Paeds · nephrology-urology-fluids-and-electrolytes
Renal tubular acidosis
Also known as Renal tubular acidosis · Distal RTA · Proximal RTA · Type 4 RTA · Hyperchloraemic metabolic acidosis · Normal anion gap acidosis · dRTA · Fanconi syndrome acidosis
Fellowship guide to renal tubular acidosis in children: a normal anion gap (hyperchloraemic) metabolic acidosis from impaired renal acid-base handling, classified into distal (type 1) with hypokalaemia and nephrocalcinosis, proximal (type 2) with bicarbonate wasting and Fanconi syndrome, and type 4 with hypoaldosteronism and hyperkalaemia, the urine anion gap as a surrogate for urinary ammonium, and alkali therapy that differs in dose between distal (1 to 4 mEq per kg per day) and proximal (10 to 20 mEq per kg per day) RTA.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
Every day the body generates acid from protein metabolism, and the kidney disposes of it in two coordinated steps. The proximal tubule reclaims the filtered bicarbonate so that almost none is lost in the urine, and the distal nephron secretes new hydrogen ion, buffered by ammonia, to regenerate the bicarbonate that buffered the day's acid load. Renal tubular acidosis is what happens when one of those two jobs fails. The result is the same biochemistry — a metabolic acidosis with a normal anion gap, because chloride rises to replace the bicarbonate that was lost — but the mechanism, the potassium, the urine pH and the treatment all depend on which part of the nephron broke. [9] [2]
The acidosis in RTA looks deceptively mild next to diabetic ketoacidosis, and that is part of the danger. It is chronic, so it leaches mineral from growing bone and silently stops a child growing. It is often found late, after nephrocalcinosis has already appeared on an ultrasound done for failure to thrive. And because the potassium is abnormal in a direction that depends on the type, a child can present with weakness, polyuria, or — in type 4 — an arrhythmia from hyperkalaemia long before anyone thinks of the kidney tubule. This page builds the model around three questions the bedside forces: is the anion gap normal, what does the potassium do, and is the kidney excreting ammonium. [1] [5]
Overview & Definition
Renal tubular acidosis is defined as a persistent metabolic acidosis with a normal anion gap and a near-normal glomerular filtration rate, caused by a specific defect in renal acid-base handling rather than by kidney failure itself. The anion gap — calculated as serum sodium minus the sum of chloride and bicarbonate — is normal (typically 8 to 12, up to about 16 mmol per litre) because the kidney does not retain unmeasured acids; instead it loses bicarbonate, and chloride rises to preserve electroneutrality. That single distinction, normal gap versus high gap, is the gate through which every metabolic acidosis must pass before RTA is even considered. [9] [2]
A high anion gap acidosis — diabetic ketoacidosis, lactic acidosis, renal failure, salicylate or methanol toxicity — is never RTA. In those states unmeasured acids accumulate, widening the gap. RTA belongs to the normal anion gap (hyperchloraemic) group, alongside the bicarbonate loss of diarrhoea and the acid load of some nutritional and drug states. Within that group, the next question is whether the kidney is doing its job: is it excreting ammonium to compensate? A kidney that cannot is in renal tubular acidosis; a kidney that can, but is overwhelmed by gastrointestinal bicarbonate loss, is not. [9] [10]
The three classical types map onto three sites. Type 1 (distal) RTA is a failure of hydrogen ion secretion by the alpha-intercalated cells of the collecting duct. Type 2 (proximal) RTA is a failure of bicarbonate reabsorption by the proximal tubule. Type 4 RTA is a failure of aldosterone action — deficiency or resistance — on the distal nephron, which impairs potassium and hydrogen secretion and ammonium production. The first two cause hypokalaemia; the third causes hyperkalaemia. That potassium direction is the most reliable bedside discriminator of the three, and the reason it is worth committing to memory before anything else. [1] [2]
Classification
The classification of RTA is built around two branching questions: which nephron segment failed, and which direction the potassium moved. Answer both and you have the type. The figure below lays the three types onto their nephron sites with their characteristic potassium, urine pH and cause, so the type can be inferred at the bedside from a chemistry panel and a urine sample. [2] [5]
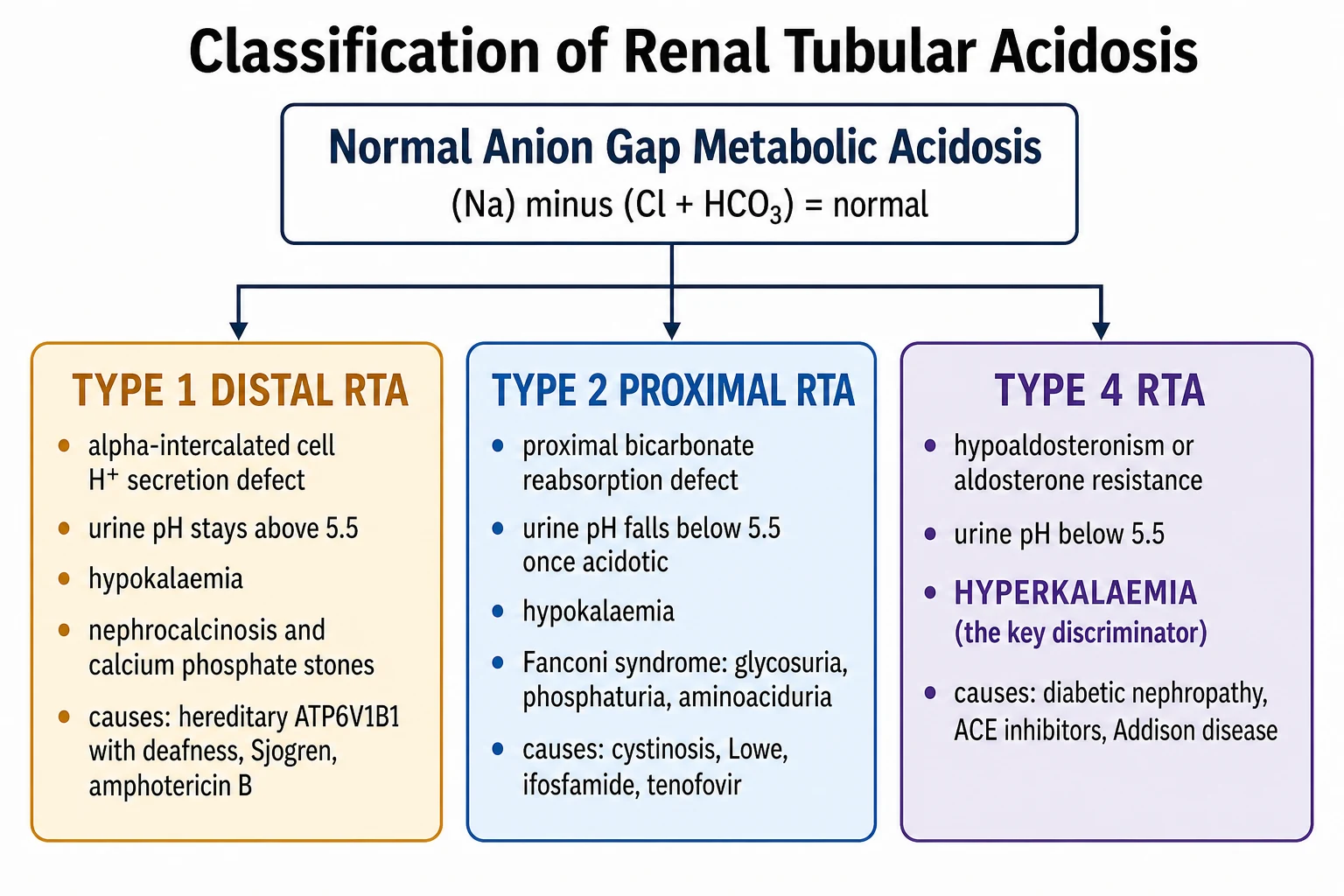
Type 1 (distal) RTA is the archetype. The alpha-intercalated cell of the collecting duct cannot secrete hydrogen ion through its H-ATPase proton pump, so the urine can never be properly acidified and the urine pH stays above 5.5 even when the blood is profoundly acidotic. The potassium is low, because increased distal sodium delivery and secondary hyperaldosteronism drive potassium secretion. And nephrocalcinosis is the calling card: chronic acidosis buffers calcium out of bone, producing hypercalciuria, while the acidotic cell reabsorbs more citrate so urinary citrate falls, and calcium phosphate precipitates in the persistently alkaline urine. [3] [7]
Type 2 (proximal) RTA is a lowered threshold for bicarbonate reabsorption. The proximal tubule normally reclaims about 85 percent of filtered bicarbonate; when it fails, bicarbonate spills into the distal nephron and is lost in the urine, taking sodium and water and driving potassium secretion with it. Once the plasma bicarbonate has fallen below the new (lower) threshold, the distal nephron reasserts control and the urine pH drops below 5.5 — so a low urine pH in a proximal RTA patient who is already acidotic is expected, not paradoxical. Proximal RTA is often part of generalised proximal tubular dysfunction, the Fanconi syndrome, with glycosuria despite normal blood glucose, phosphaturia, aminoaciduria and hypokalaemia. [6] [2]
