Paeds · neurology-neurodisability-and-neuromuscular
Absence, focal and generalised epilepsies
Also known as childhood absence epilepsy · juvenile myoclonic epilepsy · self-limited epilepsy with centrotemporal spikes · rolandic epilepsy · BECTS · focal epilepsy · generalised tonic-clonic epilepsy · ILAE classification of the epilepsies
A fellowship approach to absence, focal and generalised epilepsies: the child with staring spells whose EEG shows three-per-second spike-and-wave, the teenager whose morning jerks declare juvenile myoclonic epilepsy, and the ILAE 2017 framework that turns a seizure description into a syndrome, an aetiology, and a matched first-choice medicine.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
A seven-year-old girl is reported by her teacher to be staring blankly and not responding for ten seconds, many times a day, with rapid return to normal and no memory of the events; her EEG shows the three-per-second generalised spike-and-wave of childhood absence epilepsy. A fourteen-year-old boy presents before breakfast with a few jerks of his arms that throw his toothbrush, and the same morning a generalised tonic-clonic seizure; the combination is juvenile myoclonic epilepsy. An eight-year-old boy has nocturnal twitching of one side of the face with drooling and preserved awareness — self-limited epilepsy with centrotemporal spikes. The task in each is to classify the syndrome, confirm with EEG, image the brain where focal, and match the first-choice antiseizure medicine. [4] [6]
The five moves — Witness, Wave, Weight, Write, Watch
Overview & Definition
Epilepsy is a chronic disorder of the brain marked by an enduring predisposition to generate epileptic seizures, and it is defined operationally by the International League Against Epilepsy as at least two unprovoked seizures more than twenty-four hours apart. A single unprovoked seizure with a sixty per cent or greater recurrence probability over ten years also satisfies the definition, as does a recognised epilepsy syndrome. [3]
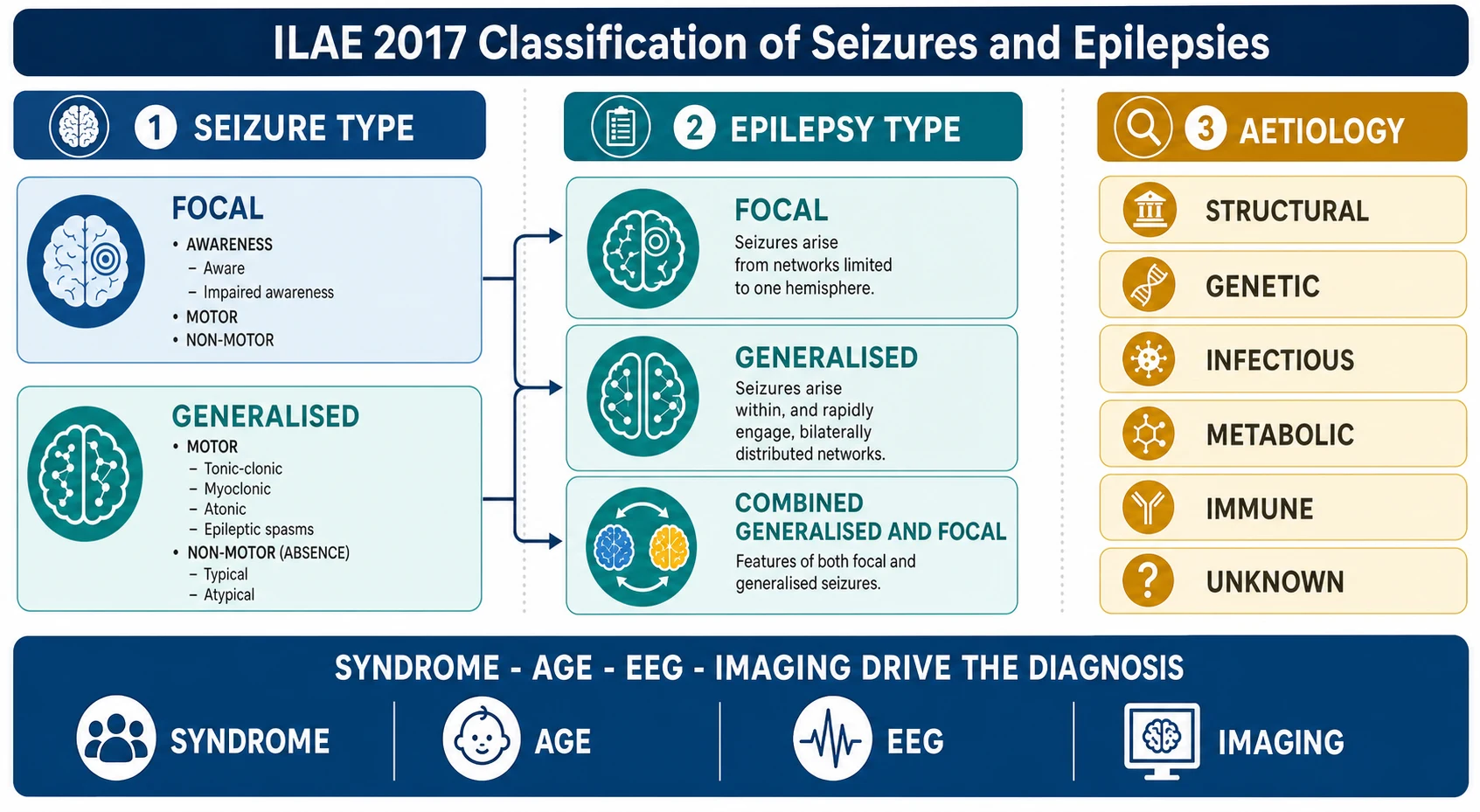
The fellowship framing rests on one distinction that governs everything that follows. A seizure is the event, while the epilepsy is the disease that produces it, and a syndrome is the pattern of seizure type, age of onset, EEG, and comorbidity that points to a cause and a treatment. Classifying the seizure is only the first step; naming the syndrome is what makes management rational. [1] [2]
The clinical importance comes from what the wrong label does to the child. An absence epilepsy labelled as focal and treated with carbamazepine worsens, and a focal epilepsy labelled as generalised and denied imaging misses an operable lesion. The ILAE 2017 framework — seizure type, then epilepsy type, then aetiology, then syndrome — is the safeguard against both errors, and it is the structure a fellowship candidate must be able to recite aloud. [1] [4]
Classification
Classification runs on two axes from the 2017 ILAE position papers. The first axis is seizure type — focal onset, with or without preserved awareness and with motor or non-motor features, or generalised onset, in which the discharge engages bilateral networks from the start and produces motor events such as tonic-clonic, myoclonic, atonic, and epileptic spasms, or non-motor absence events. The second axis is epilepsy type — focal, generalised, or combined generalised and focal — and beyond it sits aetiology and the named syndrome. [2] [1]
The named syndromes of childhood and adolescence are the fellowship currency, because each carries a seizure type, an age of onset, an EEG signature, a prognosis, and a first-choice medicine. Childhood absence epilepsy appears between four and ten years with brief staring spells and three-per-second generalised spike-and-wave. Juvenile absence epilepsy and juvenile myoclonic epilepsy declare themselves around puberty with absences, myoclonic jerks, and generalised tonic-clonic seizures on a polyspike-and-wave background. Self-limited epilepsy with centrotemporal spikes, formerly rolandic epilepsy, presents between three and fourteen years with nocturnal focal motor seizures and centrotemporal spikes on the EEG. [4] [6]
The aetiology axis runs alongside the syndrome and it is where imaging and genetics earn their place. The six ILAE aetiology categories are structural, genetic, infectious, metabolic, immune, and unknown, and naming the aetiology changes management — a structural lesion may be resected, a genetic diagnosis may guide counselling, and an immune aetiology demands immunotherapy. A focal epilepsy at any age warrants magnetic resonance imaging, because a focal onset implies a focal substrate until imaging and the clinical story prove otherwise. [1] [9]
Epidemiology & Risk Factors
