Paeds · neurology-neurodisability-and-neuromuscular
Acute disseminated encephalomyelitis and demyelinating disease
Also known as ADEM · Acquired demyelinating syndromes · MOG-antibody associated disease · Paediatric multiple sclerosis · Neuromyelitis optica spectrum disorder · Clinically isolated syndrome
A fellowship approach to the child with a first central nervous system demyelinating event. Recognise acute disseminated encephalomyelitis by the mandatory encephalopathy that separates it from the monofocal clinically isolated syndromes, then drive a unified antibody-led pathway that distinguishes the four entities a general paediatrician must tell apart - ADEM, myelin-oligodendrocyte-glycoprotein antibody-associated disease, paediatric multiple sclerosis, and neuromyelitis optica spectrum disorder - on serum MOG-IgG, aquaporin-4 IgG, MRI patterns, and the cerebrospinal fluid. Treat the acute event early with high-dose corticosteroids, step to intravenous immunoglobulin or plasma exchange when steroid-refractory, and match long-term therapy to the confirmed diagnosis, because the wrong maintenance drug - a multiple-sclerosis disease-modifying therapy given for neuromyelitis optica - can worsen the disease.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
A previously well five-year-old arrives drowsy and irritable four days after a viral upper-respiratory illness, now dragging a leg and slurring his speech. Across the corridor a twelve-year-old describes three days of pain and blurred vision in one eye. In both rooms the unifying question is the same: is this a first central nervous system demyelinating event, which entity is it, and is there a long-term treatment whose window opens only if the antibody answer is right? The fellowship task is to convert that bedside picture into a structured, antibody-led pathway that treats the acute event immediately while the confirmatory tests return, and that matches the maintenance therapy to the confirmed diagnosis rather than to a guess. [1] [4]
M · Y · E · L · I · N
Overview & Definition
The clinician's first act is to recognise that a new neurological deficit in a child can be an immune-mediated attack on central nervous system myelin, and to name the family it belongs to. The International Pediatric Multiple Sclerosis Study Group (IPMSSG) defines the acquired demyelinating syndromes of childhood as a group of monophasic or relapsing inflammatory disorders that share a presumed autoimmune attack on myelin but differ in their phenotype, their imaging, and - decisively - their antibody signature and long-term treatment. The family comprises ADEM, the clinically isolated syndromes (optic neuritis, transverse myelitis, brainstem and other focal syndromes), and, after investigation, the three named diseases: MOGAD, paediatric MS, and NMOSD. [1]
Why does this grouping matter at the bedside? Because a child who looks acutely unwell with a polysymomatic encephalopathy and a child who looks well with a single symptomatic optic nerve can both be having a first demyelinating event, yet their prognoses and their maintenance treatments are entirely different. The 2013 IPMSSG definitions deliberately anchor the first fork on a single clinical feature - encephalopathy - because encephalopathy is reproducible, observable, and separates the ADEM phenotype (by definition encephalopathic) from the clinically isolated syndromes (by definition non-encephalopathic, beyond the fever and irritability expected in a sick young child). Getting that one feature right sets the whole pathway in motion. [1] [6]
What makes this group worth knowing in depth is that the modern antibody-led framework has transformed a previously confusing overlap of names into a clean diagnostic logic, and that the long-term treatment now depends on getting the antibody answer right. A child with MOGAD is managed differently from a child with MS, and both differ from a child with NMOSD - and the wrong maintenance drug can actively harm. The general paediatrician's role is to treat the acute event immediately, request the antibody tests and the imaging in parallel, and hand over to the neuroimmunology service with the right provisional label and the confirmatory samples already in flight. [2] [4]
Classification
The acquired demyelinating syndromes are best classified by the bedside phenotype first and the antibody signature second, because the phenotype predicts the first test and the antibody signature predicts the long-term treatment. The figure below lays out the four named entities alongside the unifying antibody triad that resolves them - serum MOG-IgG, AQP4-IgG, and the CSF oligoclonal bands - because that triad is what a general paediatrician can act on at the first encounter. [1] [4]
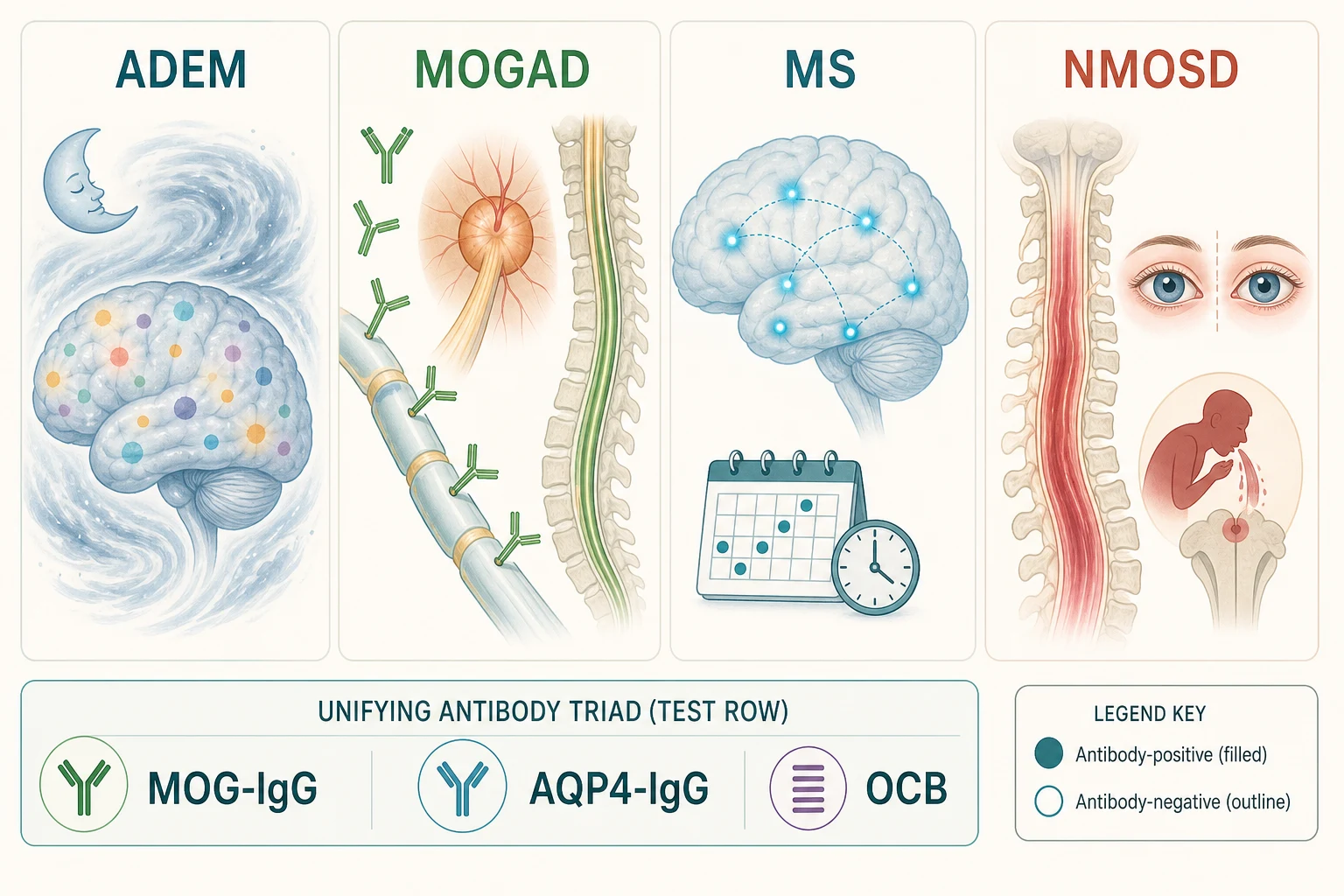
The four entities behave differently at the bedside. ADEM is polysymomatic and, by definition, encephalopathic, and it predominates in younger children, especially under six years. MOGAD is the antibody-defined disorder that most often presents as ADEM in young children and as optic neuritis or transverse myelitis in older ones; its serum MOG-IgG, detected by a cell-based assay, sets it apart. Paediatric MS is a relapsing disorder of dissemination in space and time, commoner in adolescents, with a characteristic periventricular MRI pattern and CSF oligoclonal bands. NMOSD is the aquaporin-4-IgG-mediated disorder of severe optic neuritis and longitudinally extensive transverse myelitis, with its hallmark area-postrema syndrome of intractable hiccups, nausea, and vomiting. [1] [3]
