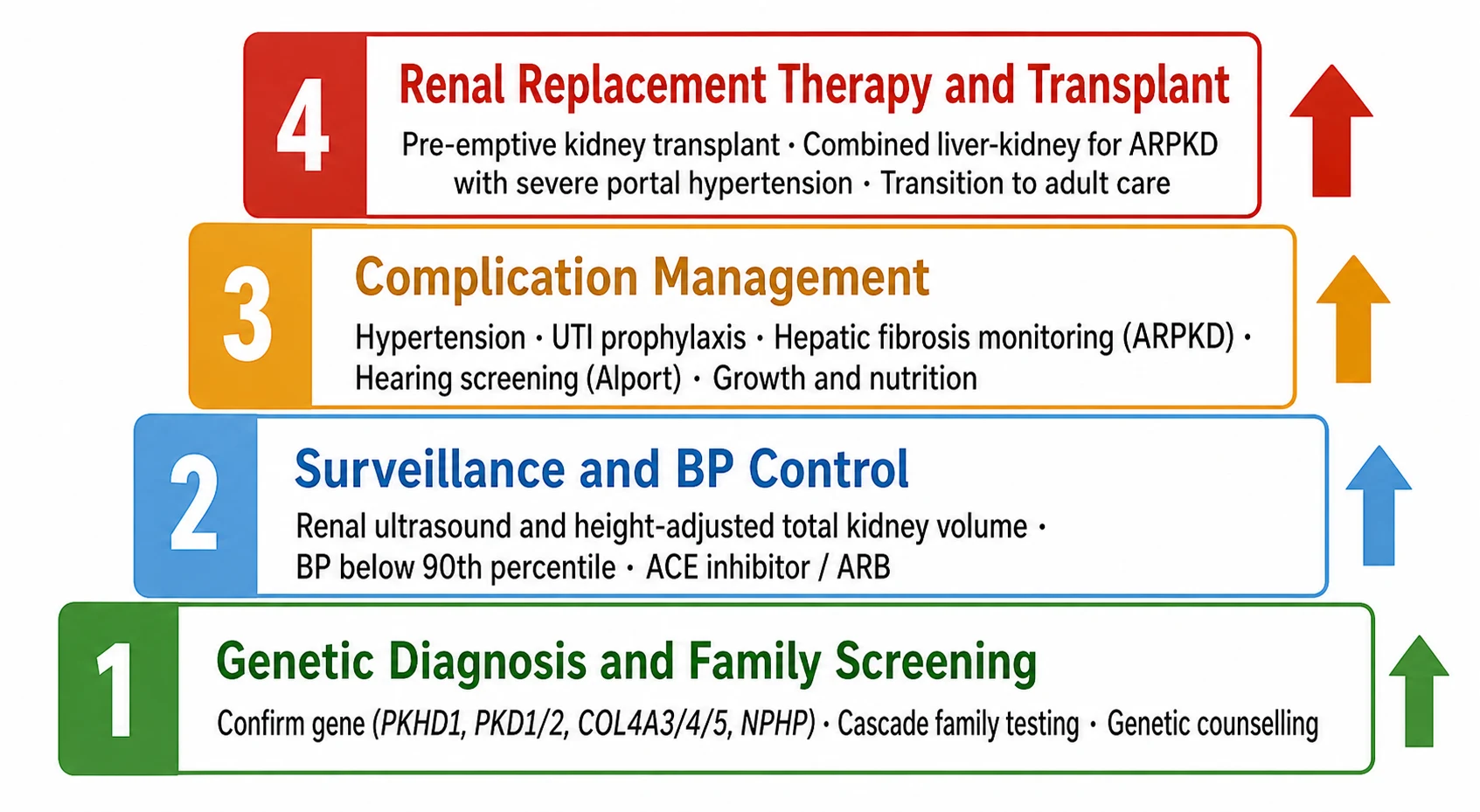
Definitive management of the inherited nephropathies is built on three pillars: genetic confirmation and family screening, nephroprotective surveillance and complication control, and timely preparation for renal replacement therapy. Genetic confirmation is the foundation, because it defines the diagnosis, predicts the course, enables cascade testing of relatives, and informs reproductive counselling. Once the family mutation is known, targeted testing of siblings, parents and at-risk relatives identifies those who need surveillance and those who are spared. Genetic counselling must accompany this process, addressing carrier status, recurrence risk and reproductive options including prenatal and preimplantation genetic diagnosis. [11]
Confirm the genetic diagnosis
Establish surveillance and nephroprotection
Manage disease-specific complications
Plan renal replacement therapy
Blood pressure control and renin-angiotensin system blockade are the cornerstone of nephroprotection across the inherited nephropathies. In Alport syndrome, the 2020 clinical practice recommendations represent a landmark shift: treatment with an ACE inhibitor is now recommended at the time of diagnosis in males with X-linked Alport syndrome and in males and females with autosomal recessive Alport syndrome, rather than waiting for overt proteinuria. In females with X-linked Alport syndrome and in males and females with autosomal dominant Alport syndrome, treatment is started at the onset of microalbuminuria. This early intervention strategy reflects the evidence that renin-angiotensin blockade delays the onset of kidney failure, with studies showing that ACE inhibitor therapy initiated before proteinuria develops can postpone dialysis by a median of over a decade. [6]
In autosomal dominant polycystic kidney disease, the disease-modifying therapy is tolvaptan, a vasopressin V2 receptor antagonist that reduces cyclic AMP signalling and slows cyst growth. The paediatric randomised controlled trial demonstrated that tolvaptan, in children and adolescents aged 4 to 17 years with ADPKD and an estimated GFR of 60 mL per minute per 1.73 square metres or more, slowed the rate of height-adjusted total kidney volume increase compared to placebo over one year. The KDIGO 2025 guideline positions tolvaptan for patients at high risk of rapid progression, identified by imaging classification using height-adjusted total kidney volume, family history of early kidney failure, or genetic profile. The drug requires careful monitoring for aquaresis, which demands adequate water intake, and for hepatotoxicity, with liver function tests monitored regularly, and it is contraindicated in pregnancy. [2]
Tolvaptan in Children and Adolescents with ADPKD
Key finding
In a randomised controlled trial of children and adolescents aged 4 to 17 years with ADPKD and eGFR 60 or more, tolvaptan slowed the annual rate of height-adjusted total kidney volume increase compared with placebo over one year, with a predictable aquaresis and liver enzyme safety profile consistent with adult trials.
Practice change
Tolvaptan is a disease-modifying option for rapidly progressive paediatric ADPKD, requiring careful patient selection by Mayo imaging class and monitoring for aquaresis and hepatotoxicity.
Complication management is disease-specific and lifelong. In ARPKD, the management extends beyond nephrology to hepatology because congenital hepatic fibrosis causes portal hypertension that may result in variceal bleeding, and the biliary abnormalities may lead to recurrent cholangitis. Children with ARPKD require surveillance for portal hypertension with abdominal ultrasound, platelet counts and endoscopic screening for varices. In severe ARPKD with both kidney and liver failure, combined liver-kidney transplantation may be necessary, and this approach addresses both the portal hypertension and the renal failure, with outcomes reported as favourable in experienced centres. In ADPKD, blood pressure control, prompt treatment of cyst infections and haemorrhage, and avoidance of nephrotoxins are the priorities, with the goal of preserving function for as long as possible and reaching transplantation in the best condition. [12]
