Paeds · neurology-neurodisability-and-neuromuscular
Ataxia in children
Also known as Acute cerebellar ataxia · Childhood ataxia · Post-infectious cerebellar ataxia · Friedreich ataxia · Opsoclonus-myoclonus syndrome · Sensory ataxia · Cerebellar ataxia in children
Fellowship guide to ataxia in children: separating the common, benign post-infectious acute cerebellar ataxia from the dangerous posterior fossa tumour and the progressive hereditary causes such as Friedreich ataxia, the gait and bedside examination that localises cerebellar from sensory ataxia, the red-flag screen that drives urgent neuroimaging, and the genetic and multidisciplinary management of chronic progressive ataxia including omaveloxolone for Friedreich ataxia.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
Overview & Definition
Picture the four-year-old who is brought to the emergency department walking as though drunk, feet wide apart and reeling, two weeks after a bout of chickenpox, and whose parents are terrified. That child carries the whole teaching point of paediatric ataxia: the unsteady gait is alarming to the family but is usually benign, the diagnosis is clinical, and the clinician's job is to confirm the safe, common pattern and to exclude the small minority that are dangerous. The decisive first question is not which drug to give but whether this is acute or chronic, and whether red flags are present, because that single distinction separates reassurance and observation from urgent imaging and oncology. [1] [2]
Ataxia is impaired coordination of voluntary movement that cannot be explained by weakness, sensory loss, or extrapyramidal rigidity alone. The lesion usually lies in the cerebellum or its connections, producing a wide-based, unsteady gait, dysmetria (past-pointing), intention tremor, dysdiadochokinesia (impaired rapid alternating movement), slurred speech, and nystagmus. A second, distinct mechanism is sensory ataxia, in which loss of proprioception from the dorsal columns deprives the brain of position sense, so the child walks unsteadily and falls in the dark and stamps the feet, with a positive Romberg sign. The clinician's first bedside task is to localise the ataxia to the cerebellum, to the sensory pathway, or to a vestibular cause, because the differential and the workup diverge from there. [1]
The framework that organises the whole topic is the acute-versus-chronic split combined with the red-flag screen. Acute ataxia, evolving over hours to days, is dominated by post-infectious acute cerebellar ataxia, drug ingestion, and the dangerous causes, posterior fossa tumour, cerebellitis, stroke, and central nervous system infection. Chronic or progressive ataxia, evolving over weeks to months, points to the hereditary degenerations, of which Friedreich ataxia is the prototype, and to the metabolic and congenital disorders. The red-flag screen, for headache, vomiting, encephalopathy, focal signs, and a progressive course, is the bridge between the bedside pattern and the imaging decision. [1] [3]
Classification
The most useful first cut in a child with ataxia is between the acute and the chronic, because it determines whether the next step is a period of observation or a genetic and imaging workup. Acute ataxia evolves over hours to days, is most often post-infectious or due to drug ingestion, and carries the burden of excluding a posterior fossa mass, a stroke, or an infection. Chronic or progressive ataxia evolves over weeks to months and points to a hereditary degeneration, a metabolic disorder, or a congenital malformation of the cerebellum. A third and critical category is the acute or subacute ataxia with a specific signature, such as opsoclonus-myoclonus syndrome, that demands a dedicated workup regardless of tempo. [1]
