Paeds · nephrology-urology-fluids-and-electrolytes
Post-infectious glomerulonephritis
Also known as PSGN · Post-streptococcal glomerulonephritis · Acute postinfectious glomerulonephritis · Diffuse endocapillary proliferative glomerulonephritis · Postinfectious GN
Fellowship guide to post-infectious glomerulonephritis (PSGN): the classic acute nephritic syndrome that follows group A streptococcal pharyngitis by one to three weeks or skin infection by three to six weeks, defined by smoky haematuria, oedema, and hypertension with a low C3 and normal C4 that recovers within eight weeks; the immune-complex and complement pathophysiology with subepithelial humps; the supportive management of fluid restriction, diuretics, and antihypertensives plus streptococcal eradication; and the excellent prognosis in children, with the atypical features that mandate renal biopsy.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
Overview & Definition
A previously well six-year-old who had a sore throat ten days ago and now arrives with puffy eyes, smoky brown urine, and a blood pressure of 130 over 85 has post-infectious glomerulonephritis. The disease is an immune-complex injury of the glomerulus that lands one to three weeks after a group A streptococcal throat infection or three to six weeks after a streptococcal skin infection. The injured filter leaks red cells and protein while leaking less water and salt, which is why the child looks oedematous, hypertensive, and passing cola-coloured urine all at once. [1]
Post-streptococcal glomerulonephritis (PSGN) is the single most common form of glomerulonephritis in children worldwide, and it is the prototype of the acute nephritic syndrome. The critical laboratory signature is a depressed C3 with a normal C4, because the injury is driven largely by the alternative complement pathway, and the C3 characteristically recovers to normal within six to eight weeks. That recovery is the single most useful confirmatory feature, because a C3 that stays low beyond eight weeks is no longer PSGN and points toward C3 glomerulopathy, membranoproliferative glomerulonephritis, or lupus nephritis. [5]
Classification
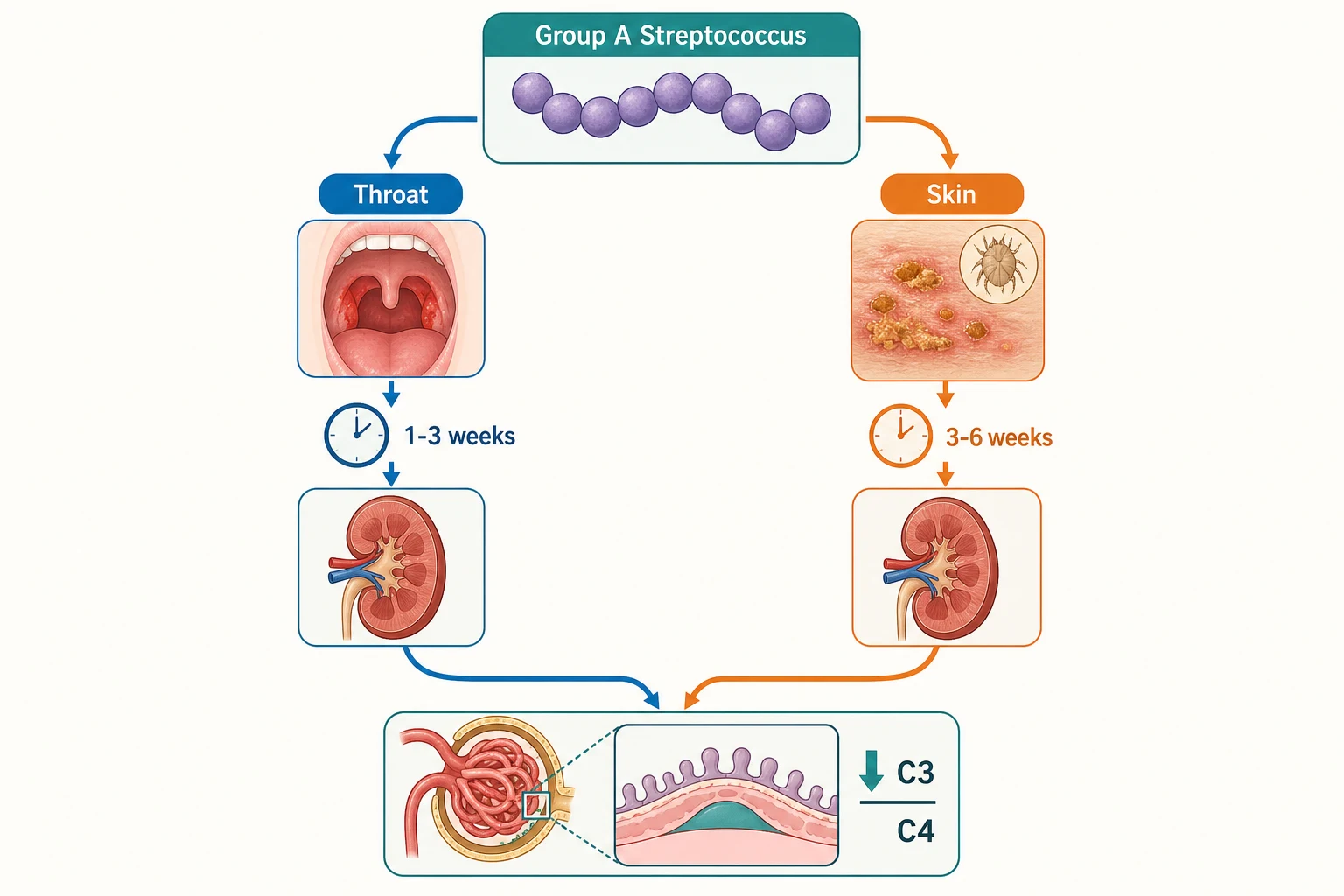
Classification matters because it determines which streptococcal serology to trust and how the latency interval reads. Pharyngeal infection is the dominant portal in temperate climates and in school-aged children, and it carries a shorter latency of one to three weeks. Skin infection with nephritogenic strains predominates in tropical and subtropical regions, in Indigenous communities, and in younger children, and it carries a longer latency of three to six weeks. A child with impetigo may never have had a sore throat, so the absence of pharyngitis does not exclude PSGN. [7]
[1]The lesion itself is histologically a diffuse endocapillary proliferative glomerulonephritis, meaning every glomerulus is hypercellular from infiltration by neutrophils and monocytes that swell the capillary tuft. Immunofluorescence shows coarse granular deposits of IgG and C3 along the capillary walls, and electron microscopy reveals the pathognomonic dome-shaped subepithelial electron-dense deposits known as humps. This constellation is what distinguishes PSGN from other infection-related glomerulonephritides, such as the staphylococcus-related form that occurs concurrently with active infection in older patients with endocarditis or diabetes. [11]
Epidemiology & Risk Factors