Epilepsy is the commonest serious neurological disorder of childhood, with an active prevalence of roughly four to five per thousand children and an incidence highest in the first year of life and again in later childhood and adolescence. The childhood syndromes dominate this topic: childhood absence epilepsy peaks between four and ten years and is slightly more common in girls, while juvenile myoclonic epilepsy appears around puberty and is one of the commonest genetic generalised epilepsies of adolescence. [4] [6]
The risk factors that matter for the fellowship answer cluster into the aetiology categories. A structural cause — malformation of cortical development, a focal lesion, hippocampal sclerosis, or a tumour — raises the risk of focal epilepsy and drives the imaging decision. A genetic cause underlies the generalised epilepsies such as childhood absence and juvenile myoclonic epilepsy, and a family history of epilepsy increases the individual risk. An infectious cause, notably neurocysticercosis in endemic regions, produces focal epilepsy and is a major global contributor. [1] [6]
The neurodevelopmental comorbidities are as important as the seizures themselves. Children with epilepsy have higher rates of learning difficulty, attention-deficit hyperactivity disorder, anxiety, and depression than their peers, and these comorbidities shape the management as much as seizure control does. A history of febrile seizures is a recognised antecedent of mesial temporal sclerosis and temporal lobe epilepsy in a minority, and prematurity, neonatal encephalopathy, and chromosomal or copy-number disorders all raise epilepsy risk. [4] [9]
Pathophysiology
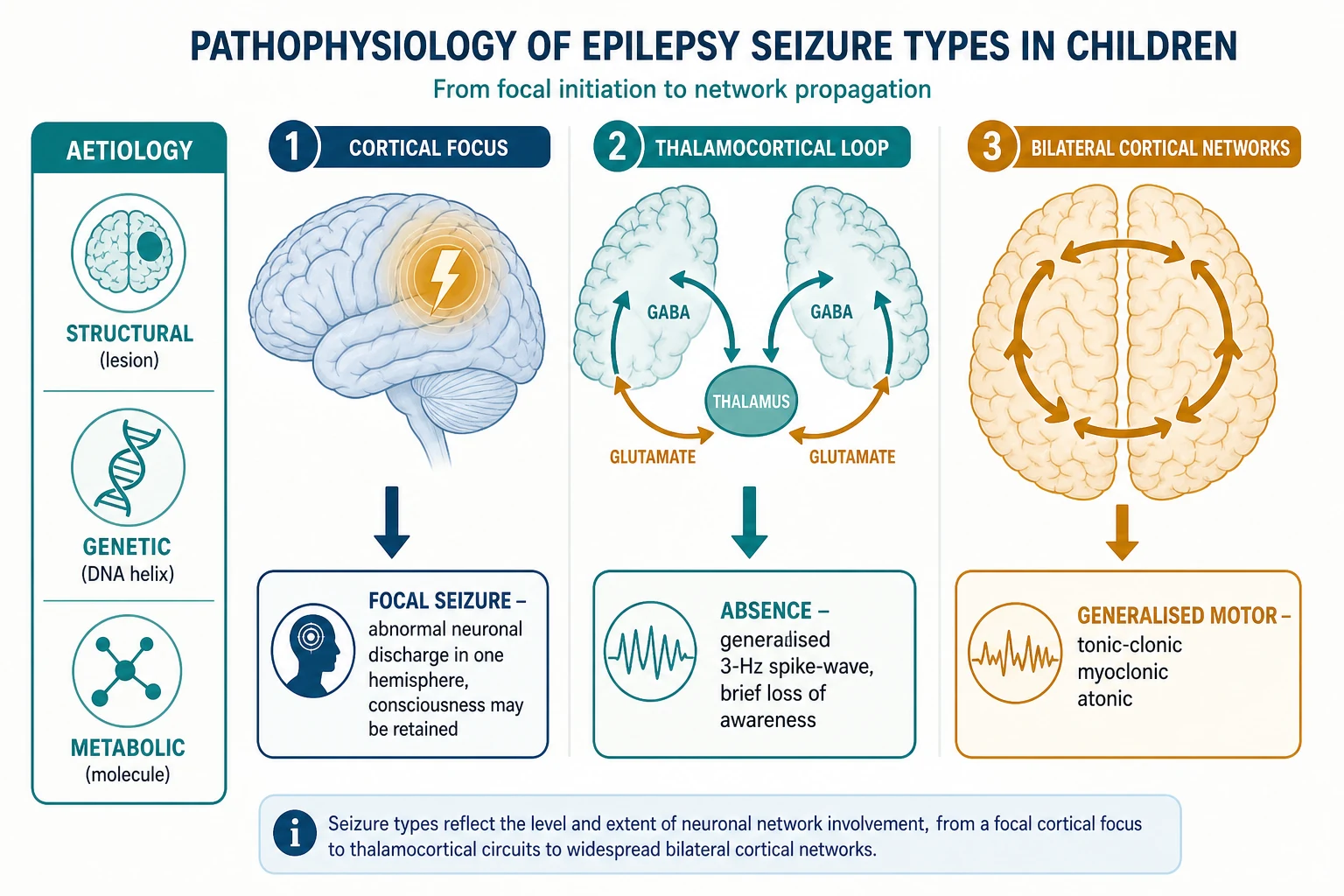
A seizure is the clinical expression of excessive and hypersynchronous neuronal discharge, and the difference between focal and generalised seizure types reflects where that discharge begins and how it spreads. A focal seizure arises from a localised cortical focus — a scarred gyrus, a dysplastic cortex, or a low-grade tumour — and the discharge recruits neighbouring cortex, producing semiology that maps to the lobe of onset. Awareness is retained or lost depending on whether the discharge reaches the networks that sustain consciousness. [2] [1]
A generalised seizure, by contrast, engages bilateral cortical and thalamocortical networks from the outset, and the archetype is the typical absence seizure. The three-per-second spike-and-wave of childhood absence epilepsy arises from an oscillation between the thalamus and the cortex, in which GABAergic thalamic reticular neurons and cortical pyramidal cells fire in lockstep, briefly blanking awareness without a convulsion. This thalamocortical loop is why absence is generalised at onset and why sodium-channel drugs cannot reach it. [7] [2]
The generalised motor seizures share the bilateral onset but add the motor pathways. A myoclonic jerk is a brief, shock-like muscle contraction driven by a cortical polyspike, and a generalised tonic-clonic seizure begins with sustained muscle contraction and ends in rhythmic jerking as the discharge exhausts the cortex. In juvenile myoclonic epilepsy the jerks cluster in the early morning, often provoked by sleep deprivation or photic stimulation, on a substrate of cortical hyperexcitability that persists for life even when seizures are controlled. [6]
The aetiology determines the substrate. In the genetic generalised epilepsies, polygenic and sometimes single-gene variants raise cortical excitability without a visible lesion, and the magnetic resonance imaging is normal. In the focal epilepsies a structural lesion is often visible — focal cortical dysplasia, a low-grade glioneuronal tumour, hippocampal sclerosis, or the residue of an old infarct or infection — and the lesion is the target if surgery is considered. The metabolic and immune aetiologies are rarer but treatable, and they must not be missed in an atypical or refractory presentation. [1] [9]
Clinical Presentation
The presentation of childhood absence epilepsy is the child who stares. The typical absence is a sudden interruption of activity lasting a few seconds to half a minute, with blank staring, sometimes subtle eyelid fluttering or automatisms, immediate recovery, and no memory of the event. The attacks occur many times a day, are provoked by hyperventilation, and interrupt schooling and play, which is why the teacher is often the first to notice. Onset is between four and ten years in an otherwise normal child, and the EEG shows the three-per-second generalised spike-and-wave. [4] [7]