In Alport syndrome, audiometric surveillance begins at about age five and continues annually, with hearing aids provided when hearing loss becomes functionally significant. Ophthalmology review monitors for anterior lenticonus, which may require corrective lenses, and cataract formation. In nephronophthisis, the management centres on supportive care for the progressive kidney failure: treating the anaemia with iron and erythropoiesis-stimulating agents, managing the salt wasting and polyuria, and monitoring growth and nutrition. There is no disease-specific therapy for nephronophthisis yet, though agents targeting the ciliary signalling pathways, including vasopressin receptor antagonists and cyclin-dependent kinase inhibitors, are under investigation. [8]
Specific Subtypes & Scenarios
Autosomal recessive polycystic kidney disease is the subtype with the widest spectrum of severity, from perinatal lethality to presentation in adulthood. The perinatal form presents with massively enlarged kidneys, pulmonary hypoplasia and respiratory failure at birth, and historically carried a very high mortality, though modern neonatal intensive care, ventilation and blood pressure management have substantially improved survival. The neonatal and infantile forms present with hypertension, kidney dysfunction and failure to thrive, often with hepatic fibrosis becoming clinically apparent later through splenomegaly, cytopenias from hypersplenism, or variceal bleeding. The management challenge is dual: controlling the renal disease and its complications while simultaneously surveilling and managing the hepatic fibrosis, which may be the dominant clinical problem even after successful kidney transplantation. [1]
Autosomal dominant polycystic kidney disease in childhood is increasingly recognised as a stage where intervention can change the trajectory, rather than simply a waiting period. Cyst growth begins early, and children with a high cyst burden at a young age are at greatest risk of rapid progression. The Mayo imaging classification, based on height-adjusted total kidney volume adjusted for age, stratifies patients into classes that predict the rate of kidney function decline, and it guides the decision to start tolvaptan. Very early-onset ADPKD, defined by symptoms before age 15, is associated with a higher burden of PKD1 mutations, particularly truncating variants, and a more rapid course, and recent systematic review confirms that these children have worse kidney outcomes than those presenting later. [10]
Alport syndrome in females is a scenario that examiners probe because it is often underestimated. Females with X-linked Alport syndrome from COL4A5 mutations are not merely carriers: about 15 to 30 percent develop kidney failure by age 60, and pregnancy can accelerate progression. They require surveillance with blood pressure, urinalysis and renal function, and they should receive ACE inhibitor therapy at the onset of microalbuminuria. The autosomal recessive form, from biallelic COL4A3 or COL4A4 mutations, produces a severe phenotype in both sexes that resembles the X-linked disease in males, with early-onset kidney failure, hearing loss and ocular features. The autosomal dominant form, from heterozygous COL4A3 or COL4A4 mutations, is increasingly recognised and produces a milder course, though some families show significant progression, blurring the boundary with thin basement membrane nephropathy. [7]