Type 4 RTA is the aldosterone problem. Aldosterone normally stimulates distal sodium reabsorption, potassium and hydrogen secretion, and ammoniagenesis. When aldosterone is deficient (Addisonian crisis, hyporeninaemic states, drug suppression) or when the distal nephron resists it (pseudohypoaldosteronism, obstructive uropathy, interstitial disease), potassium rises and ammonium excretion falls, producing a normal anion gap acidosis with hyperkalaemia out of proportion to the glomerular filtration rate. The urine pH is below 5.5 because the H-ATPase pump itself is intact — the problem is the missing aldosterone-driven ammonium buffer and potassium secretion, not the proton pump. [2] [9]
Type 1 — Distal RTA
- Alpha-intercalated cell H+ secretion failure (H-ATPase)
- Urine pH stays above 5.5 throughout
- Hypokalaemia from distal potassium wasting
- Nephrocalcinosis and calcium phosphate stones
- Causes: hereditary ATP6V1B1 with deafness, Sjogren, amphotericin B, toluene
Type 2 — Proximal RTA
- Proximal bicarbonate reabsorption failure (lowered threshold)
- Urine pH below 5.5 once acidosis established
- Hypokalaemia from bicarbonate-driven distal wasting
- Fanconi syndrome: glycosuria, phosphaturia, aminoaciduria
- Causes: cystinosis, Lowe, galactosemia, ifosfamide, tenofovir
Type 4 RTA
- Aldosterone deficiency or aldosterone resistance
- Urine pH below 5.5 (H-ATPase intact)
- Hyperkalaemia — the key discriminator
- Reduced ammonium excretion, mild-to-moderate acidosis
- Causes: diabetic nephropathy, ACE inhibitors, ARBs, Addison, pseudohypoaldosteronism
Epidemiology & Risk Factors
In children, the inherited and metabolic causes dominate, and they present differently from the adult disease. The single commonest inherited cause of proximal RTA and Fanconi syndrome in children is cystinosis, a lysosomal storage disorder that deposits cystine crystals throughout the body and progressively destroys the proximal tubule. It presents in infancy with failure to thrive, polyuria, vomiting, rickets and photophobia from corneal crystal deposition, and the proximal RTA is part of a wider Fanconi picture. Cysteamine therapy depletes the lysosomal cystine and changes the trajectory of the disease, which is why early diagnosis matters. [6] [2]
Hereditary distal RTA is the other early-childhood presentation. Autosomal recessive mutations in the genes encoding the H-ATPase proton pump — ATP6V1B1 and ATP6V0A4 — produce a failure of distal hydrogen secretion that appears in infancy with failure to thrive, vomiting, polyuria, severe hypokalaemic acidosis, nephrocalcinosis, and often sensorineural deafness. The ATP6V1B1 form carries the earlier, more severe deafness; ATP6V0A4 tends to a later-onset hearing loss. Autosomal dominant distal RTA, from SLC4A1 (the chloride-bicarbonate exchanger), is milder and presents later. All require lifelong alkali, and early therapy protects growth, hearing and kidney function. [3] [4]
Type 4 RTA is the commonest form in adults, where diabetic nephropathy produces hyporeninaemic hypoaldosteronism, but in children the picture is different. The paediatric causes are more often drug-induced (ACE inhibitors, angiotensin receptor blockers, calcineurin inhibitors, NSAIDs, potassium-sparing diuretics), the result of obstructive uropathy and tubulointerstitial damage, or the inherited pseudohypoaldosteronisms. Addisonian adrenal insufficiency is the endocrine cause, and it is one of the reasons a child with hyperkalaemia and a normal anion gap acidosis must have adrenal function assessed. [2] [9]
Drug-induced RTA spans all three types and is increasingly recognised as children with complex chronic conditions take more nephrotoxic and tubuloactive drugs. Amphotericin B and toluene (glue sniffing) produce a distal RTA by damaging the proton-secreting cell. Ifosfamide, tenofovir, outdated tetracyclines and several antiretrovirals produce proximal RTA and Fanconi syndrome by poisoning the proximal tubule. And ACE inhibitors, ARBs, NSAIDs, heparin and potassium-sparing diuretics converge on type 4 RTA by suppressing the renin-angiotensin-aldosterone axis or blocking distal potassium secretion. A careful drug history is therefore part of every RTA workup. [6] [7]
Pathophysiology
The kidney handles acid in two coordinated operations, and RTA breaks one of them. The first operation is bicarbonate reclamation. The proximal tubule reabsorbs about 85 percent of filtered bicarbonate through the sodium-hydrogen exchanger on its luminal membrane, supported by carbonic anhydrase, which converts the secreted hydrogen and intracellular bicarbonate back into carbon dioxide and water for reabsorption. The distal nephron reclaims the rest. When the proximal tubule fails at this job, as in type 2 RTA, the bicarbonate that should have been reclaimed is delivered distally, overwhelms the limited distal capacity, and is lost in the urine. [6] [9]
The second operation is new bicarbonate generation, and it happens in the distal nephron. The alpha-intercalated cell secretes hydrogen ion into the tubular lumen through the H-ATPase proton pump, working against a steep gradient. That secreted hydrogen binds to ammonia (produced by the proximal tubule from glutamine) to form ammonium, which is trapped in the urine and excreted, and for every hydrogen secreted this way a new bicarbonate molecule is returned to the blood. When the alpha-intercalated cell cannot secrete hydrogen, as in type 1 RTA, this whole regenerative loop fails, the urine stays alkaline, and the body slowly acidifies. [3] [2]
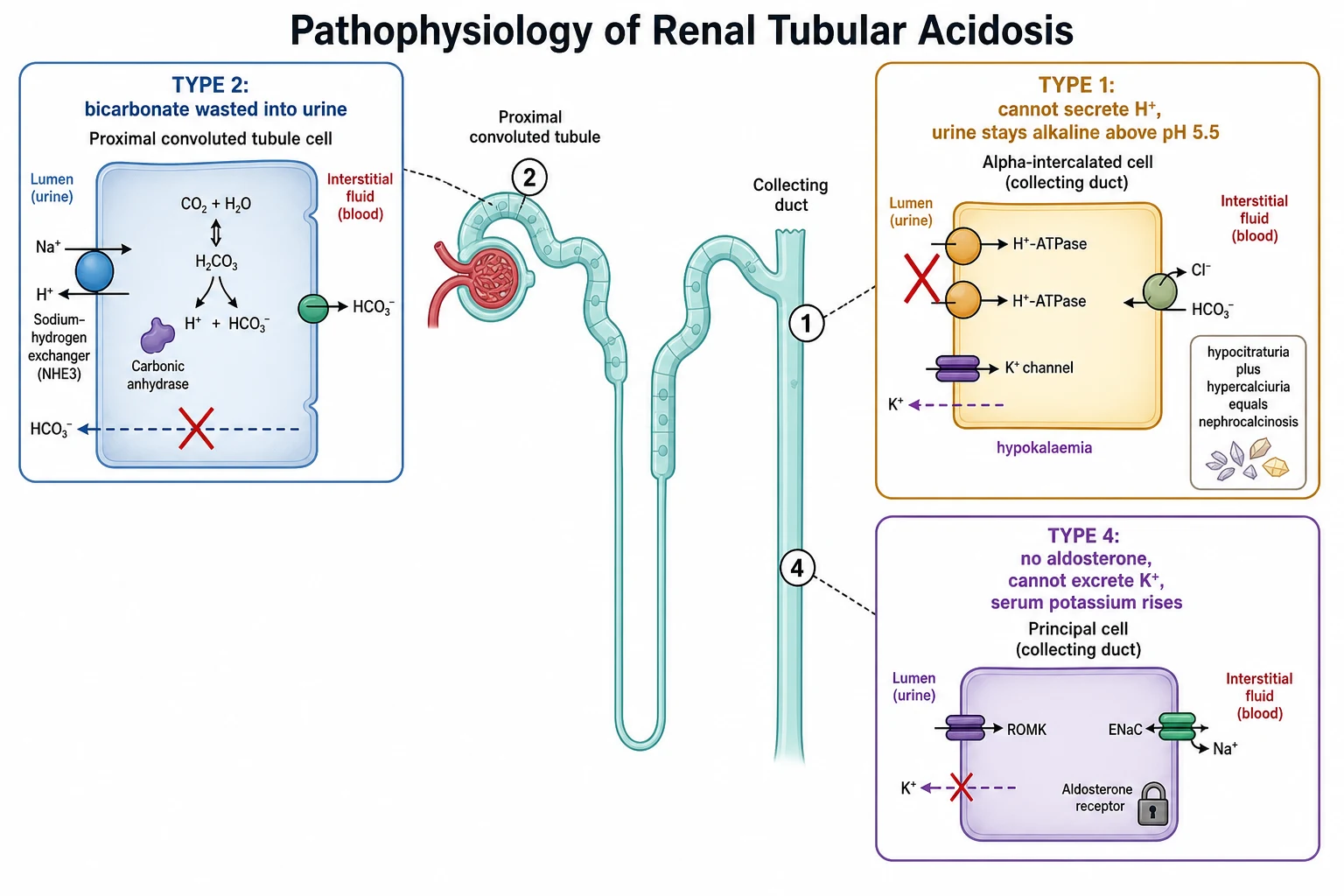
The hypokalaemia of types 1 and 2 is a distal phenomenon, not a primary one. In distal RTA the failing alpha-intercalated cell still leaves sodium to be reabsorbed by the principal cell downstream, and the resulting increased distal sodium delivery, combined with volume-depletion-driven secondary hyperaldosteronism, drives potassium secretion into the urine. In proximal RTA the spilled bicarbonate acts as a non-reabsorbable anion in the distal nephron, increasing distal sodium delivery and electrogenic potassium secretion in the same way. Both mechanisms wash potassium out, and both worsen when alkali is started — which is why potassium must be replaced alongside the alkali. [7] [2]