Juvenile myoclonic epilepsy presents around puberty with the triad of myoclonic jerks, generalised tonic-clonic seizures, and sometimes absences. The jerks cluster in the early morning, affecting the arms, and they may throw objects or cause falls; a generalised tonic-clonic seizure often follows on waking, provoked by sleep deprivation, alcohol, or photic stimulation. The EEG shows a generalised polyspike-and-wave at four to six hertz, and photosensitivity is common. The intellect is normal and the magnetic resonance imaging is normal, but the epilepsy is lifelong, with a high relapse rate if medicine is withdrawn. [6] [10]
Self-limited epilepsy with centrotemporal spikes, formerly benign rolandic epilepsy, presents between three and fourteen years with brief focal motor seizures that involve the lower face, tongue, and throat, often at night, with drooling, guttural speech, and preserved awareness that may progress to a generalised tonic-clonic seizure in sleep. The EEG shows centrotemporal spikes that are markedly activated by sleep, and the child is otherwise normal. The seizures remit spontaneously by the mid-teens, which is why treatment is often unnecessary unless the seizures are frequent or diurnal. [4] [6]
The focal epilepsies present with semiology that points to the lobe of origin. A temporal lobe seizure begins with an epigastric rising aura and progresses to orofacial automatisms and impaired awareness; a frontal lobe seizure produces bizarre hypermotor movements, often nocturnal and easily mistaken for a sleep disorder or a non-epileptic event; an occipital seizure produces visual phenomena; and a rolandic seizure produces the focal motor pattern described above. The new focal epilepsy at any age demands imaging, because the semiology declares a focal substrate. [2] [9]
Which reassuring-sounding stories must never close the search for serious epilepsy? A child labelled with behavioural staring or daydreaming may have untreated absence that is eroding learning. A teenager whose morning jerks are dismissed as clumsiness may have juvenile myoclonic epilepsy that will declare itself in a generalised tonic-clonic seizure. And a child with new focal seizures and a normal early scan may still harbour a focal cortical dysplasia or tumour that only a dedicated epilepsy-protocol magnetic resonance imaging will reveal. [4] [1]
Differential Diagnosis
The differential diagnosis of a paroxysmal event in a child is broad, and the first task is to distinguish an epileptic seizure from its non-epileptic mimics. Syncope produces a brief loss of consciousness with pallor and limp collapse, often provoked by standing, pain, or venesection, and it may include brief twitching that is not epilepsy. Breath-holding spells, prolonged sleep myoclonus of infancy, self-gratification behaviour, and shuddering attacks all mimic epilepsy in the very young, and their resolution rests on the history and the interictal EEG. [2] [3]
Non-epileptic events and functional seizures enter the differential in older children and adolescents, presenting as episodes that resemble seizures but arise from a dissociative mechanism. These events are genuine, are not feigned, and they demand a careful multidisciplinary approach rather than an antiseizure medicine. The video-EEG captures the event, and the absence of an electrographic correlate during a typical episode confirms the diagnosis. Mislabelling a functional seizure as epilepsy exposes the child to years of unnecessary medication. [2]
Within the epilepsies, the differential is between the syndromes themselves, and it is resolved by the age of onset and the EEG. Childhood absence epilepsy is separated from juvenile absence epilepsy and juvenile myoclonic epilepsy by the age band and the polyspike-and-wave pattern, and from atypical absence of a developmental and epileptic encephalopathy by the development, the slower spike-wave, and the cognitive impact. The focal epilepsies are separated from the generalised epilepsies by the semiology and the EEG, and a focal seizure with rapid secondary generalisation can be mistaken for a primary generalised tonic-clonic if the focal onset is not elicited. [4] [6]
The treatable mimics must not be missed. A paroxysmal event in a child with a metabolic disorder may be a treatable metabolic decompensation rather than an epileptic seizure, and a cardiac arrhythmia such as a long-QT syndrome can produce convulsive syncope that is indistinguishable from epilepsy without an electrocardiogram. Every child with a suspected first seizure should have an electrocardiogram alongside the EEG, because a fatal arrhythmia masquerading as epilepsy is the most costly misdiagnosis in paediatric neurology. [3] [9]
Clinical & Bedside Assessment