Nephronophthisis-related ciliopathies present the broadest phenotypic spectrum because the shared ciliary biology connects the kidney to the retina, brain, liver and skeleton. The isolated renal form progresses to kidney failure at a predictable age that clusters into infantile, juvenile and adolescent onset depending on the gene, with NPHP2 causing the infantile form and NPHP1 the classic juvenile form. The syndromic forms add their distinctive features: Senior-Loken syndrome adds retinitis pigmentosa with night blindness and visual field constriction, Joubert syndrome adds the molar tooth sign on brain imaging with hypotonia, ataxia and developmental delay, and Mainzer-Saldino syndrome adds cone-shaped epiphyses and retinal dystrophy. Management requires a coordinated multidisciplinary team, and genetic counselling is complex because the inheritance is autosomal recessive with a 25 percent recurrence risk. [8]
Complications & Pitfalls
The complications of the inherited nephropathies cluster around hypertension, progressive kidney failure and the extrarenal manifestations of each gene defect. Hypertension is common and early in ARPKD, affects a significant proportion of children with ADPKD, and develops with declining function in Alport syndrome and nephronophthisis. It must be identified and treated to the below-90th-percentile target, because uncontrolled hypertension accelerates progression and drives cardiovascular disease. Portal hypertension from congenital hepatic fibrosis in ARPKD is a unique complication that can cause life-threatening variceal bleeding, and it requires active surveillance and prophylactic intervention. [1]
The classic pitfalls span diagnosis and management. Failing to screen the family of a child newly diagnosed with an inherited nephropathy misses affected siblings who would benefit from early surveillance and treatment. Treating Alport syndrome as a benign cause of familial haematuria, rather than initiating ACE inhibitor therapy, forfeits the proven benefit of early intervention. Offering tolvaptan without the imaging classification to identify high-risk patients exposes children to the drug's aquaresis and hepatotoxicity burden without the likelihood of benefit. Missing the hepatic fibrosis component of ARPKD and focusing only on the kidneys leaves a child vulnerable to variceal bleeding. And failing to recognise the syndromic features of nephronophthisis-related ciliopathies, such as the night blindness of Senior-Loken syndrome or the neurological signs of Joubert syndrome, delays the coordinated multidisciplinary care these complex children need. [8]
Tolvaptan therapy carries specific pitfalls that demand vigilance. The aquaresis can cause dehydration and hypernatraemia if the child cannot maintain adequate water intake, and the hepatotoxicity, though rare, is potentially serious, requiring baseline and regular liver function testing, and immediate cessation if significant transaminase elevation occurs. The drug is contraindicated in pregnancy because of its teratogenic potential, which is relevant in adolescent girls, and it does not benefit patients with advanced disease where the cyst burden is already overwhelming the remaining nephrons. Patient and family education about the aquaretic effect, the need for free water access, and the importance of adherence and monitoring is essential before therapy is started. [4]
Prognosis & Disposition
The prognosis of the inherited nephropathies is determined by the gene, the specific variant and the adequacy of management. Autosomal recessive polycystic kidney disease carries the most variable prognosis, from perinatal death to survival into adulthood with preserved kidney function. Neonates who survive the respiratory and hypertensive crises of the perinatal period often stabilise, and with modern intensive care and nephroprotective management, a substantial proportion reach adulthood before requiring renal replacement therapy. Those who progress to kidney failure are managed with dialysis and transplantation, with combined liver-kidney transplantation reserved for those with severe hepatic fibrosis and portal hypertension. [1]