Post-streptococcal glomerulonephritis is the most common glomerulonephritis of childhood. It has a worldwide distribution but its burden falls heaviest where overcrowding, skin sepsis, scabies, and limited access to primary care coexist. The peak age is between five and twelve years, it is uncommon under three years, and it is rare in adults. Boys and girls are affected equally. Only a minority of group A streptococcal strains are nephritogenic, which is why most streptococcal infections do not lead to glomerulonephritis. [9]
Several factors raise the chance that a streptococcal infection proceeds to glomerulonephritis. Infection with a nephritogenic M type is the dominant factor, because only certain strains carry the antigens that lodge in the glomerulus. The classical throat nephritogenic strains include M types 1, 12, and 49, while the skin strains include M types 49, 55, 57, and 60. Young school-age children, households with multiple siblings, overcrowded living conditions, untreated skin sepsis, and poor access to early antibiotics all increase risk. Indigenous children in Australia and New Zealand carry a disproportionate burden, driven largely by scabies and streptococcal pyoderma. [7]
Pathophysiology
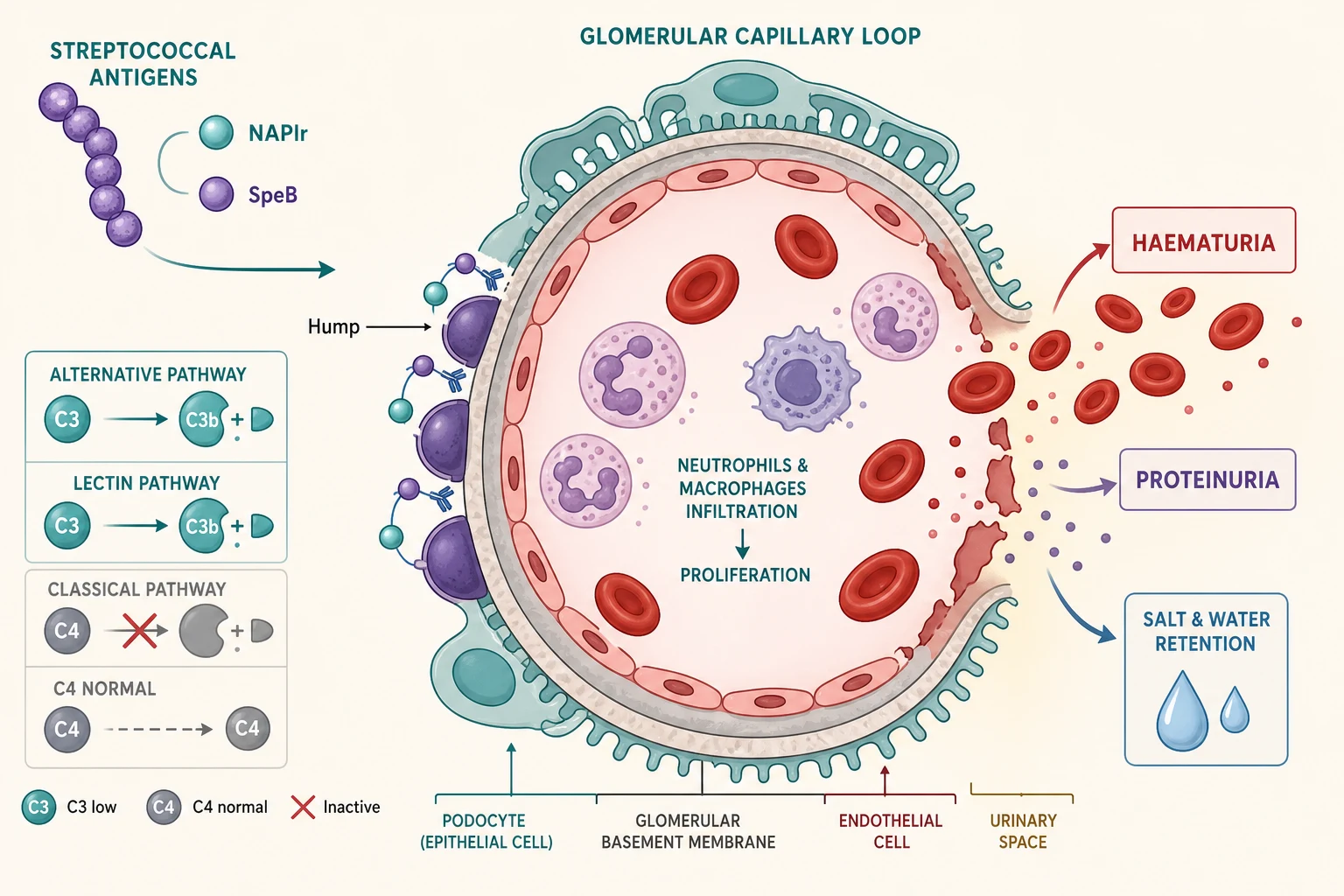
The mechanism is an immune-complex injury in which streptococcal antigens lodge in the glomerulus and ignite complement. Two antigens dominate the modern understanding. The nephritis-associated plasmin receptor, which is a glyceraldehyde-3-phosphate dehydrogenase on the streptococcal surface, binds to glomerular mesangium and podocytes and activates the plasmin system, which in turn drives alternative-pathway complement activation and C3-dominant injury. The streptococcal pyrogenic exotoxin B and its precursor are the other key antigen, and antibodies to it colocalise precisely with the glomerular deposits. [4]
Complement activation is what produces the defining low C3. Both the alternative pathway and the lectin pathway are engaged, while the classical pathway is relatively spared, which is why C4 is typically normal. This pattern, a depressed C3 with a preserved C4, is the serological fingerprint of PSGN and reflects consumption of C3 downstream of the alternative-pathway C3 convertase. Hisano and colleagues demonstrated lectin-pathway activation in glomeruli of children with PSGN, supporting the idea that more than one complement route contributes to the injury. [5]
HUMP
Once complement is activated, the inflamed glomerular capillary wall leaks. Neutrophils and macrophages infiltrate the tuft, producing the endocapillary hypercellularity seen on light microscopy. Red blood cells cross the damaged barrier, giving dysmorphic red cells and red cell casts in the urine, and the brown smoky colour from haemoglobin breakdown. Protein escapes with the red cells, usually in mild to moderate amounts. The same inflammation reduces the filtration surface and triggers salt and water retention, which is the direct cause of the oedema, the hypertension, and occasionally the pulmonary oedema and acute kidney injury that bring children to hospital. [2]
An autoimmune component adds to the injury. Rodriguez-Iturbe emphasised that antibodies directed against streptococcal antigens can cross-react with glomerular components, and that anti-IgG rheumatoid-factor-like activity contributes to the granular IgG deposition. This explains why the deposits persist for a time even after the organism has been cleared, and it links the immune-complex and autoantibody mechanisms rather than treating them as alternatives. [3]
In Australia and New Zealand, PSGN remains endemic in many remote Indigenous communities, where streptococcal pyoderma and scabies are the predominant drivers. Public health programmes that treat scabies and skin sepsis have reduced but not eliminated the burden, and outbreaks continue to occur in household and school clusters. Access to early serology and urinalysis in these settings shortens the time to diagnosis. [7]