The nephrocalcinosis of distal RTA is worth understanding because examiners reward the mechanism, and because it is the finding that brings the child to attention. Chronic acidosis is buffered in part by bone, which releases calcium and phosphate into the blood and then the urine, producing hypercalciuria. At the same time, intracellular acidosis in the proximal tubule increases citrate reabsorption, so urinary citrate falls — and citrate is the main inhibitor of calcium crystallisation in the urine. Calcium phosphate is more soluble in acid and precipitates in alkali, so the persistently alkaline urine of distal RTA is the perfect medium for stones. Hypocitraturia plus hypercalciuria plus alkaline urine equals nephrocalcinosis, and it is almost unique to distal RTA. [8] [3]
Type 4 RTA is the mirror image. Aldosterone normally does three things in the distal nephron: it reabsorbs sodium (creating the lumen-negative potential that drives potassium and hydrogen secretion), it directly stimulates the sodium-potassium ATPase and potassium channels, and it stimulates ammoniagenesis. When aldosterone is absent or resisted, all three fail. Potassium accumulates, giving the characteristic hyperkalaemia; ammonium production falls, so the acid the kidney can still secrete has no buffer to carry it into the urine, and the patient acidifies. The urine pH stays below 5.5 because the proton pump itself works — what is missing is the aldosterone-driven ammonium buffer and potassium secretion. [2] [9]
Clinical Presentation
Distal RTA presents in the infant or young child with the consequences of chronic acidosis: failure to thrive, anorexia, vomiting, polyuria and polydipsia from nephrogenic diabetes insipidus-like concentrating defects, constipation, and muscle weakness from hypokalaemia. Bone pain and rickets may appear from chronic bone buffering. The finding that often triggers the diagnosis is nephrocalcinosis on an abdominal ultrasound done for failure to thrive or recurrent vomiting — and in an infant with this combination, hereditary distal RTA is the diagnosis until proven otherwise. Sensorineural deafness, when present, points to the ATP6V1B1 form. [4] [8]
Proximal RTA presents with failure to thrive and the wider Fanconi picture. The parents describe a child who is small, drinks and urinates excessively, and may have bone deformity from hypophosphataemic rickets. The urine shows glycosuria despite a normal blood glucose, and the serum shows hypokalaemia, hypophosphataemia and a metabolic acidosis. Cystinosis, the commonest inherited cause, adds photophobia and a failure to thrive that is relentless despite feeding, and the diagnosis is confirmed by measuring leukocyte cystine and finding corneal crystals on slit-lamp examination. [6] [2]
Type 4 RTA presents less with acidosis symptoms and more with hyperkalaemia. The child may have muscle weakness, or the hyperkalaemia is found incidentally on bloods drawn for another reason and is notable for being out of proportion to the degree of renal impairment. The history points to a cause: diabetes (hyporeninaemic hypoaldosteronism), obstructive uropathy, an ACE inhibitor or ARB started for renoprotection, chronic NSAID use, or adrenal insufficiency with fatigue, weight loss and hyperpigmentation. The acidosis is usually mild and the anion gap normal. [2] [9]
Several atypical presentations deserve attention because they are the way RTA is missed. An older child or adolescent with recurrent calcium phosphate kidney stones and a normal plasma bicarbonate may have incomplete distal RTA, where the kidney cannot acidify the urine after an acid load but maintains a near-normal bicarbonate at rest. An adolescent with an autoimmune disease — Sjogren syndrome, systemic lupus erythematosus — who develops a normal anion gap acidosis has autoimmune distal RTA, and the hypokalaemia may even be the presenting feature of the autoimmune disease. And any child with unexplained nephrocalcinosis, short stature, or a family history of deafness with kidney stones deserves a bicarbonate and an anion gap. [12] [4]
Differential Diagnosis
The first differential question is not "which RTA" but "is this an RTA at all". The anion gap decides. A high anion gap acidosis — from lactic acidosis, diabetic ketoacidosis, renal failure, or toxins such as salicylate, methanol and ethylene glycol — is never RTA, and treating it as RTA with alkali without addressing the cause will fail. The anion gap is calculated as serum sodium minus the sum of chloride and bicarbonate, and a gap above roughly 16 mmol per litre with an acidosis should redirect the differential to the high-gap causes before any RTA thinking begins. [9] [2]
Within the normal anion gap acidoses, the urine anion gap splits renal from gastrointestinal causes. A negative urine anion gap indicates a high ammonium excretion — the kidney is doing its job, so the cause is gastrointestinal bicarbonate loss (diarrhoea, ureterosigmoidostomy, fistula, laxative abuse). A positive urine anion gap indicates a low ammonium excretion — the kidney is failing, so the cause is renal tubular acidosis or renal failure. This single bedside calculation turns a long differential into a branch, and it is one of the most examined concepts in paediatric electrolyte medicine. [10] [11]
The three RTA types are then separated from one another by a small set of features. The serum potassium is low in types 1 and 2 and high in type 4. The urine pH is above 5.5 in type 1 (the kidney cannot acidify) and below 5.5 in types 2 and 4 once the patient is acidotic (the distal H-ATPase works when the filtered bicarbonate falls below the lowered threshold in proximal RTA, and the pump is intact in type 4). The Fanconi features (glycosuria, phosphaturia, aminoaciduria) point to proximal RTA. Nephrocalcinosis points to distal RTA. Hyperkalaemia out of proportion to the renal function points to type 4. [2] [6]
GOLD MARK
Three mimics must not be missed. Chronic diarrhoea produces a normal anion gap acidosis with a negative urine anion gap, because the kidney compensates appropriately — confusing this with RTA wastes time and leads to the wrong treatment. Primary hyperparathyroidism produces hypercalcaemia and calcium stones but no acidosis, so it does not explain the metabolic picture. And the tubular dysfunction of advancing chronic kidney disease produces a metabolic acidosis that is partly normal-gap, but it is driven by failing glomerular filtration and ammonium excretion rather than a discrete tubular defect, and it is not classical RTA — the distinction matters for treatment and prognosis. [9] [10]
Clinical & Bedside Assessment