The antibody signature is the pivot of the whole classification, and it is what the lower band of the figure is teaching. ADEM with persistent MOG-IgG is MOGAD and relapses; ADEM without antibodies is usually monophasic. A clinically isolated syndrome with MOG-IgG is MOGAD, with AQP4-IgG is NMOSD, and with neither antibody but with dissemination on imaging or oligoclonal bands is paediatric MS. Holding the antibody triad in mind prevents the cardinal error of the field: treating all demyelinating events as if they were multiple sclerosis, when three of the four entities require a different long-term drug and one of them is made worse by the multiple-sclerosis drug. [2] [4]
The four entities at a glance - phenotype, antibody, MRI hallmark, long-term treatment
- ADEM: polysymomatic, encephalopathic, younger child. Antibody: usually negative (MOG-IgG positive in a substantial minority). MRI: diffuse, poorly demarcated, bilateral white and deep grey matter lesions. Long-term treatment: usually none - monophasic; acute steroids; prolonged taper if MOG-positive.
- MOGAD: often ADEM in the young, optic neuritis or transverse myelitis in the older child. Antibody: serum MOG-IgG positive (cell-based assay). MRI: bilateral optic nerve enhancement with disc swelling; longitudinally extensive but better-resolving cord lesions. Long-term treatment: maintenance immunotherapy only if relapsing.
- Paediatric MS: relapsing, dissemination in space and time, older child and adolescent. Antibody: MOG- and AQP4-negative; CSF oligoclonal bands. MRI: periventricular, juxtacortical, infratentorial, and spinal lesions; dissemination on serial imaging. Long-term treatment: a multiple-sclerosis disease-modifying therapy.
- NMOSD: severe optic neuritis, longitudinally extensive myelitis, area-postrema syndrome. Antibody: AQP4-IgG positive. MRI: longitudinally extensive transverse myelitis spanning three or more segments; optic nerve lesions. Long-term treatment: a NMOSD-specific maintenance drug, never a multiple-sclerosis drug. [1] [3]
Epidemiology & Risk Factors
Each of the four entities is uncommon, but together they are the commonest cause of acquired non-infective neurological deficit in previously well children beyond the neonatal period, and the annual incidence of a first acquired demyelinating syndrome in children is estimated at around one to two per hundred thousand. ADEM predominates in younger children - the median age is around five to eight years, and it is the leading phenotype under six - while paediatric MS declares itself later, with a sharp rise after ten years of age and a female predominance that emerges in adolescence. MOGAD and NMOSD span childhood, though NMOSD has a bimodal distribution with a paediatric and an adult peak and a strong female and non-European predilection in many series. [5] [6]
The strongest environmental risk factors are shared across the group and cluster around immune activation. A preceding infection within the prior one to four weeks - most often a non-specific viral upper-respiratory or gastrointestinal illness - is reported in the majority of ADEM and a substantial fraction of MOGAD cases, and a recent vaccination is reported in a minority; the consensus is that the small attributable risk of vaccination is vastly outweighed by the risk of the natural infection it prevents, and vaccination is not a contraindication to subsequent immunisation. Paediatric MS carries the classic MS risk architecture: genetic susceptibility (the HLA-DRB1 locus), female sex after puberty, European ancestry, low ultraviolet exposure and low vitamin D, prior Epstein-Barr virus infection, and childhood obesity. [5] [10]
NMOSD has its own epidemiology that reshapes the pre-test probability. AQP4-IgG-positive NMOSD is over-represented in non-European populations - African, East Asian, South Asian, and Latin American ancestries carry a higher burden - and it frequently coexists with other systemic autoimmunity, most commonly systemic lupus erythematosus, Sjogren syndrome, and thyroid autoimmunity, so a child with a connective-tissue disease and a new optic neuritis or myelitis carries a higher prior probability of NMOSD. MOGAD, by contrast, is less associated with systemic autoimmunity and has a more even sex distribution, and in children the MOG antibody can be transient - a feature that has real implications for who relapses and for how the test is interpreted over time. [3] [12]
Phenotype and age at a glance
Pathophysiology
A demyelinating event is, at its core, an immune-mediated attack on central nervous system myelin - the multilamellar sheath that oligodendrocytes wrap around axons to speed conduction and to support axonal metabolism - and the mechanism of the attack is what separates the four entities and defines their treatment. The cascade runs from a trigger, usually an infection, through a loss of immune tolerance, to a specific effector mechanism that injures myelin, oligodendrocytes, and the underlying axon; the mechanism - antibody-mediated complement attack, T-cell-driven cellular autoimmunity, or innate macrophage inflammation - is what the antibody test and the MRI read, and what the maintenance drug is matched to. [4] [6]
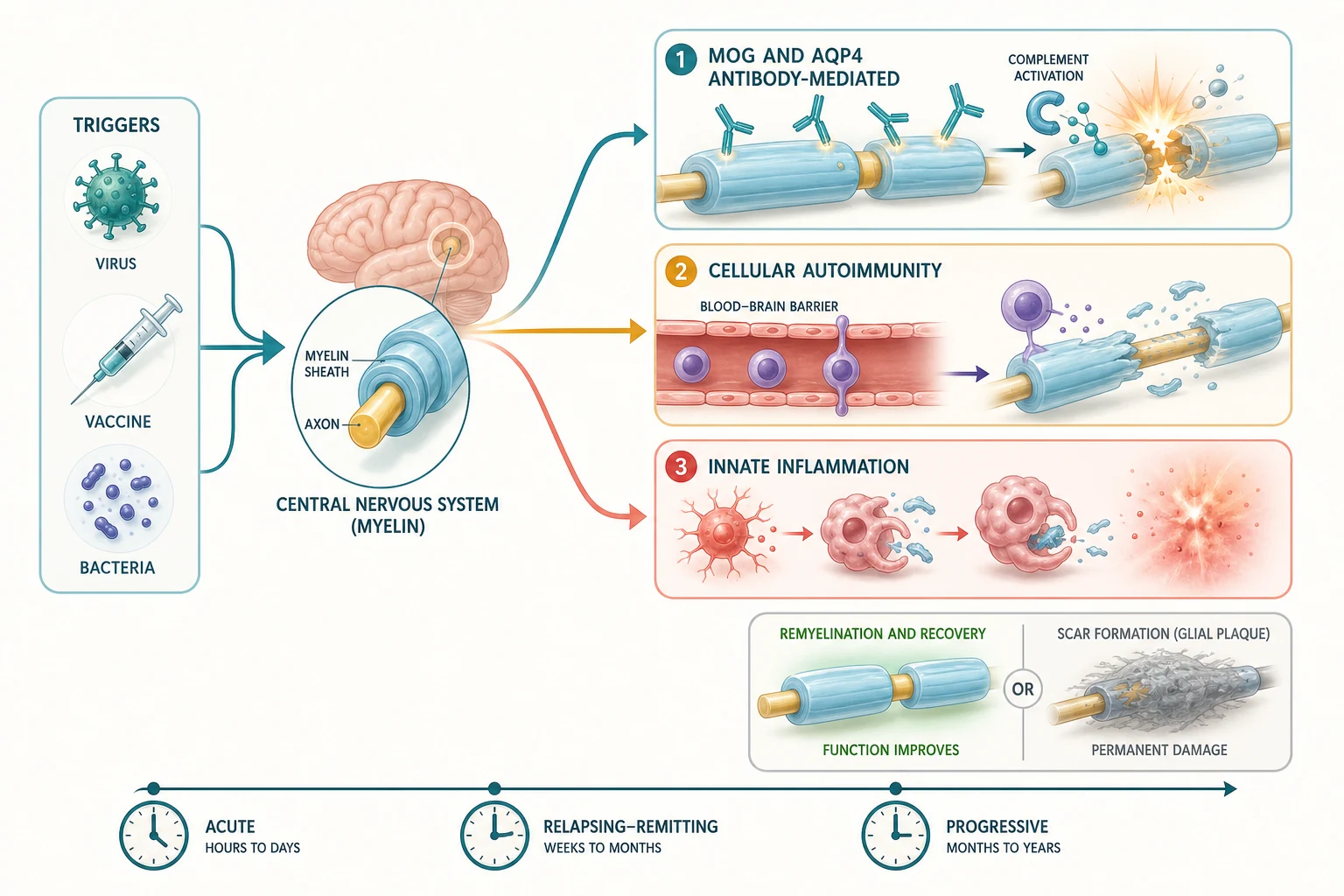