Severity
Severe neonatal ARPKD
Bilateral massively enlarged kidneys with pulmonary hypoplasia and oligohydramnios. Critical neonatal management with ventilation, blood pressure control and fluid management. Mortality remains significant in the perinatal period despite modern intensive care.
Severity
Progressive ADPKD in childhood
High cyst burden with rapidly increasing height-adjusted total kidney volume indicating rapid progression. Tolvaptan therapy indicated with careful monitoring. Blood pressure control and nephroprotection essential to delay kidney failure.
Severity
Alport syndrome with early ACE inhibitor therapy
Persistent haematuria with preserved kidney function and no hearing loss yet. ACE inhibitor initiated at diagnosis. Prognosis for delaying kidney failure is substantially improved with early renin-angiotensin blockade, though progressive disease remains likely in males with X-linked disease.
Autosomal dominant polycystic kidney disease in childhood has an excellent short-term prognosis but a predictable long-term trajectory. With blood pressure control, avoidance of nephrotoxins, and tolvaptan where indicated, progression can be slowed, and many patients preserve kidney function well into middle age. The goal of paediatric management is to reach adulthood in the best possible condition, delaying the need for renal replacement therapy as long as possible and ensuring a smooth transition to adult nephrology. In Alport syndrome, the prognosis is stratified by sex and genotype: males with X-linked disease almost universally progress to kidney failure, with the age of onset modified by the specific COL4A5 variant and the timing of ACE inhibitor initiation, while the autosomal forms follow a course determined by whether one or two pathogenic alleles are present. [6]
Nephronophthisis universally progresses to kidney failure, typically within the first two decades of life, and the management goal is to support the child through this progression with attention to anaemia, growth, nutrition and psychosocial wellbeing, and to plan pre-emptive transplantation. Kidney transplantation is curative for the renal disease, and recurrence does not occur because the genetic defect is kidney-intrinsic rather than systemic, which is an important difference from glomerular diseases. Disposition across all four conditions follows the stage and rate of progression, with stable children managed in joint general paediatric and nephrology clinics and those approaching kidney failure managed in specialist paediatric nephrology centres with access to dialysis, transplant assessment and a multidisciplinary team. [8]
Special Populations
Neonates and infants are the population in whom ARPKD has its greatest impact, and they present unique management challenges. The combination of massively enlarged kidneys, respiratory compromise, severe hypertension and electrolyte disturbance demands coordinated neonatal and nephrology care, often in a tertiary centre with capacity for advanced ventilation and renal support. Sodium supplementation, adequate nutrition and careful fluid management are central to growth and stability in infants with the polyuric, salt-wasting phenotype, and these practical measures may matter more than any drug in the early months. Hepatic fibrosis surveillance must begin early, because portal hypertension may declare itself with variceal bleeding before the kidney disease becomes the dominant problem. [1]