Begin with growth, because chronic acidosis is a growth disorder in children. Plot weight, height and weight-for-height, and ask whether the child has fallen away from their centiles — a child who was growing normally and has stopped, or who has always been small, is the classic presentation of untreated RTA. Assess hydration and volume status, because the polyuria of distal RTA (from a concentrating defect) and the bicarbonate-driven diuresis of proximal RTA both produce dehydration that raises the urgency of assessment and the care with which any intravenous fluid must be given. [5] [1]
Examine for the features that point to a type and a cause. Nephrocalcinosis is felt as a finding on imaging rather than at the bedside, but the infant with a distal RTA is often wasted, hypotonic, and dehydrated with sunken eyes and dry mucous membranes. Rickets, with widened wrists, frontal bossing and bowing of the legs, points to proximal RTA and the phosphate wasting of Fanconi syndrome. Hearing should be assessed, because the ATP6V1B1 form of hereditary distal RTA carries sensorineural deafness that may be the only clue to the genetic diagnosis. [3] [8]
Examine for the underlying cause. The photophobia and corneal crystals of cystinosis, the cataracts and hypotonia of Lowe syndrome (oculocerebrorenal syndrome), the hepatosplenomegaly of the storage disorders, and the dry eyes, dry mouth and salivary enlargement of Sjogren syndrome are all physical signs that point to a specific cause of the RTA. In type 4 RTA, look for the hyperpigmentation, postural drop and weight loss of adrenal insufficiency, and the signs of the underlying diabetic or obstructive uropathy. [6] [2]
The history identifies precipitants and inherited patterns. Ask about consanguinity and a family history of deafness, kidney stones, failure to thrive, or infant deaths. Take a careful drug history — amphotericin B, ifosfamide, tenofovir, ACE inhibitors, ARBs, calcineurin inhibitors, NSAIDs, potassium-sparing diuretics — and ask about solvent or toluene exposure. Ask about chronic diarrhoea, which would redirect the differential toward a gastrointestinal cause with a negative urine anion gap. And assess the volume status carefully before any intravenous bicarbonate is given, because over-rapid correction can precipitate hypokalaemia, hypocalcaemic tetany and hypernatraemia. [1] [6]
Investigations
The core panel is drawn from one cannula and answers the first three questions at once. Send serum electrolytes (sodium, potassium, chloride, bicarbonate) with the calculated anion gap, a venous blood gas for pH and bicarbonate, urea and creatinine for the glomerular filtration rate, glucose, calcium, phosphate and magnesium, and a urinalysis with a urine pH. The anion gap and the potassium together give the first branch of the diagnosis; the urea and creatinine exclude renal failure as the cause of the acidosis; and the urine pH begins to separate the types. [1] [2]
The anion gap is calculated as serum sodium minus the sum of chloride and bicarbonate, with a normal reference range around 8 to 12 and up to about 16 mmol per litre. A normal gap with an acidosis confirms the hyperchloraemic (non-anion-gap) group, to which RTA and diarrhoea both belong; a high gap redirects to the high-anion-gap causes. The anion gap should be adjusted for albumin in the sick child: it falls by about 2.5 mmol per litre for every 10 g per litre reduction in serum albumin, so a "normal" gap in a hypoalbuminaemic child may hide a high-gap acidosis. [9] [10]
The urine anion gap is the tool that splits the normal anion gap acidoses into renal and gastrointestinal. It is calculated as the sum of urine sodium and potassium minus urine chloride, and it acts as a surrogate for urinary ammonium, which is hard to measure directly. Because ammonium is excreted mainly with chloride, a high ammonium excretion raises urine chloride and makes the gap negative, while a low ammonium excretion leaves the gap positive. The rule to remember: a negative urine anion gap means the kidney is excreting acid appropriately (think diarrhoea); a positive urine anion gap means it is not (think RTA). [10] [11]
The urine anion gap has limits, and the good candidate knows them. It is unreliable when the urine sodium is below 25 mmol per litre, because distal sodium delivery is then insufficient to generate the chloride-ammonium excretion the gap depends on — a volume-depleted patient should be rehydrated before the test is trusted. It is also unreliable when unmeasured urinary anions are present, such as ketoacids in ketoacidosis or hippurate in toluene toxicity, which distort the chloride balance. In these settings the urine osmolal gap (the difference between measured and calculated urine osmolality, largely attributable to ammonium) or direct ammonium measurement is more accurate. [11] [10]
Targeted tests follow the screen and are guided by the type suspected. For distal RTA, send urine calcium and citrate (hypercalciuria and hypocitraturia are expected), a renal ultrasound for nephrocalcinosis, an autoimmune screen (anti-nuclear antibodies, anti-Ro/La for Sjogren) in older children, and genetic testing for ATP6V1B1 and ATP6V0A4 where hereditary disease is likely. For proximal RTA, run a Fanconi screen — urine glucose with a normal blood glucose, urine amino acids, and urine phosphate — and measure leukocyte cystine if cystinosis is possible. For type 4 RTA, measure plasma renin and aldosterone (low renin and aldosterone in hyporeninaemic states; high renin with low aldosterone in adrenal insufficiency) and review the drug list. [2] [6]
Management — Resuscitation
Severe symptomatic metabolic acidosis — a pH below 7.1 or a bicarbonate below about 8 mmol per litre — needs treatment, but the correction must be cautious. Intravenous sodium bicarbonate is given to raise the bicarbonate only partially, typically to around 12 mmol per litre, rather than to normalise it, because the body has adapted to the chronic acidosis and rapid alkalinisation is dangerous. The reasons for caution are three: potassium shifts into cells as the pH rises, worsening the hypokalaemia of types 1 and 2; ionised calcium falls as the pH rises, precipitating tetany and seizures; and the sodium load can cause hypernatraemia and fluid overload. [9] [2]