The antibody-mediated disorders are biologically distinct and this is why the antibody test governs treatment. In MOGAD, antibodies bind the extracellular domain of myelin-oligodendrocyte glycoprotein on the outermost myelin lamella and on the oligodendrocyte surface, recruiting complement and producing a demyelinating lesion that often spares the axon - which explains the better recovery and the relapsing optic-neuritis phenotype when the antibody persists. In NMOSD, AQP4-IgG antibodies bind the aquaporin-4 water channel on astrocyte end-feet at the blood-brain barrier, producing a complement-mediated astrocytopathy with secondary demyelination and a characteristic perivascular lesion in the optic nerve, the spinal cord, and the area postrema of the brainstem - the last explaining the pathognomonic hiccups, nausea, and vomiting. The target cell differs - the oligodendrocyte in MOGAD, the astrocyte in NMOSD - and that biological difference is why a multiple-sclerosis drug, which works on T-cell and lymphocyte traffic, does not control either antibody-mediated disorder and may worsen NMOSD. [3] [4]
Paediatric MS carries a different biology again, and in children it is conspicuously more inflammatory than its adult counterpart. The lesion is T-cell- and B-cell-driven, with perivenular inflammatory cuffs of macrophages and lymphocytes producing the classic sharply demarcated demyelinated plaque, and the paediatric variant shows more frequent involvement of the brainstem, cerebellum, and optic nerves, a higher relapse rate, and a larger inflammatory lesion burden on MRI - features that make the acute MRI look more aggressive even as the long-term degenerative phase emerges more slowly. ADEM, by contrast, is a broadly inflammatory, often post-infectious process with a perivenular sleeve of demyelination and a diffuse, poorly demarcated lesion pattern that favours recovery because the axon is relatively spared and remyelination is efficient in the young brain. [5] [6]
Clinical Presentation
The presenting complaint is the acute or subacute onset of neurological deficit in a previously well child, and the pattern of the deficit points to the entity. The first pattern is the polysymomatic encephalopathy of ADEM. A child - most often under ten and frequently under six - develops fever, irritability, and drowsiness over hours to days, usually one to four weeks after a viral illness, and then accrues multifocal neurological deficits that can include hemiparesis, ataxia, cranial-nerve palsies, speech disturbance, seizures, and even a depressed conscious level that may require intensive-care support. The defining feature is encephalopathy - an altered conscious level or behaviour that is not explained by fever alone - and its presence is what separates ADEM from every other first demyelinating event. [1] [6]
The second pattern is the monofocal clinically isolated syndrome, and within it the eye and the cord do most of the localising. Optic neuritis presents with subacute unilateral (or in MOGAD and NMOSD, often bilateral) pain on eye movement, reduced visual acuity, a relative afferent pupillary defect, a swollen optic disc, and colour desaturation; bilateral severe optic neuritis with marked disc swelling in a child raises MOGAD and NMOSD over MS. Acute transverse myelitis presents with bilateral leg weakness, a sensory level, sphincter disturbance, and back pain, evolving over hours to days; a longitudinally extensive lesion spanning three or more vertebral segments on spinal MRI raises NMOSD and MOGAD over MS, which typically produces short, peripheral, focal cord lesions. A brainstem syndrome produces diplopia, ataxia, and cranial-nerve palsies. [8]
The third pattern is the syndrome-complex that is pathognomonic for NMOSD and that every candidate must recognise. The area-postrema syndrome - intractable hiccups, nausea, and vomiting - reflects a lesion at the floor of the fourth ventricle and is a hallmark of AQP4-IgG-positive disease that is frequently mislabelled as gastroenteritis before the optic neuritis or myelitis declares itself. Severe bilateral optic neuritis, a longitudinally extensive transverse myelitis producing paraplegia and sphincter loss, and an area-postrema syndrome in any combination, especially in a non-European child or one with coexisting autoimmunity, is NMOSD until the antibody is excluded, and it is the one presentation in which a multiple-sclerosis drug must never be started. [3]
The fourth pattern is the relapsing course of paediatric MS. An older child or adolescent presents with discrete episodes of focal deficit separated by weeks to months - an optic neuritis that recovers, a sensory cord event that recovers, a brainstem episode that recovers - and it is the recurrence, with dissemination in space and time on imaging or CSF oligoclonal bands, that converts a clinically isolated syndrome into multiple sclerosis. The acute attack itself resembles any demyelinating event, so the diagnosis rests on the accrual of events and on the MRI, not on the single presentation. [5] [10]
Differential Diagnosis
The differential of an acute neurological deficit with white-matter change on imaging is broad, and several mimics must be actively excluded because each changes the trajectory if missed. The first group is the infective and post-infective encephalopathies - viral encephalitis, mycoplasma, and, in the unimmunised or immunocompromised, progressive multifocal leukoencephalopathy - which can produce a similar acute encephalopathy with white-matter change and are excluded by the infective screen and the CSF. Autoimmune encephalitis, dominated by NMDA-receptor antibody encephalitis, produces a subacute psychiatric and dyskinetic encephalopathy that overlaps ADEM and is excluded by the autoimmune panel and the movement-disorder phenotype. [1]