The recognition move is to take a meticulous eyewitness account of the event, because the semiology is the diagnosis before the EEG confirms it. Ask what the child was doing beforehand, how the event started, whether awareness was retained, how long it lasted, how it ended, and how the child was afterwards. A focal onset with an aura and a gradual spread differs from a generalised onset with instantaneous loss of consciousness, and that single distinction changes the imaging decision and the medicine. [2] [4]
The history must also establish the developmental trajectory, the family history of epilepsy, the antecedents such as febrile seizures or neonatal encephalopathy, and the triggers. Sleep deprivation and photic stimulation provoke juvenile myoclonic epilepsy, hyperventilation provokes absence, and fever or intercurrent illness lower the threshold in any epilepsy. A developmental plateau or regression alongside new seizures raises a developmental and epileptic encephalopathy and demands urgent specialist referral. [5] [1]
The bedside examination in a child with new-onset epilepsy is usually normal, and a focal neurological sign is the red flag that demands imaging. Examine the skin for the ash-leaf macules, adenoma sebaceum, and shagreen patches of tuberous sclerosis, the cafe-au-lait patches of neurofibromatosis, and the port-wine stain of Sturge-Weber syndrome, because a neurocutaneous syndrome may declare itself through the seizures. Cut the examination short only after these have been excluded. [1] [9]
Which findings must never be dismissed? A child with new focal seizures and a hemiparesis or a visual field defect has a structural lesion until imaging proves otherwise. A child with epilepsy and developmental regression has an encephalopathy that demands an electroencephalogram, urgent metabolic and genetic workup, and specialist input. And a child whose first seizure follows a head injury or a febrile illness has a treatable cause that must be sought before an idiopathic label is applied. [5] [2]
Investigations
The electroencephalogram is the single most important investigation in suspected epilepsy, and its proper use is syndrome-specific. A routine awake EEG with activation procedures — hyperventilation and photic stimulation — reveals the three-per-second generalised spike-and-wave of childhood absence epilepsy, the polyspike-and-wave of juvenile myoclonic epilepsy, and the centrotemporal spikes activated by sleep of self-limited epilepsy with centrotemporal spikes. A sleep-deprived EEG or a sleep recording increases the yield in focal epilepsy. A normal interictal EEG does not exclude epilepsy, and a repeat study is often worthwhile. [2] [4]
Magnetic resonance imaging is mandatory in focal epilepsy and in any epilepsy with an atypical syndrome, developmental delay, or a focal neurological sign, because the focal onset implies a structural substrate. A dedicated epilepsy-protocol study with thin coronal slices through the temporal lobes detects focal cortical dysplasia, hippocampal sclerosis, low-grade tumours, and the residue of prior infarction or infection. In the genetic generalised epilepsies the magnetic resonance imaging is normal, and imaging is requested only to exclude a structural mimic when the semiology or the EEG is atypical. [1] [9]
The electrocardiogram is the investigation that is forgotten and that saves lives. Every child with a suspected first seizure should have a twelve-lead electrocardiogram to exclude a long-QT syndrome or another channelopathy that can produce convulsive syncope and sudden death. Genetic testing is increasingly part of the workup, particularly in the early-onset epilepsies and the developmental and epileptic encephalopathies, where a gene panel or a chromosomal microarray may identify a treatable or a counselling-relevant diagnosis. [5] [6]
Management — Resuscitation
Resuscitation is rarely the primary need in the chronic epilepsies, because the seizure self-terminates within minutes and the child recovers. The exception is the prolonged seizure or the cluster of seizures that does not stop, which is status epilepticus and which is owned by the sibling topic on status epilepticus. For the chronic epilepsies on this page, the acute task is to ensure the airway is safe during the seizure, to time the event, and to arrange review if the seizure exceeds five minutes. [3] [9]
The first generalised tonic-clonic seizure in a child is managed by assessment of the airway, breathing, and circulation, a bedside glucose, a careful history and examination, and the investigations above. Admission is required if recovery is incomplete, if the seizure was prolonged or focal, if there is a fever without an obvious source, or if the child is under twelve months. Most first seizures can be investigated as an outpatient, with the electroencephalogram and the electrocardiogram arranged urgently and the magnetic resonance imaging requested when a focal or atypical element is present. [3] [2]