Clinical Presentation
The classic story is a school-aged child who was apparently recovering from a sore throat, or whose scabies and impetigo were healing, and who then develops smoky brown urine, morning periorbital swelling, and is found to be hypertensive. The parents may describe the urine as looking like cola, tea, or washing-up water, and may have noticed reduced urine output. The latency is the key discriminator from IgA nephropathy: in PSGN the haematuria follows the infection by one to three weeks, whereas in IgA nephropathy it occurs during the infection, so-called synpharyngic haematuria. [2]
The full acute nephritic syndrome comprises haematuria, oedema, and hypertension. Periorbital oedema is often the first sign and is most noticeable in the morning, settling through the day as the child stands. Hypertension is common and may be the presenting feature, discovered only when the child presents with a complication. Around half of children have a mild to moderate acute kidney injury with oliguria and a rising creatinine, and systemic features such as malaise, lethargy, abdominal or flank pain, and low-grade fever are frequent. [9]
A minority of children present not with the nephritic triad but with one of its complications. Hypertensive encephalopathy may announce the disease with headache, vomiting, visual disturbance, or a seizure, and PSGN is an important cause of new-onset seizures in a school-aged child with occult hypertension. Pulmonary oedema presents as breathlessness and respiratory distress from salt and water overload. A small number have nephrotic-range proteinuria or a rapidly progressive course with falling renal function, and these atypical presentations should prompt urgent nephrology referral and consideration of biopsy. [2]
Differential Diagnosis
The first task is to separate PSGN from the other glomerulonephritides that produce haematuria with a low C3, because the prognosis and the management differ. IgA nephropathy is the most common mimic of recurrent visible haematuria, but it has a normal C3 and produces haematuria during rather than after an infection. The complement pattern and the latency interval therefore do most of the diagnostic work. [1]
[11]C3 glomerulopathy and membranoproliferative glomerulonephritis are the conditions to exclude when the C3 fails to normalise. Both produce persistently low C3 with a similar proliferative lesion, and C3 glomerulonephritis can even follow an apparent PSGN. Lupus nephritis lowers both C3 and C4, and a low C4 in a child with glomerulonephritis should always trigger an autoimmune screen. Shunt nephritis and endocarditis-associated glomerulonephritis produce immune-complex injury with low complement in a child with a foreign body or a systemic infection, and the staphylococcus-related glomerulonephritis described by Glassock and colleagues occurs concurrently with active infection rather than after it. [10]
Non-glomerular causes of dark urine must not be forgotten. Urinary tract infection, renal calculi, and hypercalciuria can cause red or brown urine but do not produce the nephritic syndrome with hypertension and oedema. Pigment from myoglobin or bilirubin can darken the urine without red cells, and a positive dipstick for blood with no red cells on microscopy points to rhabdomyolysis or haemolysis rather than glomerular bleeding. [2]
Clinical & Bedside Assessment