The metabolic and genetic leukoencephalopathies are the great structural mimics and must be excluded whenever the imaging is atypical or the course is progressive rather than relapsing-remitting. X-linked adrenoleukodystrophy in a boy with a contrast-enhancing parieto-occipital lesion, metachromatic leukodystrophy and Krabbe disease with symmetric confluent white-matter change, mitochondrial disorders with a lactate peak on spectroscopy, and the small-vessel vasculitides can all resemble a demyelinating event on a single scan. A progressive rather than relapsing course, a lesion pattern that does not fit, or a family history should redirect the workup toward the metabolic and genetic causes, and the safeguard is the serial MRI and the metabolic and genomic assays that the neurology service directs. [5]
A focused differential also includes the vascular and surgical causes. An acute ischaemic or haemorrhagic stroke produces a sudden focal deficit that is distinguished by its vascular territory and its diffusion restriction on MRI; a cerebral sinovenous thrombosis produces headache and seizures; and a spinal-cord tumour or abscess produces a subacute myelopathy. Posterior reversible encephalopathy syndrome can mimic ADEM on imaging. The point of the differential is not to memorise every mimic but to hold the discipline of excluding the treatable and the dangerous before committing to a long-term immune label. [1] [6]
Clinical & Bedside Assessment
The single most informative bedside act is to establish the tempo, the focality, and - above all - whether there is encephalopathy. Ask exactly when the child was last well, whether there was a preceding illness, how fast the deficit evolved, and whether the child has been drowsy, irritable, or behaviourally altered in a way that fever alone does not explain. A polysymomatic deficit with an altered conscious level is ADEM; a single focussed deficit with a normal conscious level is a clinically isolated syndrome; and that one distinction sets the first provisional label and the urgency of the MRI. [1] [6]
Three questions frame every assessment. First, is the central nervous system involved at more than one site - are there signs referable to the brain, the brainstem, the optic nerve, and the cord together, which favours ADEM or NMOSD, or to a single site, which favours a clinically isolated syndrome? Second, is there encephalopathy, and is it real - distinguished from the lethargy of fever by its depth and its dissociation from the temperature? Third, is there a relevant systemic clue - a coexisting autoimmune disease, a recent infection or vaccination, a non-European ancestry, or a family history of MS - that reshapes the pre-test probability toward one entity? [3] [5]
The examination is then directed and recorded with the myelin distribution in mind. Assess the conscious level and behaviour first. Examine the eyes - acuity, colour plates, pupillary responses for a relative afferent defect, eye movements, and the fundus for disc swelling or pallor. Perform a full motor, sensory, and coordination examination, looking for the hemiparesis, ataxia, and cranial-nerve signs of ADEM and the spinal sensory level and sphincter disturbance of a transverse myelitis. Check for the area-postrema signs of hiccups and vomiting. The discriminating neuro-ophthalmic and spinal signs localise the lesion and point to the entity before any test is sent, which is why a focused neurological examination is the cornerstone of the first encounter. [8]
Investigations
The investigation strategy runs in parallel with the acute treatment, and it is structured to answer two questions in sequence: is this a demyelinating event, and which entity is it? The first investigation is a brain and spinal MRI with gadolinium, performed urgently, because the pattern of the lesions is both diagnostic and prognostic. ADEM shows diffuse, poorly demarcated, often bilateral white-matter lesions with deep grey-matter (thalamic) involvement and frequent enhancement; paediatric MS shows the periventricular, juxtacortical, infratentorial, and spinal lesions that, in the right number and distribution, meet dissemination in space; NMOSD shows longitudinally extensive transverse myelitis spanning three or more segments and optic-nerve lesions; and MOGAD shows bilateral optic-nerve enhancement with disc swelling and longitudinally extensive but better-resolving cord lesions. [1] [4]
The second investigation is the antibody panel, and it is the pivot of the whole workup. Send serum for MOG-IgG and AQP4-IgG by a live cell-based assay - the assay matters, because older fixed-cell or ELISA assays produce false positives - and send cerebrospinal fluid, obtained by lumbar puncture once raised intracranial pressure is excluded, for oligoclonal bands, cell count, protein, glucose, and an infective and autoimmune screen. CSF oligoclonal bands unique to the CSF (and absent from a paired serum sample) support paediatric MS and can, in the 2017 McDonald criteria, substitute for dissemination in time; a pleocytosis with more than fifty white cells favours MOGAD and NMOSD over MS. [3] [12]
The parallel workup
- Recognise the event - establish the tempo, focality, and presence or absence of encephalopathy at the bedside.