The decision to start a daily antiseizure medicine is not made after every first seizure, because the recurrence risk after a single unprovoked seizure in a normal child is roughly forty per cent, and the side effects of treatment may outweigh the benefit. Treatment is started after two unprovoked seizures, after one seizure with a high recurrence risk, or when the diagnosis is an established epilepsy syndrome with frequent seizures such as childhood absence epilepsy. The first-choice medicine is always syndrome-driven. [3] [8]
Management — Definitive & Stepwise
The definitive management of the chronic epilepsies is the syndrome-matched antiseizure medicine, chosen by the seizure type and the EEG rather than by habit. For childhood absence epilepsy the Childhood Absence Epilepsy trial proved ethosuximide and valproate superior to lamotrigine for seizure freedom, and ethosuximide was superior to valproate on attentional outcomes, which is why ethosuximide is first-line when the child has absence alone and no generalised tonic-clonic seizures. If generalised tonic-clonic seizures coexist, valproate is preferred in males and lamotrigine in females. [7] [8]
Juvenile myoclonic epilepsy is managed with valproate as the most effective single agent in males, with levetiracetam and lamotrigine as alternatives in females of childbearing potential. Self-limited epilepsy with centrotemporal spikes often needs no treatment, because the seizures are infrequent, nocturnal, and self-remitting; when treatment is required for frequent or diurnal seizures, levetiracetam or carbamazepine is effective. The focal epilepsies are managed with levetiracetam or carbamazepine as first-line, with the SANAD trial supporting lamotrigine and oxcarbazepine as alternatives. [6] [9]
The valproate rule is the fellowship-critical constraint. Valproate is teratogenic, raising the risk of neural tube and other malformations, and it carries a dose-dependent risk of neurodevelopmental impairment in exposed offspring. The Medicines and Healthcare products Regulatory Agency restricts its use in girls and women of childbearing potential to the situations in which no effective alternative exists, with a pregnancy prevention programme and annual specialist review. Levetiracetam and lamotrigine are the preferred alternatives in female adolescents with generalised epilepsy, with lamotrigine titrated slowly to avoid rash. [8] [6]

The four syndromes and their first-choice medicine
Specific Subtypes & Scenarios
Childhood absence epilepsy is the archetype of a treatable, age-defined syndrome, and it is the highest-yield scenario for the fellowship examination. The child is between four and ten years, otherwise normal, with dozens of brief staring spells a day and a pathognomonic EEG. Ethosuximide controls the absences in the majority with a favourable cognitive profile, and the epilepsy remits in most children by adolescence, with a small proportion later developing generalised tonic-clonic seizures that change the medicine. The long-term outcome trial showed that the risk of later generalised tonic-clonic seizures is concentrated in those whose absences coexisted with such seizures from the start. [4] [10]
Juvenile myoclonic epilepsy is the subtype that demands lifelong framing. The seizures are controlled by valproate in the great majority, but the underlying cortical hyperexcitability persists, and withdrawal of medicine is followed by relapse in most patients. The fellowship skill is to set this expectation early, to address the lifestyle triggers — adequate sleep, avoidance of alcohol and sleep deprivation, caution with photic stimulation — and to apply the valproate rule when the patient is female. The comorbid anxiety and depression are common and they merit active management alongside the seizures. [6] [10]
Self-limited epilepsy with centrotemporal spikes is the subtype that teaches restraint. The seizures are nocturnal, infrequent, and self-remitting by adolescence, and the EEG abnormalities are common in otherwise normal children, which means that the diagnosis must not be over-called and the medicine must not be started without a clear clinical seizure. When treatment is necessary, it is effective, and it is withdrawn in the teenage years. The atypical rolandic presentations — frequent seizures, daytime attacks, or cognitive or language difficulties — are a different entity, sometimes an epileptic encephalopathy, and they demand specialist assessment rather than reassurance. [4] [6]