Assessment runs alongside resuscitation and answers three questions: how severe is the nephritic syndrome, what is the likely streptococcal portal, and are there complications that need urgent treatment. The history establishes the latency. Ask specifically about a sore throat or fever in the preceding one to three weeks, and about skin sores, scabies, or impetigo in the preceding three to six weeks, because parents rarely volunteer the skin history unless asked. Record urine output and any symptoms of encephalopathy such as headache, visual change, or seizures. [2]
Examination focuses on the fluid state and the complications of hypertension. Look for periorbital and peripheral oedema, weigh the child, and assess for pulmonary oedema with crackles, a gallop rhythm, and hepatomegaly. Measure the blood pressure with an appropriate cuff and plot it against centiles, because occult hypertension is common and may cause encephalopathy. Examine the skin for healing impetigo and scabies burrows and the throat for residual pharyngitis, since the portal of entry confirms the streptococcal link. [9]
Neurological examination is essential in any hypertensive child. Look for altered consciousness, irritability, and focal deficits, and remember that a seizure in a school-aged child with hypertension is hypertensive encephalopathy until proven otherwise. Fluid balance is the most error-prone assessment, because the child is total-body salt and water overloaded from the nephritic syndrome yet may be intravascularly depleted if vomiting has preceded presentation, and the net status guides whether to restrict or to give a cautious bolus. [2]
Investigations
The diagnosis rests on three groups of tests: the urinalysis that shows glomerular bleeding, the complement profile that shows the low-C3-normal-C4 pattern, and the streptococcal serology that confirms the preceding infection. Urinalysis shows dysmorphic red blood cells, red cell casts, and proteinuria. The proteinuria is usually mild to moderate, but nephrotic-range proteinuria can occur and is an atypical feature that should lower the threshold for biopsy. A urine culture excludes urinary tract infection. [1]
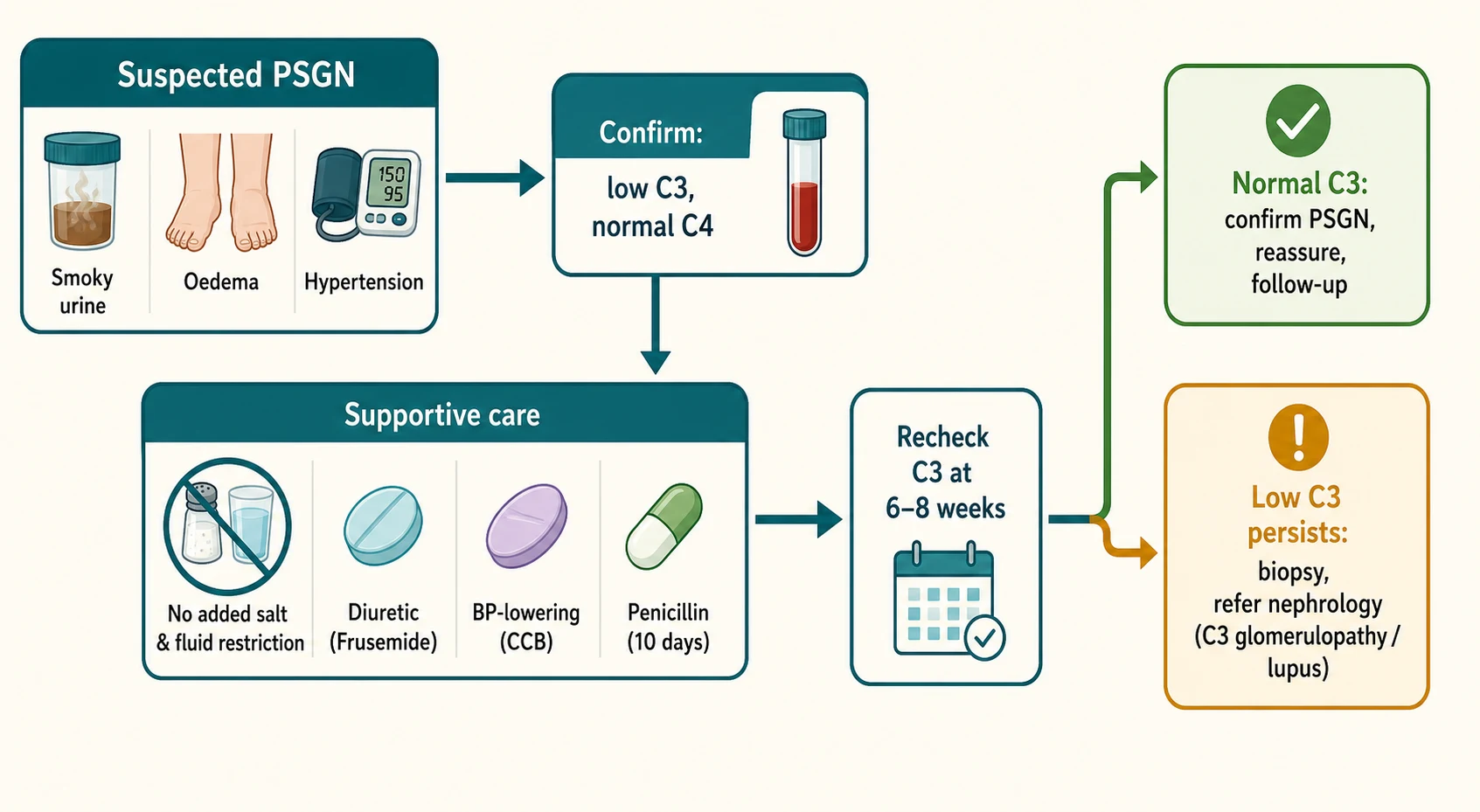
The complement profile is the single most important blood test. A low C3 with a normal C4 at presentation, recovering to normal within six to eight weeks, is diagnostic of typical PSGN in the right clinical context. The C3 should be rechecked at six to eight weeks; if it has normalised the diagnosis is confirmed, and if it remains low the child has an alternative or atypical complement-mediated glomerulonephritis and needs referral and biopsy. A low C4 in addition to a low C3 should always prompt an autoimmune screen for systemic lupus erythematosus, because lupus nephritis is the classic low-C3-low-C4 glomerulonephritis. [5]
Streptococcal serology confirms the preceding infection. The antistreptolysin O titre rises in around 80 percent of children after pharyngitis but is less reliable after skin infection, where the anti-DNase B antibody is more sensitive because it stays elevated for many months. A single raised titre confirms past exposure, and a rising titre between acute and convalescent samples is more specific. Throat and skin swabs may grow group A streptococcus but are often negative by the time PSGN declares itself, so a negative culture does not exclude the diagnosis. Bloods should also check renal function, electrolytes, albumin, and a full blood count, and a renal ultrasound excludes obstruction and other structural disease. [1]