- MRI brain and spine with gadolinium - urgent; the lesion pattern names the phenotype and assesses dissemination in space.
- Serum MOG-IgG and AQP4-IgG by live cell-based assay - the antibody pivot; the result governs the long-term treatment.
- Lumbar puncture and cerebrospinal fluid - cell count, protein, glucose, oligoclonal bands on paired CSF and serum, and an infective and autoimmune screen.
- Ancillary tests - visual evoked potentials for occult optic neuritis, optical coherence tomography for retinal nerve-fibre-layer thinning, a metabolic and infective screen to exclude the mimics, and vitamin D for the MS risk profile. [1] [8]
The 2017 McDonald criteria for multiple sclerosis apply directly to children aged eleven years and older with a non-ADEM onset, and they allow a diagnosis of MS at the first attack when the MRI demonstrates both dissemination in space (lesions in at least two of the periventricular, juxtacortical, infratentorial, and spinal regions) and dissemination in time (a new lesion on a later scan, or simultaneous enhancing and non-enhancing lesions), or when CSF oligoclonal bands substitute for dissemination in time. In children younger than eleven and in any child with an ADEM onset, the criteria are less specific and a more conservative, serial-imaging approach is taken - a practical point because applying the adult criteria to a young ADEM child overdiagnoses MS. [1] [10]
Management — Resuscitation
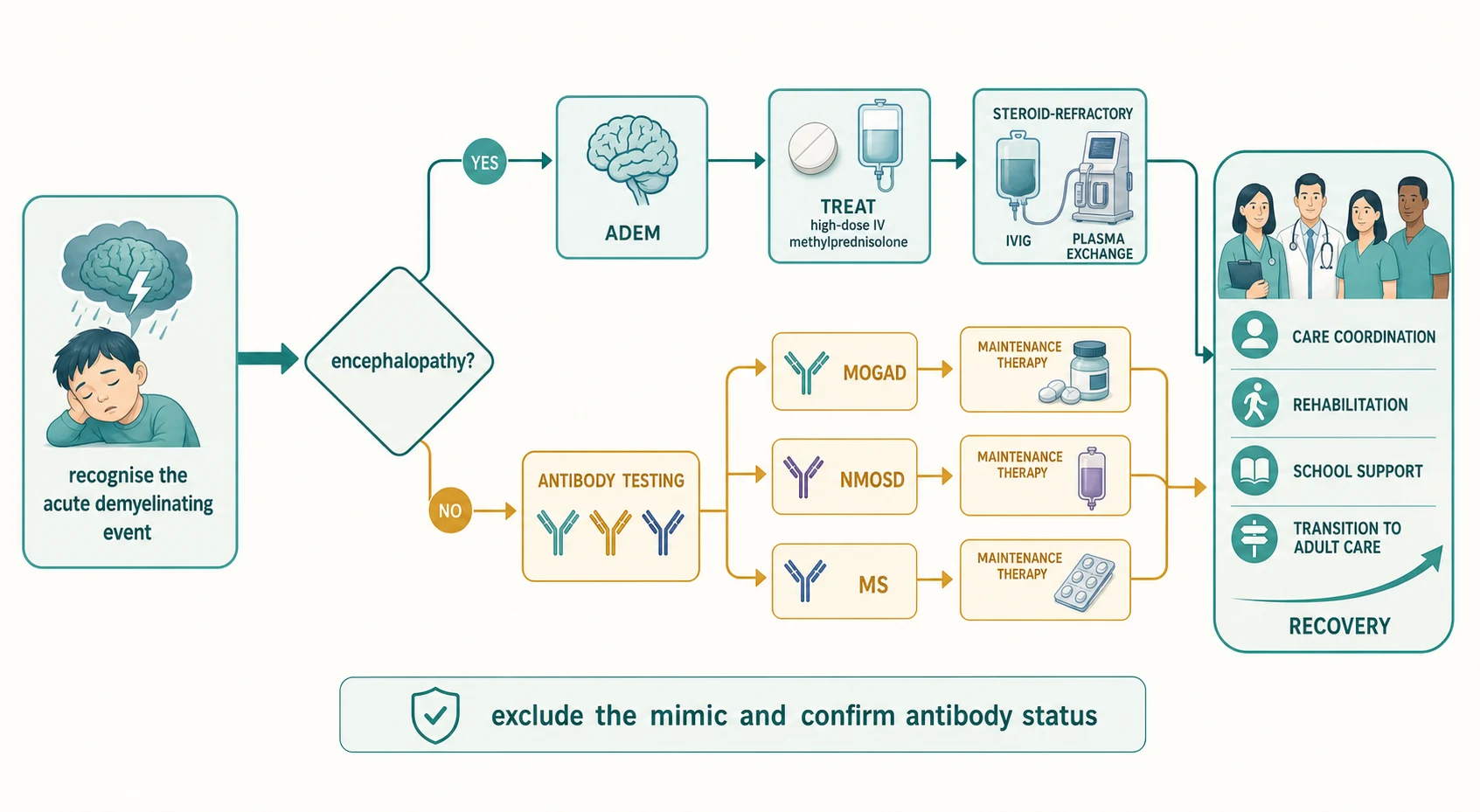
When the demyelinating event presents as acute encephalopathy, a rapidly progressing myelopathy, or visual loss, the response is resuscitation first and the diagnostic workup in parallel. Secure the airway, breathing, and circulation; treat seizures; monitor for raised intracranial pressure; and move immediately to the acute immunotherapy that every demyelinating event shares, because the outcome is better the earlier treatment begins. The general paediatrician owns the first hours, with paediatric neurology and neuroimmunology taking over as the diagnosis firms. [6] [9]
First-line acute therapy for every acquired demyelinating syndrome is high-dose intravenous methylprednisolone. The standard paediatric regimen is 20 to 30 mg/kg per day to a maximum of 1 g per day, given over three to five days, and it is begun as soon as infection is reasonably excluded - the workup proceeds in parallel rather than waiting for every result. A short oral prednisolone taper over two to four weeks follows, and in MOGAD - where relapse often occurs as the steroid is weaned - the taper is deliberately slow and prolonged over four to six weeks or longer. The principle is that the acute attack is treated identically across the four entities, because the acute mechanism is inflammatory in all of them. [9] [11]
Acute corticosteroid regimen for the first demyelinating event
Escalation is the rule, not the exception, for the severe or steroid-refractory event. If the deficit or the encephalopathy has not improved after forty-eight to seventy-two hours of high-dose corticosteroids, the next step is intravenous immunoglobulin at a total dose of 2 g/kg (given over two to five days) or plasma exchange, typically five to seven exchanges of one to one-and-a-half plasma volumes on alternate days; plasma exchange is favoured for the most severe AQP4-IgG-positive attacks because it removes the pathogenic antibody directly. These escalation decisions are shared with the neuroimmunology service, and the principle is that a worsening event on steroids is a signal to change strategy, not to repeat it. [3] [9]
Management — Definitive & Stepwise
Definitive management means naming the entity on the antibody result and the MRI, and matching the long-term therapy to the confirmed diagnosis - because the maintenance treatment diverges sharply across the four entities and the wrong drug harms. The figure below lays out the staged pathway from the acute event, through the encephalopathy fork and the antibody triad, to the named diagnosis and its matched maintenance therapy, converging on the multidisciplinary care that every child with a chronic demyelinating disorder needs. [1] [11]
For ADEM that is antibody-negative and monophasic, no maintenance therapy is required beyond the acute attack treatment and surveillance - the long-term management is reassurance, a structured follow-up MRI, and vigilance for a relapse that would reclassify the diagnosis. For relapsing MOGAD, maintenance immunotherapy is reserved for children with a relapsing course, and the European paediatric MOG consortium recommends a stepwise approach in which mycophenolate mofetil, azathioprine, intravenous immunoglobulin, and rituximab are used according to relapse frequency and severity, with rituximab and intravenous immunoglobulin increasingly favoured for the most relapsing courses. The principle is that maintenance is triggered by relapse, not by the first attack, because many children with MOGAD are monophasic. [7] [9]
For paediatric MS, the modern approach is to start a high-efficacy disease-modifying therapy early, because the paediatric disease is more inflammatory and early control of relapses preserves long-term cognitive and motor function. The licensed and used agents include the injectables (interferon-beta, glatiramer acetate), the oral agents (fingolimod, dimethyl fumarate, teriflunomide), and the monoclonal antibodies (natalizumab, ocrelizumab, ofatumumab, rituximab), with the anti-CD20 agents and natalizumab increasingly used first-line for highly active disease. The choice is individualised by activity, by the John Cunningham virus status that governs natalizumab safety, and by adherence, and it is owned by the paediatric MS service. [10] [11]