The drug-resistant focal epilepsy is the subtype that opens the door to surgery. A focal epilepsy that fails two appropriately chosen and tolerated antiseizure medicines should be referred to a specialist epilepsy centre for presurgical evaluation, because the chance of a further medicine achieving seizure freedom falls sharply after two failures, and an operable lesion may cure the epilepsy. The evaluation seeks the epileptogenic zone through video-EEG monitoring, magnetic resonance imaging, positron emission tomography, and sometimes intracranial electrodes, and resection offers a chance of seizure freedom in a well-selected child. [9] [1]
Complications & Pitfalls
The complications of uncontrolled epilepsy span seizures, development, and risk. The acute complication is status epilepticus, owned by the sibling topic, and the long-term complication is the cumulative effect of recurrent seizures on learning, behaviour, and mental health. Children with epilepsy have higher rates of attention-deficit hyperactivity disorder, anxiety, depression, and educational underachievement than their peers, and these comorbidities are part of the disease rather than incidental findings, deserving active screening and management. [4] [9]
The pitfall of the wrong drug for the syndrome is the error that causes the most preventable harm. Carbamazepine and phenytoin prescribed for an unrecognised absence or myoclonic epilepsy worsen the seizures, may precipitate absence status, and delay the correct diagnosis. The safeguard is the EEG before the prescription, and a low threshold to reconsider the syndrome whenever a child worsens on an antiseizure medicine rather than simply increasing the dose. [8] [4]
The pitfall of valproate in a girl of childbearing potential is the error with the longest reach. A female adolescent started on valproate for a generalised epilepsy may carry the medicine into her reproductive years, and the teratogenic and neurodevelopmental risk to a future pregnancy is substantial. The safeguard is to apply the valproate rule at the point of the first prescription, choosing levetiracetam or lamotrigine whenever the syndrome permits, and to review the choice at every transition. [8] [6]
The pitfall of the missed structural lesion is the error that costs a curative operation. A focal epilepsy labelled as generalised, or a child not imaged because the first seizure was called a febrile seizure, may carry an operable focal cortical dysplasia or tumour that declares itself years later. The safeguard is the magnetic resonance imaging in every focal or atypical epilepsy, read by a radiologist familiar with epilepsy protocol, and the early referral of drug-resistant focal epilepsy for presurgical evaluation. [1] [9]
Prognosis & Disposition
The prognosis of the childhood epilepsies is generally favourable when the syndrome is correctly classified and the right medicine is given. Childhood absence epilepsy remits by adolescence in most children, self-limited epilepsy with centrotemporal spikes remits by the mid-teens, and the focal epilepsies achieve seizure freedom on the first or second medicine in the majority. The syndromes with a guarded prognosis are those associated with a structural brain abnormality, a developmental and epileptic encephalopathy, or a genetic syndrome, where seizure control is harder and the developmental outcome is driven by the underlying cause. [4] [9]
Juvenile myoclonic epilepsy is the exception to the remission rule, because the seizures are controlled but the predisposition persists, and withdrawal of medicine is followed by relapse in most patients. The prognosis is framed for the patient as a treatable, lifelong condition in which adherence, adequate sleep, and the avoidance of triggers keep the seizures at bay, and in which the medicine may be lifelong. The comorbid anxiety and depression are part of the long-term picture and they merit ongoing attention. [6] [10]
The disposition is shared between the general paediatrician and the paediatric neurologist. The general paediatrician owns the first seizure, the initiation of the first-choice medicine in a straightforward syndrome, the developmental and educational surveillance, and the long-term relationship with the family. The paediatric neurologist is consulted for the atypical or drug-resistant epilepsy, the presurgical evaluation, the developmental and epileptic encephalopathies, and the transition to adult care, which is a structured handover that catches the young person who would otherwise be lost to follow-up. [1] [9]
Special Populations
Children with developmental disability and neurodiversity are over-represented among the epilepsies, and their seizures may be atypical, drug-resistant, and entangled with their behaviour and communication. The assessment adapts to the child — a longer consultation, a caregiver who interprets the baseline, a lower threshold for video-EEG to capture the event — and the management balances seizure control against the cognitive and behavioural effects of the medicines. The general paediatrician is often the coordinator, holding the relationship with the family and the school across the many services involved. [5] [9]