Renal biopsy is not needed in a typical case but is indicated when features are atypical: a rapidly progressive course with rising creatinine and oliguria suggesting crescents, nephrotic-range proteinuria, a low C4 or no streptococcal evidence, a C3 that fails to recover by eight weeks, or persistent hypertension. The AsPNA guideline for infection-related glomerulonephritis reinforces biopsy in these atypical presentations to distinguish PSGN from C3 glomerulopathy and lupus nephritis, which have fundamentally different management. [6]
Management — Resuscitation
The overriding principle is supportive care, because typical PSGN resolves spontaneously and no immunosuppression changes its course. Resuscitation begins with airway, breathing, and circulation, with immediate attention to the two life-threatening complications: severe hypertension with encephalopathy, and fluid overload with pulmonary oedema. A child with hypertensive encephalopathy needs controlled blood-pressure lowering with an intravenous antihypertensive, and a child in pulmonary oedema needs oxygen, sitting upright, and urgent fluid restriction with a loop diuretic. [2]
Restrict salt and water
Treat oedema
Control blood pressure
Eradicate streptococcus
Manage acute kidney injury
Fluid and salt balance is the cornerstone. Most children are overloaded rather than dehydrated, so the default is restriction rather than boluses. Restrict fluids to insensible losses plus urine output, give a no-added-salt diet, and use the daily weight to guide the rate of fluid removal. Oedema and the volume-dependent hypertension respond well to a loop diuretic, and frusemide given orally at 1 to 2 mg per kg per dose will usually relieve the oedema and lower the blood pressure within a day or two. A child who is genuinely intravascularly depleted and still passing urine may need a cautious isotonic bolus, but this is the exception and must be watched closely. [2]
Hypertension is treated in a stepwise way. For sustained hypertension a calcium channel blocker is first line: amlodipine 0.1 to 0.2 mg per kg once daily, starting at the lower end and titrating up to a maximum of 10 mg per day, or nifedipine modified-release at 0.25 to 0.5 mg per kg per day. For a hypertensive emergency with encephalopathy, lower the pressure in a controlled fashion with an intravenous agent such as a labetalol infusion or a nicardipine infusion, aiming to reduce the pressure by no more than 25 percent in the first hours to avoid organ hypoperfusion. Acute kidney injury is managed expectantly with treatment of hyperkalaemia and acidosis, and dialysis is reserved for refractory hyperkalaemia, severe metabolic acidosis, or pulmonary oedema unresponsive to diuretics. [2]
Management — Definitive & Stepwise
Definitive management is the supportive care described above plus eradication of the nephritogenic streptococcus. Treating the organism does not change the course of the established glomerulonephritis, because the immune injury is already underway, but it clears the strain, prevents spread to contacts, and removes the source of ongoing antigen. The standard regimen is phenoxymethylpenicillin (penicillin V) orally for 10 days, dosed as 250 mg twice daily for children under 20 kg and 500 mg twice daily for those over 20 kg. Where adherence is uncertain, a single intramuscular dose of benzathine penicillin is effective, 600,000 units for children under 27 kg and 1.2 million units for those 27 kg and above. A penicillin-allergic child receives a macrolide such as erythromycin or clarithromycin for 10 days. [6]
Phenoxymethylpenicillin (penicillin V)
Dose
Children under 20 kg: 250 mg orally twice daily for 10 days. Children 20 kg and over: 500 mg orally twice daily for 10 days. Alternative single-dose benzathine penicillin intramuscularly: 600,000 units under 27 kg, 1.2 million units at 27 kg and above.