For NMOSD, the maintenance therapy is a NMOSD-specific drug and never a multiple-sclerosis drug. The established agents - rituximab, mycophenolate mofetil, and azathioprine - are now joined by the licensed monoclonal antibodies satralizumab (an interleukin-6 receptor blocker), eculizumab and ravulizumab (complement blockers), and ensprutifrug (a CD19 B-cell-depleting agent), chosen by attack severity, coexisting autoimmunity, and access. The care plan is multidisciplinary and lifelong: rehabilitation (physiotherapy, occupational therapy, vision support), educational and psychological support, a sick-day and vaccination plan that avoids live vaccines on immunosuppression, vitamin D supplementation, and structured transition to adult neurology before the eighteenth birthday. [3] [11]
Specific Subtypes & Scenarios
ADEM is the prototypic paediatric demyelinating event and every candidate must know its course. A young child - median age five to eight years - develops encephalopathy one to four weeks after a viral illness, accrues multifocal deficits over days, and shows the diffuse, bilateral, poorly demarcated white and deep grey matter lesions on MRI. The IPMSSG definition requires encephalopathy as a core criterion, distinguishes a single ADEM event from multiphasic ADEM (a second event at least three months after the first and at least four weeks after completing steroids, with a new or enlarged lesion or the same phenotype), and separates both from an ADEM-ON or ADEM followed by optic neuritis that, when MOG-IgG is positive, is MOGAD. Most antibody-negative ADEM is monophasic and recovers fully or near-fully over weeks to months. [1] [6]
MOGAD is the antibody-defined disorder that straddles the ADEM and clinically isolated syndrome phenotypes and that has reshaped paediatric neuroimmunology. In young children it most often presents as ADEM; in older children and adolescents it presents as optic neuritis (often bilateral, with severe disc swelling, and with better recovery but a higher relapse rate than MS optic neuritis), transverse myelitis, or a brainstem syndrome. The 2023 International MOGAD Panel criteria require a clinical demyelinating event plus serum MOG-IgG detected by a cell-based assay, supported by at least one of a compatible MRI, a compatible CSF, or a characteristic optic-neuritis phenotype, and the diagnosis is excluded if AQP4-IgG is positive. The serum MOG antibody in children can be transient - high initial titres tend to persist and predict relapse, whereas seroreversion is associated with a monophasic course - which is why the titre is rechecked over time and interpreted with the clinical course. [2] [12]
Paediatric MS is a relapsing disorder of dissemination in space and time and is the entity that the maintenance disease-modifying therapy targets. It is rare under ten and rises sharply in adolescence, with a female predominance that emerges with puberty; the acute attacks are more inflammatory than in adults, with frequent brainstem, cerebellar, and optic-nerve involvement, a higher relapse rate, and a larger lesion burden on MRI, even as the long-term degenerative phase emerges more slowly. The diagnosis rests on the accrual of events with dissemination on imaging or on CSF oligoclonal bands per the 2017 McDonald criteria, and the management is early high-efficacy disease-modifying therapy to preserve long-term function. [5] [10]
NMOSD is the aquaporin-4-IgG-mediated disorder that every candidate must distinguish from MS because the treatment is entirely different. Its core clinical characteristics are severe optic neuritis (often bilateral and with poor recovery), acute longitudinally extensive transverse myelitis (spanning three or more vertebral segments and producing paraplegia and sphincter loss), the area-postrema syndrome (intractable hiccups, nausea, vomiting), and brainstem, diencephalic, and cerebral syndromes. The IPND 2015 criteria confirm the diagnosis with AQP4-IgG plus at least one core clinical characteristic and a compatible MRI, or, in the antibody-negative case, two or more core characteristics (one of which must be optic neuritis, longitudinally extensive myelitis, or area-postrema) with specific MRI requirements. The maintenance drug is NMOSD-specific, never a multiple-sclerosis drug. [3]
Complications & Pitfalls
The most consequential complications are the consequences of delay and of the wrong maintenance drug. A delayed acute attack treatment prolongs the deficit and, in the antibody-mediated disorders, allows complement-mediated injury to become irreversible - which is why the steroid course is begun as soon as infection is reasonably excluded rather than waiting for every result. The wrong maintenance drug is the cardinal avoidable harm: a multiple-sclerosis disease-modifying therapy given to a child with NMOSD can precipitate a devastating relapse, and several of the drugs have been associated with worsening attack frequency and severity in AQP4-IgG-positive disease. The safeguard is the antibody test before any long-term drug. [3] [11]
Over-reliance on a single negative antibody result is a second pitfall, and it cuts both ways. A negative MOG-IgG in the acute phase does not exclude MOGAD if the assay is insensitive or the titre is low, and a positive MOG-IgG on a non-cell-based assay can be a false positive that mislabels a child; the assay and the laboratory matter, and equivocal results are repeated and interpreted with the clinical course and the MRI. A third pitfall is applying the adult multiple-sclerosis criteria to a young child with an ADEM onset, which overdiagnoses MS and exposes a child to an unnecessary disease-modifying therapy; in children under eleven and in any ADEM-onset child, a more conservative serial-imaging approach is taken. [1] [2]
A fourth pitfall is underestimating the cognitive and educational burden, which is substantial even after a good motor recovery. Children with ADEM and paediatric MS show measurable effects on attention, processing speed, and executive function that emerge over months and that are missed if the follow-up focuses only on the motor examination, which is why a formal neuropsychological assessment and an individualised education plan are part of the management. A fifth pitfall is the steroid wean in MOGAD - relapse clusters around the taper, so the wean is slow and prolonged, and an early relapse on a fast wean is reclassified as relapsing MOGAD and triggers maintenance therapy. [7] [9]