Within the acute group, post-infectious acute cerebellar ataxia is by far the commonest cause in a child under about eight years, typically following a viral illness by one to three weeks, with varicella, Epstein-Barr virus, enteroviruses, and Mycoplasma among the recognised triggers. Drug ingestion is the other high-frequency cause, especially in toddlers with access to antiepileptics, benzodiazepines, or alcohol, and the toxicology history is part of every acute ataxia assessment. The dangerous acute causes are posterior fossa tumour and cerebellitis with obstructive hydrocephalus, cerebellar or brainstem stroke, and central nervous system infection such as meningitis or encephalitis, and the red-flag screen exists to catch them. [1]
Acute cerebellar ataxia
post-infectious
- Peak age preschool, 2 to 8 years
- Onset hours to 1 to 2 days, 1 to 3 weeks after a virus
- Truncal and gait ataxia, no encephalopathy
- Normal examination except the ataxia
- Resolves fully in weeks to months in most children
Posterior fossa tumour
red flag
- Subacute or progressive, days to weeks
- Headache, early-morning vomiting, head tilt
- Truncal ataxia, cranial nerve signs, papilloedema
- Obstructive hydrocephalus from fourth ventricle compression
- Urgent magnetic resonance imaging
Friedreich ataxia
hereditary
- Onset before 25 years, usually 5 to 15 years
- Progressive limb and gait ataxia
- Absent lower limb reflexes, extensor plantars
- Pes cavus, scoliosis, cardiomyopathy, diabetes
- GAA repeat expansion in the FXN gene
Opsoclonus-myoclonus
immune
- Acute or subacute ataxia with extreme irritability
- Opsoclonus, the dancing eyes
- Myoclonus of trunk and limbs
- Occult neuroblastoma in around half
- Immunotherapy, not observation
The chronic and progressive ataxias are a large and genetically heterogeneous group unified by a slow, relentless course. Friedreich ataxia is the prototype and the most common in European populations, and it is the one the examinations most often test, but the differential extends to ataxia telangiectasia, ataxia with oculomotor apraxia, ataxia with vitamin E deficiency, the congenital cerebellar malformations such as Joubert and Dandy-Walker syndromes, and the episodic ataxias caused by channelopathies. The unifying clinical principle is that a progressive ataxia warrants magnetic resonance imaging and a directed genetic and metabolic evaluation, because the specific diagnosis changes the counselling, the surveillance, and, in a growing minority, the treatment. [5] [9]
Epidemiology & Risk Factors
Acute cerebellar ataxia is the single most common cause of acute ataxia in children, and the peak age is the preschool years, with most cases occurring between two and eight years of age. The classic study by Connolly and colleagues of the course and outcome of acute cerebellar ataxia established the benign natural history that still anchors practice, showing that the large majority of children recover fully and that the ataxia resolves over weeks to months without specific treatment. The condition is post-infectious in most cases, and a history of a viral illness in the preceding one to three weeks is the rule rather than the exception. [2] [1]
Posterior fossa tumours are the second most common group of brain tumours in children after supratentorial tumours, and medulloblastoma is the most common malignant brain tumour of childhood. The systematic review and meta-analysis by Wilne and colleagues examined the features at presentation of childhood central nervous system tumours and found that headache, nausea and vomiting, abnormalities of gait and coordination, and educational or behavioural change are among the commonest reported features. The review highlighted that the interval from symptom onset to diagnosis is often long, because the early signs are non-specific and overlap with far commoner benign conditions, which is why a persistent or progressive ataxia is never dismissed. [3]
The numbers that anchor your viva