Immobilisation, salt restriction, and antihypertensives are continued until the oedema and blood pressure settle, which is usually within one to two weeks. There is no role for corticosteroids or other immunosuppression in typical PSGN, and giving them is both unsupported and harmful because it exposes a self-limiting disease to serious side effects. The AsPNA guideline reserves immunosuppression for the biopsy-proven atypical cases, such as crescentic PSGN with rapidly progressive renal failure, which are managed in partnership with a paediatric nephrologist. [6]
Household and close contacts should be screened for streptococcal infection in outbreak settings, particularly in remote communities and institutional environments, and treated if positive. Bedside teaching for the family explains that the disease is self-limiting, that the C3 will recover within eight weeks, and that long-term follow-up is needed because a small minority develop chronic kidney disease. [8]
Specific Subtypes & Scenarios
Typical childhood PSGN is the dominant subtype and has an excellent prognosis, but several atypical variants change both the management and the counselling. The most important is the atypically persistent hypocomplementaemic case, in which the C3 fails to normalise by eight weeks. This is not simply a slow recovery; it raises C3 glomerulonephritis, which can follow an apparent streptococcal illness and behaves as a chronic complement-mediated disease rather than a self-limiting one, and it requires biopsy and long-term nephrology care. [10]
Crescentic PSGN is the rapidly progressive form in which the biopsy shows substantial crescent formation and the child develops oliguria and a rising creatinine. Unlike typical PSGN, this form may benefit from immunosuppression in addition to supportive care, and it must be discussed urgently with a paediatric nephrologist rather than managed with restraint alone. The presence of nephrotic-range proteinuria, of a depressed C4, or of any clinical feature pointing to lupus should each prompt the same response: confirm the diagnosis histologically before committing to treatment. [6]
Predictors of chronicity after PSGN — Utari et al 2024
Key finding
In a paediatric cohort, the factors that independently predicted progression to chronic glomerulonephritis were heavier proteinuria at onset, persistent hypertension, persistent hypocomplementaemia, crescents on biopsy, and higher presenting creatinine. Children with none of these features recovered completely.
Practice change
Long-term follow-up should be weighted toward children who carry these risk factors, while typical uncomplicated cases can be reassured.
The Indigenous-community setting deserves specific attention. In remote Australia, PSGN presents against a background of high rates of streptococcal pyoderma and scabies, often in household clusters, and the skin rather than the throat is the usual portal. The latency is correspondingly longer, and the public-health response of treating scabies and skin sepsis and screening contacts is central to prevention. These children also carry a higher lifetime risk of chronic kidney disease, which makes surveillance after recovery especially important. [7]
Complications & Pitfalls
The acute complications of PSGN are the direct consequences of salt and water retention and acute kidney injury, and they are what bring children to hospital. Hypertensive encephalopathy, presenting with headache, vomiting, visual disturbance, or seizures, is the most dangerous acute complication and may be the presenting feature of the disease. Pulmonary oedema from volume overload presents with breathlessness and respiratory distress. Hyperkalaemia and metabolic acidosis accompany the acute kidney injury in the more severe cases, and a small number require dialysis for refractory hyperkalaemia, severe acidosis, or fluid overload. [2]
Severity
Typical PSGN
Smoky haematuria, mild oedema, controlled blood pressure, normal or mildly impaired renal function. Managed with restriction, diuretics, and penicillin. Prognosis excellent.
Severity
Moderate PSGN
Significant hypertension needing antihypertensives, oedema needing diuretics, and a moderate acute kidney injury. Needs admission and monitoring but recovers fully.