Adolescents with inherited nephropathies face the twin challenges of disease progression and the transition to adult care. Non-adherence to medication and monitoring rises in adolescence and is a leading cause of graft loss after transplantation, which is why structured transition programmes, peer support and honest conversations about fertility, substance use and self-management are essential. Young women with ADPKD need counselling about pregnancy, which is generally safe with preserved kidney function but carries risks of hypertension and function decline in those with advanced disease, and tolvaptan must be stopped before conception because of teratogenicity. Young women with Alport syndrome need to understand their reproductive risk and the implications of their COL4A5 mutation for their children, and genetic counselling should be integrated into transition. [5]
Children from Indigenous and socioeconomically disadvantaged backgrounds face disproportionate barriers to the diagnosis and management of inherited nephropathies. Access to genetic testing, specialist nephrology services, audiology, ophthalmology and transplant services may be limited, and the lifelong surveillance that these conditions require demands continuity of care that is harder to maintain across geographical and socioeconomic disadvantage. Recognising and addressing these inequities through culturally safe care, outreach and care coordination is part of delivering the nephroprotective bundle equitably. Families need to understand that while these conditions cannot yet be cured, they can be managed, and early diagnosis and consistent follow-up substantially improve outcomes for the child and the wider family. [3]
Evidence, Guidelines & Regional Differences
The KDIGO 2025 Clinical Practice Guideline for the Evaluation, Management and Treatment of Autosomal Dominant Polycystic Kidney Disease is the current global standard, providing evidence-based recommendations for diagnosis, risk stratification and treatment across children and adults. It confirms the imaging-based approach to classification using height-adjusted total kidney volume, positions tolvaptan for patients at high risk of rapid progression, and addresses the management of complications including hypertension, pain and infection. For children specifically, it emphasises the importance of early diagnosis through family screening, blood pressure control to the below-90th-percentile target, and careful patient selection for tolvaptan using the Mayo imaging classification. [2]
The 2024 ERKNet, ERA and ESPN guideline on Alport syndrome provides the current European consensus on diagnosis, management and treatment, complementing the Kashtan and Gross 2020 clinical practice recommendations. Together they establish the strategy of early ACE inhibitor initiation: at diagnosis for males with X-linked and all patients with autosomal recessive disease, and at the onset of microalbuminuria for females with X-linked and all patients with autosomal dominant disease. They also provide structured approaches to genetic testing, hearing surveillance, family screening and the classification of variants of uncertain significance, addressing the diagnostic challenges that arise with increasingly comprehensive genetic panels. [7]
EARLY ACE Inhibition in Alport Syndrome
Key finding
Early initiation of ACE inhibitor therapy, before the onset of overt proteinuria, delays the progression to kidney failure in Alport syndrome. The 2020 updated recommendations extend treatment to the time of diagnosis in males with X-linked disease and in both sexes with autosomal recessive disease, based on cohort evidence that early intervention postpones dialysis by a median of over a decade.
Practice change
Start ACE inhibitor therapy at diagnosis in high-risk Alport genotypes; do not wait for overt proteinuria.