Address the potassium alongside the acidosis, because correcting one changes the other. In types 1 and 2, where the potassium is already low, starting alkali will drive it lower as it shifts into cells with the rising pH — so potassium must be replaced before or with the bicarbonate, and monitored closely. In type 4, the hyperkalaemia is part of the disease and is treated with dietary potassium restriction, potassium binders, and attention to the underlying aldosterone problem. Never correct the acidosis in a hypokalaemic patient without a plan for the potassium. [7] [2]
The route depends on the severity. Intravenous therapy is reserved for severe acidosis, dehydration, or an infant who cannot tolerate oral alkali; it is a bridge to the oral therapy that will be the long-term treatment. Once the child is stable and tolerating oral intake, transition early to oral alkali — citrate or bicarbonate preparations — which is the mainstay of chronic management and is better tolerated and safer than prolonged intravenous therapy. [2] [5]
The infant presenting with hereditary distal RTA in crisis — severe acidosis, dehydration, and sometimes hypokalaemic weakness — needs intravenous fluids with cautious bicarbonate, correction of the potassium, and early transition to oral alkali. The aim of resuscitation is not to normalise the biochemistry but to make the child safe and able to start the chronic therapy that will protect growth, hearing and the kidney. Over-correction in this setting is a common and dangerous error. [4] [8]
Sodium bicarbonate (severe acidosis resuscitation)
Dose
Intravenous, titrated to raise bicarbonate to around 12 mmol per litre, not to normalise
Management — Definitive & Stepwise
The definitive management of each type is oral alkali, but the dose is where the types diverge most sharply, and getting the dose right is the difference between a child who grows and one who does not. Distal RTA is treated with oral alkali — sodium citrate or potassium citrate, or sodium bicarbonate — at 1 to 4 mEq per kg per day, because in distal RTA the bicarbonate threshold is normal and once the plasma bicarbonate is restored only a modest maintenance dose is wasted. Children, who are growing and buffering bone, need the higher end of the range to normalise bicarbonate and restore growth. Potassium citrate is preferred when the potassium is low; sodium citrate is used when potassium is adequate. [2] [7]
Proximal RTA is treated with much larger alkali doses — 10 to 20 mEq per kg per day — because the kidney wastes bicarbonate above the lowered threshold no matter what. This dose feels enormous and is often under-prescribed, leaving the child acidotic, growth-impaired and rachitic. The bicarbonate should be given in divided doses through the day to smooth the peaks of urinary loss, and potassium must be added because the increased distal bicarbonate delivery drives potassium wasting. Treatment of the underlying cause — cysteamine for cystinosis, stopping the offending drug — is central, because no amount of alkali replaces removing the toxin. [6] [2]
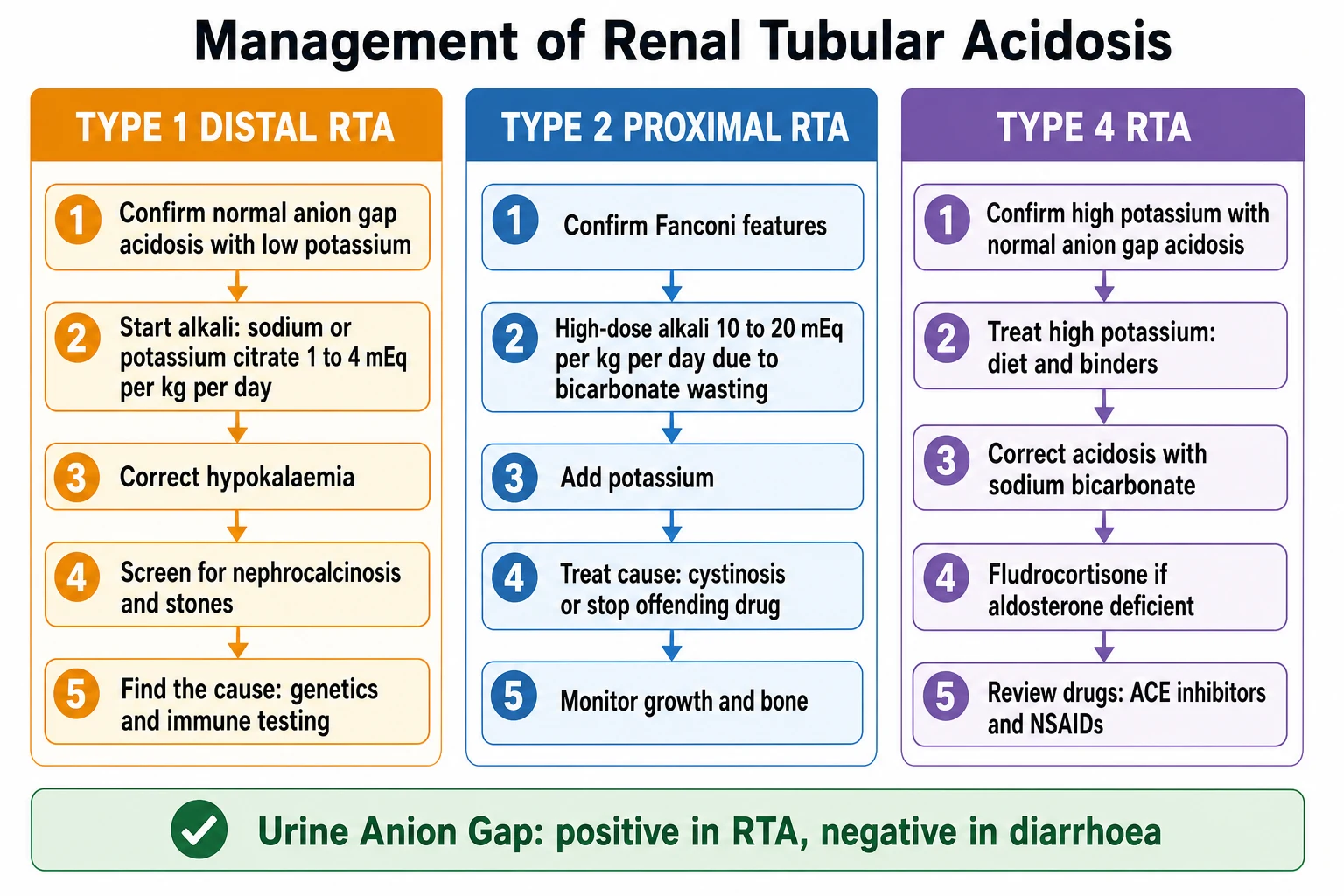
Type 4 RTA is managed differently, because the problem is not bicarbonate wasting but aldosterone lack. The hyperkalaemia is treated first, with dietary potassium restriction and potassium-binding resins; the acidosis is corrected with oral sodium bicarbonate; and mineralocorticoid replacement with fludrocortisone is given where aldosterone is deficient, as in primary adrenal insufficiency. Where the cause is a drug, withdrawal or dose adjustment is the definitive step. Where the cause is hyporeninaemic hypoaldosteronism in diabetic nephropathy, treatment is combined bicarbonate, fludrocortisone and potassium management, recognising that fludrocortisone may worsen oedema and hypertension. [2] [9]
The reason the alkali dose differs so dramatically between distal and proximal RTA is the bicarbonate threshold. In distal RTA the threshold is normal, so once the plasma bicarbonate is restored the kidney reabsorbs it and only a maintenance dose is lost — hence 1 to 4 mEq per kg per day. In proximal RTA the threshold is lowered, so the kidney continues to spill bicarbonate above that threshold no matter how much is given — hence the 10 to 20 mEq per kg per day requirement. Understanding this physiology is the key to prescribing correctly, and examiners test it directly. [2] [6]
Type 1 (distal) RTA — the alkali ladder
Confirm normal anion gap acidosis with urine pH above 5.5 and hypokalaemia
Start oral sodium or potassium citrate at 1 to 4 mEq per kg per day (children need the higher end)
Correct and maintain potassium; use potassium citrate when potassium is low
Screen for nephrocalcinosis with renal ultrasound and treat stones; check urine calcium and citrate
Identify the cause: autoimmune screen in older children, genetics (ATP6V1B1, ATP6V0A4) for hereditary forms, hearing assessment
Type 2 (proximal) RTA — the high-dose alkali ladder
Confirm Fanconi features: glycosuria with normal blood glucose, phosphaturia, aminoaciduria
Start high-dose alkali at 10 to 20 mEq per kg per day in divided doses, because the kidney wastes bicarbonate
Add potassium supplementation, because distal bicarbonate delivery drives potassium wasting
Treat the underlying cause: cysteamine for cystinosis, stop ifosfamide or tenofovir
Monitor growth, serum bicarbonate and bone mineralisation; treat rickets with phosphate and vitamin D
Type 4 RTA — the aldosterone ladder
Confirm hyperkalaemia with a normal anion gap acidosis and urine pH below 5.5
Treat hyperkalaemia: dietary potassium restriction and potassium binders
Correct acidosis with oral sodium bicarbonate
Give fludrocortisone where aldosterone is deficient (primary adrenal insufficiency)
Review and remove causative drugs: ACE inhibitors, ARBs, NSAIDs, potassium-sparing diuretics
Specific Subtypes & Scenarios
Hereditary distal RTA is the childhood archetype and deserves its own handling. Autosomal recessive mutations in ATP6V1B1 and ATP6V0A4, encoding subunits of the H-ATPase proton pump, produce a failure of distal hydrogen secretion presenting in infancy with failure to thrive, polyuria, severe hypokalaemic acidosis, nephrocalcinosis and sensorineural deafness — earlier and more severe with ATP6V1B1, later with ATP6V0A4. Autosomal dominant distal RTA, from SLC4A1 (the basolateral chloride-bicarbonate exchanger), is milder and presents later with stones and mild acidosis. All require lifelong alkali, and early and adequate therapy protects growth, hearing and kidney function by reducing the nephrocalcinosis and the chronic acid load. [3] [4]