Prognosis & Disposition
Prognosis is determined by the entity, the age, the attack severity, and - critically - the timing and accuracy of the treatment. Antibody-negative monophasic ADEM has an excellent prognosis, with the majority of children recovering fully or near-fully over weeks to months, and a low relapse risk; the task after recovery is a single surveillance MRI and vigilance for a relapse that would reclassify the diagnosis. Paediatric MS has a high relapse rate and a larger inflammatory burden than adult MS, but a slower progression to disability when the disease-modifying therapy is started early - early high-efficacy treatment preserves long-term cognitive and motor function, which is why the threshold to start is low. [5] [10]
MOGAD has a generally favourable attack-by-attack recovery - the optic neuritis and myelitis often recover well because the axon is relatively spared - but a substantial relapse rate, and the long-term task is to identify the relapsing subset early and to start maintenance therapy before accrual of deficit. The serum MOG antibody titre helps: high or persisting titres predict relapse, and seroreversion predicts a monophasic course, so the titre is monitored alongside the clinical course. NMOSD has the most guarded prognosis of the four, with severe attacks that can leave permanent visual and motor disability, and the long-term outcome is determined by the attack frequency that the maintenance drug controls - which is why early, accurate, NMOSD-specific therapy is the single biggest determinant of outcome. [3] [7]
Disposition is structured around the paediatric neurology and neuroimmunology service, with the general paediatrician holding the whole child and coordinating the multidisciplinary team. Rehabilitation - physiotherapy, occupational therapy, speech and language, vision support, and neuropsychology - is begun early and runs alongside the medical therapy rather than after it. The sick-day plan, the vaccination plan (avoiding live vaccines on immunosuppression and ensuring influenza and pneumococcal cover), vitamin D supplementation, and an individualised education plan are part of management, not afterthoughts. As the child grows, structured transition to adult neurology and immunology begins in early adolescence, with explicit handover of the diagnosis, the antibody status, the maintenance drug, and the surveillance plan before the eighteenth birthday. [11]
Special Populations
The approach differs across the age span and across the antibody strata. Infants and young children present predominantly with ADEM, where the encephalopathy is the defining feature and the MOG antibody is positive in a substantial minority - in this group the task is acute treatment, a slow steroid wean, and vigilance for a relapse that would reclassify the diagnosis as MOGAD. Older children and adolescents present with the clinically isolated syndromes and paediatric MS, where the 2017 McDonald criteria apply directly and the disease-modifying therapy is started early. Recognising the age-typical presentation sharpens the differential and the first test. [1] [5]
Non-European and Indigenous populations carry a higher burden of AQP4-IgG-positive NMOSD, and the consultation reframes the workup whenever the phenotype is severe bilateral optic neuritis or a longitudinally extensive myelitis in a child of African, East Asian, South Asian, or Latin American ancestry. Coexisting systemic autoimmunity - systemic lupus erythematosus, Sjogren syndrome, thyroid disease - raises the prior probability of NMOSD and warrants a connective-tissue screen. For rural, remote, and disadvantaged populations, equitable and culturally safe access to paediatric neuroimmunology, to the cell-based antibody assay, and to the licensed monoclonal antibodies is a real challenge, concentrated as those services are in tertiary centres, and the pathway is adapted with telehealth, a local coordinator, and retrieval networks. [3]
The technology-dependent and complex-chronic child, and the transition-age adolescent, need a plan that adapts across the lifespan. The adolescent who has lived with a demyelinating diagnosis since childhood faces new questions - adherence to the maintenance drug, transfer to adult services, reproductive decisions on teratogenic immunotherapy, and the risk of relapse with new freedoms - and a structured transition, begun in early adolescence and completed before the eighteenth birthday, is the safeguard. The immunocompromised child, whether from the disease or its treatment, needs a vaccination plan that balances infection risk against relapse prevention, and the live-vaccine contraindication on certain agents is a practical point that the general paediatrician owns. [11]
Evidence, Guidelines & Regional Differences
The conceptual backbone of the field is a set of international consensus frameworks that let the clinician act on the phenotype before the confirmatory antibody returns. The 2013 IPMSSG definitions standardised the recognition of ADEM, the clinically isolated syndromes, and paediatric MS, and anchored the first fork on encephalopathy. The 2023 International MOGAD Panel criteria formalised the diagnosis of MOG-antibody-associated disease on a clinical event plus serum MOG-IgG on a cell-based assay, with supportive features and exclusion of AQP4-IgG. The 2015 IPND criteria remain the standard for NMOSD, built on the AQP4-IgG status and the core clinical characteristics, and the 2017 McDonald criteria extend the adult multiple-sclerosis criteria to children over eleven with a non-ADEM onset, allowing oligoclonal bands to substitute for dissemination in time. [1] [2]