Severity
Severe and atypical PSGN
Hypertensive encephalopathy, pulmonary oedema, dialysis-requiring acute kidney injury, crescentic biopsy, or persistent hypocomplementaemia. Needs paediatric nephrology and possibly intensive care.
The long-term pitfall is the under-recognised minority who progress to chronic kidney disease. Most children recover completely, but a small fraction develop persistent proteinuria, hypertension, or reduced glomerular filtration rate, and some progress years later. The predictive factors identified by Utari and colleagues, namely heavier proteinuria, persistent hypertension, persistent low C3, crescents, and higher presenting creatinine, should flag the children who need closer and longer surveillance rather than discharge after the acute illness. [8]
The most common diagnostic pitfalls are avoidable. Failing to recheck the C3 at six to eight weeks misses the persistent hypocomplementaemia that unmasks C3 glomerulopathy. Assuming that a normal antistreptolysin O titre excludes the diagnosis misses the skin-associated cases where anti-DNase B is the sensitive test. Treating typical PSGN with corticosteroids exposes a self-limiting disease to harm. And forgetting that IgA nephropathy produces haematuria during rather than after infection leads to the wrong diagnosis and the wrong reassurance. [1]
Prognosis & Disposition
The prognosis of typical childhood PSGN is excellent. Over 95 percent of children recover completely, and mortality is under 1 percent in settings with modern supportive care. The C3 normalises within six to eight weeks in the great majority, the macroscopic haematuria resolves within weeks, and microscopic haematuria with low-grade proteinuria may persist for months or even one to two years before disappearing. Renal function returns to baseline, and the blood pressure settles as the oedema is mobilised. [9]
The small minority who do poorly can be identified by the predictive factors. Persistent hypertension, persistent hypocomplementaemia, heavy proteinuria, crescents on biopsy, and a higher presenting creatinine each mark a child at risk of chronic kidney disease, and end-stage kidney disease develops in a small percentage overall but concentrates in this atypical group. In resource-limited settings the outcomes are less favourable, with series such as the Ethiopian multicentre cohort reporting higher rates of acute complications and of residual kidney damage, reflecting later presentation and more severe disease rather than a different biology. [12]
Disposition depends on severity. A child with controlled blood pressure, no pulmonary oedema, and only mild renal impairment can be managed on the ward and discharged once the oedema is settling and the family understands the plan. A child with hypertensive encephalopathy, pulmonary oedema, or dialysis-requiring acute kidney injury needs a high-dependency or intensive care setting and transfer to a tertiary centre with paediatric nephrology. At discharge every child needs a plan to recheck the C3 at six to eight weeks and to monitor blood pressure and urinalysis, with longer follow-up for those who carry risk factors for chronicity. [8]
Special Populations
School-aged children between five and twelve years are the highest-risk group and account for the majority of cases. The disease is uncommon under three years, and a preschool child with a nephritic presentation deserves particular scrutiny for an alternative diagnosis such as a congenital or inherited glomerulonephritis. Adolescents present less often with typical PSGN, and when a teenager develops a hypocomplementaemic glomerulonephritis the differential tilts toward lupus nephritis and IgA nephropathy, both of which need to be excluded. [2]
Indigenous children in Australia and New Zealand carry a disproportionate burden of PSGN, driven by endemic streptococcal pyoderma and scabies in remote communities. The skin is the usual portal, the latency is longer, and presentations are often in household clusters. Public-health interventions that treat scabies and skin sepsis and that improve household crowding have reduced but not eliminated the burden, and these children carry a higher lifetime risk of chronic kidney disease that justifies careful surveillance after each episode. [7]
Children with pre-existing kidney disease who develop a superimposed episode of PSGN can suffer an acute-on-chronic decline, and a child known to have a congenital anomaly of the kidney or a dysplastic kidney should be monitored closely during and after an episode. Children with immune deficiency or nephrotic syndrome who are on immunosuppression present particular challenges, and any atypical feature in these groups should prompt early nephrology involvement and biopsy. [6]
Evidence, Guidelines & Regional Differences
The 2026 Asian Pediatric Nephrology Association clinical practice guideline on infection-related glomerulonephritis is the current paediatric reference, and it reaffirms supportive care as the cornerstone of typical PSGN, streptococcal eradication with penicillin, and biopsy for the atypical features of persistent hypocomplementaemia, nephrotic-range proteinuria, a rapidly progressive course, or absent streptococcal evidence. KDIGO places infection-related glomerulonephritis among the immune-complex glomerulonephritides and similarly recommends supportive care for the post-streptococcal form, reserving immunosuppression for crescentic disease. [6]