Cystinosis is the commonest inherited cause of proximal RTA and Fanconi syndrome in children, and it defines the proximal-RTA phenotype. Cystine accumulates in the lysosomes of every cell, but it is the proximal tubule that fails first, producing a generalised Fanconi syndrome with glycosuria, phosphaturia, aminoaciduria, hypokalaemia and acidosis, alongside failure to thrive, rickets and photophobia from corneal crystals. Without treatment it progresses to renal failure in childhood. Cysteamine depletes the lysosomal cystine and dramatically slows the disease, which is why early diagnosis by leukocyte cystine measurement is essential. The proximal RTA itself needs high-dose alkali and potassium, and phosphate and vitamin D for the rickets. [6] [2]
Autoimmune distal RTA presents in the older child or adolescent with an underlying connective tissue disease. Sjogren syndrome is the classic association, but systemic lupus erythematosus, rheumatoid arthritis and other autoimmune diseases also occur. The distal RTA may be the presenting feature, with hypokalaemic paralysis sometimes bringing the patient to attention before the autoimmune disease is recognised. The mechanism is interstitial nephritis with infiltration and destruction of the alpha-intercalated cells. Management is alkali for the RTA and immunosuppression for the underlying disease, and the prognosis depends on the autoimmune control. [7] [4]
Drug-induced RTA spans all three types and is reversible when the drug is withdrawn. Amphotericin B and toluene produce distal RTA by injuring the proton-secreting cell; ifosfamide, tenofovir, didanosine and outdated tetracyclines produce proximal RTA and Fanconi syndrome by poisoning the proximal tubule; and ACE inhibitors, ARBs, calcineurin inhibitors, NSAIDs, heparin and potassium-sparing diuretics produce type 4 RTA by suppressing aldosterone or blocking distal potassium secretion. Drug withdrawal is the definitive step where possible, with alkali and electrolyte support in the interim, and monitoring for recovery of tubular function after cessation. [6] [9]
Incomplete distal RTA is the entity that examiners probe at the corner of the topic. The plasma bicarbonate is normal at rest, but the kidney cannot acidify the urine after an acid load (the ammonium chloride load test), and the child presents with recurrent calcium phosphate stones driven by the hypocitraturia and alkaline urine of the subclinical acidification defect. The ammonium chloride load test is now rarely performed in children; the diagnosis is often made on the basis of low urinary citrate, a stone history, and a family history, and management is potassium citrate to raise the urinary citrate and reduce the stone risk. [12] [4]
Complications & Pitfalls
The cardinal pitfall is treating the wrong acidosis. A high anion gap acidosis — diabetic ketoacidosis, lactic acidosis, renal failure, a toxin — is not RTA, and treating it as RTA with alkali without addressing the cause will fail and may harm. Equally dangerous is treating an RTA with insulin and fluids as if it were diabetic ketoacidosis, because the child with distal RTA and hypokalaemia does not need insulin and the fluid will not correct the tubular defect. The anion gap and the urine anion gap must be assessed before committing to a pathway, and the potassium direction confirmed. [9] [2]
The second pitfall is over-rapid bicarbonate correction. In chronic acidosis the body has adapted: the bone has buffered acid, the ionised calcium has shifted, and the intracellular potassium has equilibrated. Rapid alkalinisation with intravenous bicarbonate reverses all three adaptations at once — potassium shifts into cells and the hypokalaemia worsens (sometimes dangerously), ionised calcium falls and tetany or seizures appear, and the sodium load causes hypernatraemia and fluid overload. The rule is to correct only partially, to a bicarbonate around 12 mmol per litre, and to replace potassium first in the hypokalaemic types. [9] [7]
The third pitfall is misusing the urine anion gap. It is unreliable when the urine sodium is below 25 mmol per litre, because distal sodium delivery is then insufficient to generate the chloride-ammonium excretion the gap depends on — a volume-depleted child should be rehydrated before the test is trusted. It is also unreliable when unmeasured urinary anions are present, as in ketoacidosis or toluene toxicity. A falsely positive or normal gap in these settings can mislead the clinician away from a true RTA or a true gastrointestinal cause, so the gap must always be interpreted in context, with the urine osmolal gap or direct ammonium measurement as the fallback. [11] [10]
The fourth pitfall is undertreating proximal RTA. The 10 to 20 mEq per kg per day dose feels counterintuitive and is often under-prescribed, leaving the child acidotic, growth-impaired and rachitic despite a diagnosis that has been made correctly. The dose must be titrated to the plasma bicarbonate and the growth, given in divided doses through the day, and accompanied by potassium. The fifth pitfall is missing type 4 RTA, because the hyperkalaemia can cause arrhythmia and the acidosis and aldosterone deficiency point to treatable causes — drug withdrawal, Addisonian crisis, obstructive uropathy — that should not be overlooked. [6] [2]
Prognosis & Disposition
Prognosis in RTA depends on four things: the type, the underlying cause (hereditary versus reversible drug or autoimmune), the age at diagnosis, and the adequacy and timeliness of alkali therapy. Reversible causes — drug-induced proximal or type 4 RTA, autoimmune distal RTA caught early — can recover fully with removal of the trigger or treatment of the disease. Hereditary forms require lifelong therapy, and their prognosis is governed by how early adequate alkali is started and how well adherence is maintained. Growth recovery is the key paediatric marker: a child whose growth recovers on alkali is being treated adequately; one who does not is under-treated. [5] [2]