Regional differences exist mainly in access to genetic testing, tolvaptan and transplant services. Systems with universal health coverage and established rare disease networks, such as the European Reference Network for Rare Kidney Diseases (ERKNet), provide coordinated genetic diagnosis, surveillance and management from childhood through to adult transition. In Australia and Aotearoa New Zealand, paediatric inherited nephropathies are managed through specialist paediatric nephrology centres linked to the ANZDATA registry, with access to genetic testing, tolvaptan through specialist programmes, and a national transplant system that prioritises pre-emptive living-donor transplantation. The International Pediatric Nephrology Association provides guidance and support for resource-limited settings, emphasising the high-yield interventions of blood pressure control, family screening and timely referral that are achievable even where genetic testing and tolvaptan are not universally available. [5]
Exam Pearls
The inherited nephropathies are classified by their defective protein and gene. Autosomal recessive polycystic kidney disease is caused by PKHD1 encoding fibrocystin, with an incidence of 1 in 20,000 live births, presenting with bilateral enlarged echogenic kidneys and congenital hepatic fibrosis. Autosomal dominant polycystic kidney disease is caused by PKD1 in about 85 percent and PKD2 in about 15 percent, encoding polycystin 1 and 2, with bilateral cysts and tolvaptan-responsive progression. Alport syndrome is caused by COL4A5 in the X-linked form (about 85 percent) or COL4A3 and COL4A4 in autosomal forms, presenting with persistent haematuria, progressive kidney failure and high-frequency sensorineural hearing loss. [3]
The management pillars are genetic confirmation and family screening, nephroprotective surveillance with blood pressure below the 90th percentile, and timely preparation for transplantation. In Alport syndrome, ACE inhibitor therapy should start at diagnosis in males with X-linked disease and in all with autosomal recessive disease, because early intervention delays kidney failure by years. In ADPKD, tolvaptan slows cyst growth in rapidly progressive disease identified by height-adjusted total kidney volume, with monitoring for aquaresis and hepatotoxicity. In ARPKD, combined liver-kidney transplantation is indicated for severe portal hypertension with kidney failure. Kidney transplantation is curative for the renal disease in nephronophthisis, with no recurrence, because the genetic defect is kidney-intrinsic. [6]
References
- [1]Guay-Woodford LM, Bissler JJ, Braun MC, Bockenhauer D, et al Consensus expert recommendations for the diagnosis and management of autosomal recessive polycystic kidney disease: report of an international conference. J Pediatr, 2014.PMID 25015577
- [2]Torres VE, Ahn C, Barten TRM, Brosnahan G, et al KDIGO 2025 clinical practice guideline for the evaluation, management, and treatment of autosomal dominant polycystic kidney disease (ADPKD): executive summary. Kidney Int, 2025.PMID 39848746
- [3]Bergmann C, Guay-Woodford LM, Harris PC, Horie S, et al Polycystic kidney disease. Nat Rev Dis Primers, 2018.PMID 30523303
- [4]Mekahli D, Guay-Woodford LM, Cadnapaphornchai MA, Greenbaum LA, et al Tolvaptan for Children and Adolescents with Autosomal Dominant Polycystic Kidney Disease: Randomized Controlled Trial. Clin J Am Soc Nephrol, 2023.PMID 36719158
- [5]Dachy A, Van Loo L, Mekahli D Autosomal Dominant Polycystic Kidney Disease in Children and Adolescents: Assessing and Managing Risk of Progression. Adv Kidney Dis Health, 2023.PMID 37088526
- [6]Kashtan CE, Gross O Clinical practice recommendations for the diagnosis and management of Alport syndrome in children, adolescents, and young adults-an update for 2020. Pediatr Nephrol, 2021.PMID 33159213
- [7]Torra R, Lipska-Zietkiewicz B, Acke F, et al Diagnosis, management and treatment of the Alport syndrome - 2024 guideline on behalf of ERKNet, ERA and ESPN. Nephrol Dial Transplant, 2025.PMID 39673454
- [8]Wolf MTF, Bonsib SM, Larsen CP, Hildebrandt F Nephronophthisis: a pathological and genetic perspective. Pediatr Nephrol, 2024.PMID 37930417
- [9]Gimpel C, Avni EF, Breysem L, et al Imaging of Kidney Cysts and Cystic Kidney Diseases in Children: An International Working Group Consensus Statement. Radiology, 2019.PMID 30599104
- [10]Kapogiannis C, Dimakopoulos G, Kapogiannis A, et al Comparison of genetic profile and kidney outcomes between very early-onset and non-very-early-onset autosomal dominant polycystic kidney disease in children: a systematic review and meta-analysis. BMC Nephrol, 2026.PMID 42401882
- [11]Savige J, Lipska-Zietkiewicz BS, Watson E, et al Guidelines for Genetic Testing and Management of Alport Syndrome. Clin J Am Soc Nephrol, 2022.PMID 34930753
- [12]Brinkert F, Lehnhardt A, Montoya C, et al Combined liver-kidney transplantation for children with autosomal recessive polycystic kidney disease (ARPKD): indication and outcome. Transpl Int, 2013.PMID 23582048