The pathophysiological evidence has converged on a small number of streptococcal antigens. The nephritis-associated plasmin receptor drives C3-dominant glomerular injury through plasmin activation of the alternative pathway, and the streptococcal pyrogenic exotoxin B colocalises with the deposits. The lectin complement pathway contributes alongside the alternative pathway, which explains the low C3 with preserved C4. These mechanisms underpin both the serological pattern that clinicians rely on and the rationale against immunosuppression in typical disease. [4]
In Australia and New Zealand, PSGN remains endemic in remote Indigenous communities, where it is managed according to regional guidelines that emphasise streptococcal and scabies eradication, household contact screening, and long-term renal surveillance. Access to early complement testing and urinalysis in remote settings shortens the diagnostic interval, and the public-health response to skin sepsis is central to prevention. [7]
Regional differences matter most for prognosis. In high-income settings with early presentation and supportive care, mortality is under 1 percent and over 95 percent of children recover fully. In resource-limited settings with later presentation and more severe disease at presentation, the rates of acute complications and residual kidney damage are higher, reflecting access to care rather than biology. The IPNA and AsPNA guidelines are framed to be applicable across both settings, and they prioritise the simple, low-cost interventions of fluid and salt restriction, diuretics, antihypertensives, and penicillin that change outcomes wherever they are delivered. [12]
Exam Pearls
Post-streptococcal glomerulonephritis is the most common glomerulonephritis in children and the prototype of the acute nephritic syndrome, presenting with smoky haematuria, oedema, and hypertension after group A streptococcal pharyngitis by one to three weeks or skin infection by three to six weeks. The defining serology is a low C3 with a normal C4 that recovers within six to eight weeks, and the defining histology is a diffuse endocapillary proliferative glomerulonephritis with subepithelial humps on electron microscopy. [1]
The high-yield discriminations are worth memorising. IgA nephropathy produces haematuria during the infection with a normal C3, whereas PSGN produces it after with a low C3. Lupus nephritis lowers both C3 and C4, so a low C4 in a child with glomerulonephritis always triggers an autoimmune screen. A C3 that has not normalised by eight weeks is no longer typical PSGN and points to C3 glomerulopathy or membranoproliferative glomerulonephritis. The antistreptolysin O titre is unreliable after skin disease, where anti-DNase B is the sensitive test. [5]
Management is supportive, and the two errors that examiners probe are treating typical PSGN with steroids and failing to give penicillin. Streptococcal eradication with phenoxymethylpenicillin for 10 days does not change the glomerulonephritis but clears the nephritogenic strain and prevents spread. Hypertension is controlled with a calcium channel blocker such as amlodipine, and oedema is treated with frusemide. The prognosis is excellent, with over 95 percent full recovery, and the children who do less well are those with heavy proteinuria, crescents, persistent hypertension, or persistent hypocomplementaemia. [2]
References
- [1]Rodriguez-Iturbe B, Musser JM The current state of poststreptococcal glomerulonephritis J Am Soc Nephrol, 2008.PMID 18667731
- [2]Brant Pinheiro SV, de Freitas VB, de Castro GV, Rufino Madeiro BC, de Araujo SA, et al Acute Post-Streptococcal Glomerulonephritis in Children: A Comprehensive Review Curr Med Chem, 2022.PMID 35702785
- [3]Rodriguez-Iturbe B Autoimmunity in Acute Poststreptococcal GN: A Neglected Aspect of the Disease J Am Soc Nephrol, 2021.PMID 33531351
- [4]Yoshizawa N, Yamada M, Fujino M, Oda T Nephritis-Associated Plasmin Receptor (NAPlr): An Essential Inducer of C3-Dominant Glomerular Injury and a Potential Key Diagnostic Biomarker of Infection-Related Glomerulonephritis (IRGN) Int J Mol Sci, 2022.PMID 36077377
- [5]Hisano S, Matsushita M, Fujita T, Takeshita M, Iwasaki H Activation of the lectin complement pathway in post-streptococcal acute glomerulonephritis Pathol Int, 2007.PMID 17539966
- [6]Meena J, Sinha A, Krishnasamy S, Alba AA, Aziz MA, et al AsPNA Clinical Practice Guidelines for the management of infection-related glomerulonephritis Pediatr Nephrol, 2026.PMID 41627401
- [7]Blyth CC, Robertson PW, Rosenberg AR Post-streptococcal glomerulonephritis in Sydney: a 16-year retrospective review J Paediatr Child Health, 2007.PMID 17535174
- [8]Utari IALA, Adhi S, Hermawan K, Arguni E Predictive factors of progression to chronic glomerulonephritis in pediatric patients with post streptococcal acute glomerulonephritis Pediatr Neonatol, 2024.PMID 38649317
- [9]Bajracharya P, Khadgi A, Shrestha S, Silwal R, Tandukar A Acute Post-streptococcal Glomerulonephritis in a Pediatric Population: A Five-Year Retrospective Study Cureus, 2024.PMID 38618409
- [10]Glassock RJ, Alvarado A, Prosek J, Hebert C, Parikh S, et al Staphylococcus-related glomerulonephritis and poststreptococcal glomerulonephritis: why defining post is important in understanding and treating infection-related glomerulonephritis Am J Kidney Dis, 2015.PMID 25890425
- [11]Nadasdy T, Hebert LA Infection-related glomerulonephritis: understanding mechanisms Semin Nephrol, 2011.PMID 21839370
- [12]Moges TA, Dagnew SB, Yayeh YT, Wondm SA, Dagnew FN, et al Outcomes and Predictors of Acute Post-Streptococcal Glomerulonephritis in Hospitalized Children in Northwest Ethiopia: A Multicenter Retrospective Cohort Study Health Sci Rep, 2026.PMID 42382504