Hereditary distal RTA carries a risk of progressive chronic kidney disease from nephrocalcinosis and recurrent stones, and of progressive sensorineural hearing loss in the ATP6V1B1 form. Early and adequate alkali therapy reduces the hypercalciuria and hypocitraturia that drive the stones, and it protects growth and bone. Cystinosis, without cysteamine, progresses to end-stage renal disease in childhood; with cysteamine, the progression is dramatically slowed, and renal survival into adulthood is achievable. The prognosis is therefore inseparable from the timeliness of the specific diagnosis and the adherence to the specific therapy. [4] [6]
Disposition follows the severity. The child with severe symptomatic acidosis, dangerous hyperkalaemia in type 4, or dehydration needs admission, with high-dependency or intensive care for severe acidosis or arrhythmia. The stable child, once the acidosis is partially corrected and oral therapy is established, goes to the ward for titration and then home with outpatient nephrology follow-up. Most RTA management is outpatient: oral alkali titration, serial electrolytes and growth monitoring, and the surveillance specific to the cause. [2] [1]
The monitoring plan covers growth, biochemistry, bone and the cause-specific risks. Serial electrolytes, bicarbonate and the urine calcium-to-creatinine ratio confirm adequate therapy and surveillance for nephrocalcinosis; height and weight plotted over time confirm growth recovery; bone age and renal ultrasound monitor bone and kidney; and a hearing assessment is part of the surveillance for hereditary distal RTA. Adherence to alkali is the single biggest determinant of outcome in the hereditary forms, and it is the thing most often lost in adolescence — which is why transition planning matters. [5] [8]
On discharge, give the family a clear safety-net. Explain the lifelong nature of the inherited forms, the importance of alkali adherence even when the child feels well, the sick-day rules (increase fluids, never stop the alkali, seek help for vomiting or diarrhoea that prevents the tablets being kept down), and the warning signs — severe weakness, palpitations, collapse. Flag the diagnosis and the medications to any new doctor or dentist, and ensure the family knows whom to contact. [1] [2]
Special Populations
Infants dominate the paediatric RTA presentation, and they need special handling. Hereditary distal RTA and cystinosis present in the first year, severe acidosis and failure to thrive are the rule, and the alkali dose must be weight-based and revised frequently as the infant grows. The polyuria of distal RTA produces dehydration that intercurrent illness worsens, and the family must be taught sick-day management early. Oral alkali in divided doses, frequent growth and biochemistry monitoring, and early involvement of a paediatric nephrologist and a metabolic team (for cystinosis) are the foundations of care. [4] [6]
The child with diabetic nephropathy or obstructive uropathy developing type 4 RTA reflects tubulointerstitial damage with hyporeninaemic hypoaldosteronism. The hyperkalaemia and acidosis are managed with sodium bicarbonate, fludrocortisone where appropriate, and potassium restriction, always balanced against the risk of oedema and hypertension from the mineralocorticoid. The child on chemotherapy receiving ifosfamide, or antiretroviral therapy with tenofovir, is at risk of proximal RTA and Fanconi syndrome; electrolyte and bicarbonate monitoring during and after treatment is essential, and drug cessation is the definitive step where the disease allows it. [9] [6]
Children with complex chronic conditions are often on multiple drugs at once — a diuretic, an ACE inhibitor, an NSAID, a calcineurin inhibitor — and any combination can produce or worsen an RTA. A medication review is part of every assessment, and changes should be made one at a time with follow-up electrolytes, because the contribution of each drug to the potassium and acid-base balance is hard to predict in combination. The adrenal-insufficient child on replacement therapy who develops a normal anion gap acidosis and hyperkalaemia may be under-replaced, and the stress-dose hydrocortisone and fludrocortisone should be reviewed. [2] [9]
The inherited forms — hereditary distal RTA, cystinosis, Lowe syndrome — require a structured transition to adult nephrology care, because the disease does not stop at 18 and adherence is the thing most often lost in adolescence. Genetic counselling for the family is part of the plan, because the autosomal recessive forms carry recurrence risks. Attention to growth, bone density, hearing, renal function and adherence through the teenage years is the work of the transition clinic, and a young person who arrives in adult care with a stable regimen, a documented diagnosis, and an understanding of their disease is a transition done well. [3] [5]
Evidence, Guidelines & Regional Differences
The urine anion gap is the most examined concept in RTA, and its evidence base is well summarised by Batlle and colleagues and by Rehman and colleagues. The gap is a reliable surrogate for urinary ammonium in most normal anion gap acidoses, distinguishing the high-ammonium (negative gap) state of gastrointestinal loss from the low-ammonium (positive gap) state of RTA and renal failure. It fails when the urine sodium is low, when unmeasured anions are present, and in advanced renal failure, where the osmolal gap or direct ammonium measurement is preferred. The lesson for practice is to use the gap as a first branch but to know its limits. [10] [11]
The understanding of distal RTA pathophysiology has advanced through genetics. The identification of the H-ATPase subunit genes ATP6V1B1 and ATP6V0A4, and the chloride-bicarbonate exchanger SLC4A1, has explained the hereditary forms and linked the early sensorineural deafness to the shared expression of the proton pump in the inner ear. Wagner and colleagues, in their 2023 Nature Reviews Nephrology synthesis, laid out the cellular and genetic basis of distal RTA, and Giglio and colleagues provided a systematic diagnostic-to-treatment framework. These reviews underpin the current paediatric approach. [3] [4]