Indigenous, migrant, refugee, and asylum-seeking children face the additional burden of access and equity. The incidence of epilepsy is higher in many remote and Indigenous communities, driven by the higher prevalence of acquired causes such as infection and perinatal injury, and the distance to a specialist and an electroencephalogram delays the diagnosis and the treatment. A newly arrived migrant or refugee child should have a seizure history sought actively, because a past epilepsy may be undocumented, and the workup may need to begin from the first principles. [4] [1]
Adolescent girls on valproate are the special population that demands the most deliberate transition. The valproate rule applies from the onset of reproductive potential, and the transition from a paediatric to an adult neurologist is the moment to review the medicine, to introduce contraception and pregnancy planning, and to involve the general practitioner and the adult service in a structured handover. The young woman who is lost to follow-up at this transition is the one who presents in early pregnancy on a teratogenic medicine, and the structured transition is the safeguard against that outcome. [8] [6]
Socioeconomic disadvantage compounds all of these. Adherence to a daily medicine is harder when the family is under stress, the appointments are missed, and the epilepsy is uncontrolled, which deepens the educational and the developmental disadvantage. The fellowship answer acknowledges the social determinants and builds the supports — a clear seizure action plan, a school liaison, a reliable supply of the medicine, and a single named coordinator — that keep the child safe and in care. [9] [4]
Evidence, Guidelines & Regional Differences
The 2017 ILAE position papers are the global standard for the classification of seizure types and epilepsies, and they are the framework that the fellowship candidate must recite. The 2014 ILAE practical clinical definition of epilepsy extended the diagnostic boundary to a single unprovoked seizure with a high recurrence risk, and it anchored the syndrome as a diagnostic category. The 2022 ILAE syndrome position papers — on neonatal and infant onset, childhood onset, and variable-age onset — codified the diagnostic criteria for the named syndromes that decide the medicine. [1] [2] [3]
The evidence for the medicines rests on a small number of landmark trials. The Childhood Absence Epilepsy trial established ethosuximide and valproate as superior to lamotrigine for seizure freedom, with ethosuximide superior to valproate on attention. The SANAD trial compared the standard and the newer antiepileptic drugs for focal and generalised epilepsy and supported lamotrigine and oxcarbazepine for focal and valproate for generalised epilepsy. The 2013 ILAE evidence review graded the initial monotherapy evidence by seizure type and syndrome, and it remains the reference for first-choice selection. [7] [9] [8]
The regional differences matter for the valproate rule. In the United Kingdom and Europe the Medicines and Healthcare products Regulatory Agency and the European Medicines Agency restrict valproate in girls and women of childbearing potential through a pregnancy prevention programme, while in Australia the Therapeutic Goods Administration advises caution without the same regulatory structure, and in North America the emphasis is on informed shared decision-making. The fellowship answer applies the most cautious standard, because the teratogenic and neurodevelopmental risk is the same in every region. [8] [6]
The controversies are few but real. The age at which an antiseizure medicine may be withdrawn after a remission varies by syndrome and by clinician, with the lifelong nature of juvenile myoclonic epilepsy set against the remitting nature of childhood absence and self-limited focal epilepsy. The role of generics, the management of the comorbidities, and the timing of the surgery referral are the further judgement calls that the fellowship answer names without pretending that a single algorithm resolves them. [9] [4]
Exam Pearls
The fellowship answer turns on five facts. Epilepsy is two unprovoked seizures more than twenty-four hours apart, or one with a high recurrence risk, or a recognised syndrome. The seizure is focal or generalised by the eyewitness semiology, confirmed by the EEG. The syndrome is named from the age of onset and the EEG — childhood absence at four to ten years with three-per-second spike-and-wave, juvenile myoclonic at puberty with polyspike-and-wave, rolandic at three to fourteen years with centrotemporal spikes. The medicine is syndrome-driven, and valproate is avoided in girls of childbearing potential. [1] [4]