In Australia and Aotearoa New Zealand, paediatric neuroimmunology services and the live cell-based MOG-IgG and AQP4-IgG assays are concentrated in tertiary paediatric centres, with regional and remote access via telehealth and retrieval networks. Funded access to the licensed NMOSD monoclonal antibodies - satralizumab, eculizumab and ravulizumab, and the B-cell-depleting agents - and to the paediatric multiple-sclerosis disease-modifying therapies is governed by national and jurisdictional programmes that evolve over time, and the antibody assay may have a turnaround of weeks in centres without on-site neuroimmunology. State the local panel and the local funded pathway rather than assuming a universal list, treat the acute attack on the phenotype, and involve the regional service early. [3] [11]
The strength of evidence varies across the field. The evidence for high-dose corticosteroids as first-line acute therapy is mature and consistent across the entities. The evidence for plasma exchange in the severe AQP4-IgG-positive attack, for rituximab and the newer monoclonals in NMOSD maintenance, and for early high-efficacy disease-modifying therapy in paediatric MS is strong and growing. The evidence for the optimal maintenance strategy in relapsing MOGAD is still maturing - the European paediatric MOG consortium provides a stepwise framework, but randomised trials in children are few, and the choice is individualised by relapse frequency and severity. Uncertainty changes counselling: a family should be told what is known, what is likely, and what is genuinely unknown, so that decisions rest on real information rather than false reassurance. [7] [9]
Exam Pearls
Remember the two principles that frame every consultation. First, treat the acute attack immediately with high-dose methylprednisolone 20 to 30 mg/kg per day to a maximum of 1 g for three to five days, and escalate to intravenous immunoglobulin or plasma exchange if it is steroid-refractory - the acute mechanism is inflammatory across all four entities, so the acute treatment is shared. Second, never start a multiple-sclerosis disease-modifying therapy before the aquaporin-4 and MOG antibody status is known, because the wrong drug can precipitate a devastating relapse in NMOSD. The diagnostic rate-limiting step is the antibody answer, and the long-term treatment is matched to the confirmed diagnosis, never to a guess. [1] [11]
Self-test: a six-year-old with encephalopathy and a hemiparesis
A previously well six-year-old presents four days after a viral illness with drowsiness, irritability, a right hemiparesis, and slurred speech. Brain MRI shows diffuse, bilateral, poorly demarcated white-matter lesions with thalamic involvement and patchy enhancement. What is the most likely diagnosis, what is the first-line treatment, and what test governs the long-term management? [1] [6]
Answer: This is acute disseminated encephalomyelitis until proved otherwise - the polysymomatic deficit with encephalopathy, the post-infectious timing, and the diffuse bilateral white and deep grey matter lesions on MRI are the classic phenotype. First-line treatment is high-dose intravenous methylprednisolone 20 to 30 mg/kg per day to a maximum of 1 g for three to five days, with a short oral taper. The serum MOG-IgG and AQP4-IgG (by cell-based assay) and the CSF oligoclonal bands govern the long-term management: a persisting MOG-IgG reclassifies the case as MOGAD and prolongs the wean and the surveillance, an AQP4-IgG reclassifies it as NMOSD, and antibody negativity with a single event supports monophasic ADEM and no maintenance therapy. [2] [9]
References
- [1]Krupp LB, Tardieu M, Amato MP, Banwell B, Chitnis T, Dale RC, et al. International Pediatric Multiple Sclerosis Study Group criteria for pediatric multiple sclerosis and immune-mediated central nervous system demyelinating disorders: revisions to the 2007 definitions. Mult Scler, 2013.PMID 23572237
- [2]Banwell B, Bennett JL, Marignier R, Kim HJ, Brilot F, Flanagan EP, et al. Diagnosis of myelin oligodendrocyte glycoprotein antibody-associated disease: International MOGAD Panel proposed criteria. Lancet Neurol, 2023.PMID 36706773
- [3]Wingerchuk DM, Banwell B, Bennett JL, Cabre P, Carroll W, Chitnis T, et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology, 2015.PMID 26092914
- [4]Marignier R, Hacohen Y, Cobo-Calvo A, Probstel AK, Aktas O, Alexopoulos H, et al. Myelin-oligodendrocyte glycoprotein antibody-associated disease. Lancet Neurol, 2021.PMID 34418402
- [5]Waldman A, Ness J, Pohl D, Simone IL, Anlar B, Amato MP, et al. Pediatric multiple sclerosis: Clinical features and outcome. Neurology, 2016.PMID 27572865
- [6]Tenembaum S, Chitnis T, Ness J, Hahn JS, International Pediatric MS Study Group. Acute disseminated encephalomyelitis. Neurology, 2007.PMID 17438235
- [7]Ramanathan S, Mohammad S, Tantsis E, Nguyen TK, Merheb V, Fung VSC, et al. Clinical course, therapeutic responses and outcomes in relapsing MOG antibody-associated demyelination. J Neurol Neurosurg Psychiatry, 2018.PMID 29142145
- [8]Petzold A, Fraser CL, Abegg M, Alroughani R, Alshowaeir D, Alvarenga R, et al. Diagnosis and classification of optic neuritis. Lancet Neurol, 2022.PMID 36179757
- [9]Bruijstens AL, Wendel EM, Lechner C, Bartels F, Finke C, Breu M, et al. E.U. paediatric MOG consortium consensus: Part 5 - Treatment of paediatric myelin oligodendrocyte glycoprotein antibody-associated disorders. Eur J Paediatr Neurol, 2020.PMID 33176999
- [10]Kornbluh AB, Kahn I. Pediatric Multiple Sclerosis. Semin Pediatr Neurol, 2023.PMID 37451754
- [11]Margoni M, Preziosa P, Rocca MA, Filippi M. Anti-CD20 Therapies in Pediatric Acquired Demyelinating Syndromes: Evidence Across MS, AQP4-IgG-Positive NMOSD and MOGAD. CNS Drugs, 2026.PMID 42334795
- [12]Wendel EM, Thonke HS, Bertolini A, Baumann M, Blaschek A, Merkenschlager A, et al. Temporal Dynamics of MOG Antibodies in Children With Acquired Demyelinating Syndrome. Neurol Neuroimmunol Neuroinflamm, 2022.PMID 36229191