ANZ and UK paediatric nephrology practice follows the international reviews (Pelletier 2017, Alexander and Bitzan 2019) for the alkali dosing and the choice of citrate versus bicarbonate preparations, with local unit differences in formulation and in the target plasma bicarbonate. The citrate preparations available differ between regions — potassium citrate mixtures and sodium citrate alternatives — and the choice depends on the potassium and the tolerance. Always confirm the local paediatric nephrology protocol before prescribing, and document the guideline you are following. [1] [2]
The international consensus, as reflected in the KDIGO-aligned reviews and the paediatric nephrology literature, treats distal RTA with 1 to 4 mEq per kg per day of alkali and proximal RTA with 10 to 20 mEq per kg per day, and manages type 4 RTA with bicarbonate, potassium control and fludrocortisone where indicated. Regional differences exist in the citrate formulations, the fludrocortisone dosing conventions in paediatric type 4 RTA, and the surveillance intervals for nephrocalcinosis, all of which should be checked against the local protocol. [6] [2]
The evidence is weakest in three places. The long-term renal outcome of hereditary distal RTA despite apparently adequate alkali — whether nephrocalcinosis and chronic kidney disease are fully preventable — remains uncertain and is drawn from observational cohorts. The role of citrate in preventing nephrocalcinosis progression, beyond its effect on urinary citrate and calcium, is inferred rather than proven. And the optimal fludrocortisone dosing in paediatric type 4 RTA, balanced against oedema and hypertension, is extrapolated from adult and small-cohort paediatric experience. These are the corners where specialist nephrology discussion is most valuable. [4] [9]
The incomplete distal RTA entity is itself controversial in children. The ammonium chloride load test, the historical standard for diagnosis, is rarely performed because it is unpleasant and poorly tolerated, and the diagnosis is now often made on the basis of low urinary citrate, a stone history, and a family history, as Alonso-Varela and colleagues discussed. Whether potassium citrate changes the long-term stone risk in these children is inferred rather than proven, and the threshold for treatment varies between units. [12] [4]
Exam Pearls
Commit the potassium direction first, because it names the type before any urine test. Types 1 and 2 RTA are hypokalaemic; type 4 is hyperkalaemic. The urine pH then refines it: above 5.5 throughout points to distal RTA (the kidney cannot acidify), while below 5.5 once the patient is acidotic points to proximal or type 4 (the distal H-ATPase is intact). Nail this two-step branch and you have answered the spine of every RTA viva. [1] [2]
The urine anion gap is the classic RTA tool, and the rule is worth memorising exactly. It is calculated as the sum of urine sodium and potassium minus urine chloride, and it is a surrogate for urinary ammonium. A positive gap means low ammonium (the kidney is not excreting acid — think RTA or renal failure); a negative gap means high ammonium (the kidney is doing its job — think diarrhoea). The mnemonic a negative gap is a normal kidney response helps, but the cleanest statement is: positive gap equals renal cause, negative gap equals gut cause. [10] [11]
Nephrocalcinosis is the calling card of distal RTA, and the mechanism is examiner gold. Chronic acidosis buffers calcium out of bone, producing hypercalciuria; intracellular acidosis increases proximal citrate reabsorption, so urinary citrate falls; and calcium phosphate precipitates in the persistently alkaline urine. Hypocitraturia plus hypercalciuria plus alkaline urine equals nephrocalcinosis, and the combination is almost unique to distal RTA. [8] [3]
The alkali dose separates proximal from distal RTA at the bedside, and it is one of the most reliable single-answer stems in the exam. Distal RTA needs 1 to 4 mEq per kg per day, because the bicarbonate threshold is normal and little is wasted once the plasma level is restored. Proximal RTA needs 10 to 20 mEq per kg per day, because the lowered threshold means the kidney keeps spilling bicarbonate no matter what. The dose is the consequence of the physiology, and knowing the physiology makes the dose unforgettable. [2] [6]
Finally, never forget that a high anion gap acidosis is never RTA. Calculate the gap, confirm it is normal and hyperchloraemic, and only then enter the RTA pathway. Cystinosis is the commonest inherited cause of proximal RTA and Fanconi syndrome in children; hereditary distal RTA (ATP6V1B1, ATP6V0A4) presents in infancy with failure to thrive, nephrocalcinosis and deafness; and type 4 RTA is the hyperkalaemic one with hypoaldosteronism. These four facts, with the urine anion gap rule, answer the corners. [9] [3]
References
- [1]Pelletier J; Gbadegesin R; Staples B Renal Tubular Acidosis. Pediatr Rev, 2017.PMID 29093127
- [2]Alexander RT; Bitzan M Renal Tubular Acidosis. Pediatr Clin North Am, 2019.PMID 30454739
- [3]Wagner CA; Unwin R; Lopez-Garcia SC; Kleta R The pathophysiology of distal renal tubular acidosis. Nat Rev Nephrol, 2023.PMID 37016093
- [4]Giglio S; Montini G; Trepiccione F; Gambaro G Distal renal tubular acidosis: a systematic approach from diagnosis to treatment. J Nephrol, 2021.PMID 33770395
- [5]Santos F; Gil-Pena H; Alvarez-Alvarez S Renal tubular acidosis. Curr Opin Pediatr, 2017.PMID 28092281
- [6]Finer G; Landau D Clinical Approach to Proximal Renal Tubular Acidosis in Children. Adv Chronic Kidney Dis, 2018.PMID 30139461
- [7]Valles PG; Batlle D Hypokalemic Distal Renal Tubular Acidosis. Adv Chronic Kidney Dis, 2018.PMID 30139458
- [8]Al-Beltagi M; Saeed NK; Bediwy AS; Elbeltagi R Renal calcification in children with renal tubular acidosis: What a paediatrician should know. World J Clin Pediatr, 2023.PMID 38178934
- [9]Kraut JA; Madias NE Metabolic acidosis: pathophysiology, diagnosis and management. Nat Rev Nephrol, 2010.PMID 20308999
- [10]Batlle D; Ba Aqeel SH; Marquez A The Urine Anion Gap in Context. Clin J Am Soc Nephrol, 2018.PMID 29311217
- [11]Rehman MZ; Melamed M; Harris A; Shankar M Urinary Ammonium in Clinical Medicine: Direct Measurement and the Urine Anion Gap as a Surrogate Marker During Metabolic Acidosis. Adv Kidney Dis Health, 2023.PMID 36868734
- [12]Alonso-Varela M; Gil-Pena H; Santos F Incomplete distal renal tubular acidosis in children. Acta Paediatr, 2020.PMID 32212394