The examiner probes three traps. The first is the absence epilepsy worsened by carbamazepine — the EEG comes before the prescription. The second is the focal epilepsy denied imaging — the focal onset demands a magnetic resonance imaging to seek an operable lesion. The third is the female adolescent started on valproate without a pregnancy prevention plan — the valproate rule applies from the onset of reproductive potential, and the transition to adult care is the moment to review it. [8] [2]
The examiner rewards the candidate who frames the epilepsies as syndromes with comorbidities, not as seizure counts. The learning, the behaviour, and the mental health are part of the disease, and they are screened for and managed alongside the seizures. The family is given a seizure action plan, the school is engaged, and the young person is prepared for a transition that protects them into adult care. A candidate who shows this breadth demonstrates the systems thinking that the fellowship demands. [4] [9]
References
- [1]Scheffer IE, Berkovic S, Capovilla G, Connolly MB, French J, Guilhoto L, Hirsch E, Jain S, Mathern GW, Moshe SL, Nordli DR, Perucca E, Tomson T, Wiebe S, Zhang YH, Zuberi SM. ILAE classification of the epilepsies: Position paper of the ILAE Commission for Classification and Terminology. Epilepsia, 2017.PMID 28276062
- [2]Fisher RS, Cross JH, French JA, Higurashi N, Hirsch E, Jansen FE, Lagae L, Moshe SL, Peltola J, Roulet Perez E, Scheffer IE, Zuberi SM. Operational classification of seizure types by the International League Against Epilepsy: Position Paper of the ILAE Commission for Classification and Terminology. Epilepsia, 2017.PMID 28276060
- [3]Fisher RS, Acevedo C, Arzimanoglou A, Bogacz A, Cross JH, Elger CE, Engel J Jr, Forsgren L, French JA, Glynn M, Hesdorffer DC, Lee BI, Mathern GW, Moshe SL, Perucca E, Scheffer IE, Tomson T, Watanabe M, Wiebe S. ILAE official report: a practical clinical definition of epilepsy. Epilepsia, 2014.PMID 24730690
- [4]Specchio N, Wirrell EC, Scheffer IE, Nabbout R, Riney K, Samia P, Guerreiro M, Guber D, Jovic N, Nagaraddi G, Jennings T, Bodin MP, Manganaro J, Smith G, Tokarz A, Triki C, Wiebe S, Yozawitz E, Pressler R, Zuberi SM, Moshe SL, Perucca E, Tinuper P, Wiebe S. International League Against Epilepsy classification and definition of epilepsy syndromes with onset in childhood: Position paper by the ILAE Task Force on Nosology and Definitions. Epilepsia, 2022.PMID 35503717
- [5]Zuberi SM, Wirrell E, Yozawitz E, Wilmshurst JM, Specchio N, Riney K, Pressler R, Auvin S, Samia P, Hirsch E, Watanabe K, Teng T, Guber D, Wiebe S, Jovic N, Nabbout R, Guerreiro M, Smith G, Bodin MP, Jennings T, Nagaraddi G, Triki C, Manganaro J, Tokarz A, Moshe SL, Perucca E, Tinuper P, Scheffer IE. ILAE classification and definition of epilepsy syndromes with onset in neonates and infants: Position statement by the ILAE Task Force on Nosology and Definitions. Epilepsia, 2022.PMID 35503712
- [6]Riney K, Bogacz A, Somerville E, Hirsch E, Nabbout R, Scheffer IE, Zuberi SM, Alber M, Auvin S, Cross H, Jain S, Jennings T, Kamate M, Manganaro J, Mushtaq N, Nair PP, Nguyen A, Rios L, Samia P, Teng T, Tokarz A, Triki C, Wiebe S, Wong V, Guerreiro M, Pressler R, Yozawitz E, Wilmshurst J, Specchio N, Wirrell E, Smith G, Moshe SL, Perucca E, Tinuper P. International League Against Epilepsy classification and definition of epilepsy syndromes with onset at a variable age: position statement by the ILAE Task Force on Nosology and Definitions. Epilepsia, 2022.PMID 35503725
- [7]Glauser TA, Cnaan A, Shinnar S, Hirtz DG, Dlugos D, Masur D, Clark PO, Capparelli EV, Adamson PC; Childhood Absence Epilepsy Study Team. Ethosuximide, valproic acid, and lamotrigine in childhood absence epilepsy. N Engl J Med, 2010.PMID 20200383
- [8]Glauser T, Ben-Menachem E, Bourgeois B, Cnaan A, Guerreiro C, Kalviainen R, Mattson R, French JA, Perucca E, Tomson T; ILAE Subcommission on AED Guidelines. Updated ILAE evidence review of antiepileptic drug efficacy and effectiveness as initial monotherapy for epileptic seizures and syndromes. Epilepsia, 2013.PMID 23350722
- [9]Marson AG, Appleton R, Baker GA, Chadwick DW, Doughty J, Eaton B, Eldridge K, Fitzsimons M, Greenhalgh J, Haywood R, Hughes E, Kim L, Lorigan P, Park B, Smith DF, Smith PEM, Smith CT, Vanoli A, Williamson PR; SANAD Trial group. A randomised controlled trial examining the longer-term outcomes of standard versus new antiepileptic drugs. The SANAD trial. Health Technol Assess, 2007.PMID 17903391
- [10]Shinnar S, Cnaan A, Hu F, Clark P, Dlugos D, Hirtz DG, Glauser TA; Childhood Absence Epilepsy Study Team. Long-term outcomes of generalized tonic-clonic seizures in a childhood absence epilepsy trial. Neurology, 2015.PMID 26311751