Friedreich ataxia is the most common inherited ataxia in populations of European descent, with a prevalence of roughly one in fifty thousand, and it is inherited in an autosomal recessive pattern. The risk factor is therefore carrier status in both parents, and the condition is caused by a GAA trinucleotide repeat expansion in the FXN gene on chromosome nine, which reduces the production of frataxin, a mitochondrial protein involved in iron-sulphur cluster assembly. Larger repeat sizes correlate with earlier onset and more severe disease, and the natural history study by Rummey and colleagues documented the heterogeneity of neurological progression, which has direct consequences for clinical trial design and for counselling an individual family. [5] [6]
Pathophysiology
The cerebellum coordinates the timing and precision of voluntary movement, and a lesion within it or its connections produces the characteristic cerebellar syndrome. The midline vermis governs posture, balance, and gait, so a midline lesion causes a truncal and gait ataxia with a wide-based, staggering walk, the pattern most often seen in alcohol intoxication and in childhood acute cerebellar ataxia. The cerebellar hemispheres govern the coordination of the limbs on the same side, so a hemispheric lesion causes an appendicular ataxia with dysmetria, intention tremor, and dysdiadochokinesia on the affected side. Understanding this topography lets the clinician localise the lesion from the bedside examination alone. [1]
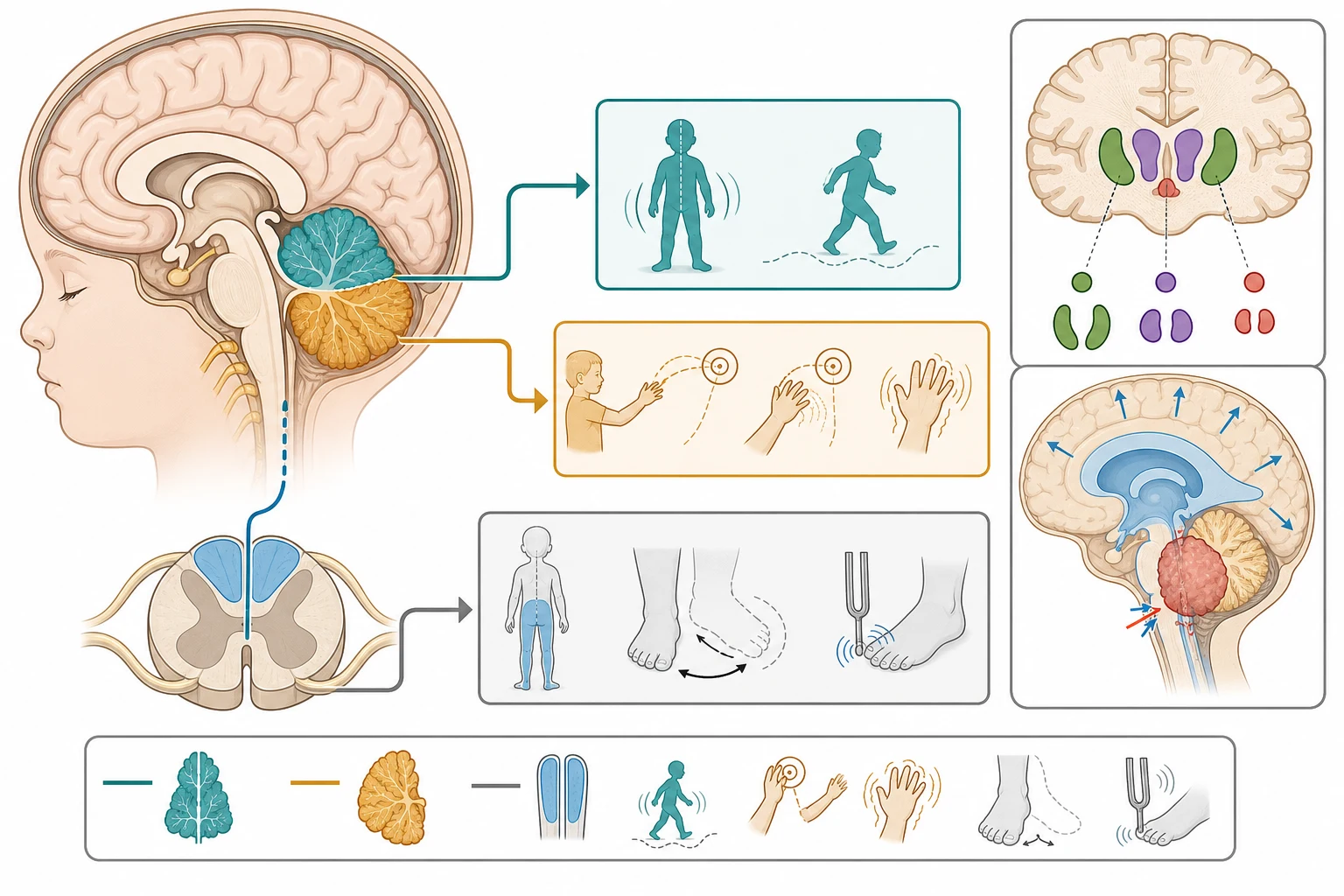
The mechanism of post-infectious acute cerebellar ataxia is thought to be an autoimmune or molecular mimicry process, in which antibodies or immune responses triggered by a preceding viral infection cross-react with cerebellar tissue, producing a transient cerebellar dysfunction without permanent neuronal loss. This explains the latency of one to three weeks after the infection, the self-limited course, and the occasional finding of a mild cerebellar swelling or enhancement on magnetic resonance imaging that the literature terms acute cerebellitis. The distinction matters clinically, because the molecular mimicry mechanism has no specific treatment beyond supportive care, and the prognosis is driven by the natural history rather than by the intervention. [1]
Friedreich ataxia has a defined molecular mechanism that is worth understanding because it explains the multisystem phenotype and now drives therapy. The GAA repeat expansion in the FXN gene reduces frataxin, a protein that assembles iron-sulphur clusters in the mitochondrion, so cellular iron metabolism is disrupted and oxidative phosphorylation is impaired, with the greatest damage falling on the metabolically demanding dorsal root ganglia, the dorsal columns, the spinocerebellar tracts, and the heart. This single mechanism produces the sensory ataxia, the loss of position and vibration sense, the cardiomyopathy, and the diabetes, and it is the rationale for omaveloxolone, which activates nuclear factor erythroid 2-related factor 2 and restores mitochondrial function. [5] [11]
[5] [7] [12]The mechanism of opsoclonus-myoclonus-ataxia syndrome is autoimmune or paraneoplastic, and in children it is driven by an immune response that is often directed against an occult neuroblastoma, so that antibodies cross-react with cerebellar and brainstem neurons to produce the characteristic syndrome. This mechanism explains why the motor symptoms may improve with immunotherapy while the cognitive and behavioural sequelae persist, and why every child is investigated for a neural crest tumour even when no mass is palpable. Ataxia telangiectasia, by contrast, is caused by biallelic mutations in the ATM gene, whose protein product coordinates the cellular response to DNA double-strand breaks, so the unifying mechanism is a failure of DNA repair that produces progressive cerebellar degeneration, immunodeficiency, and a predisposition to malignancy. [8] [9]
Clinical Presentation
The typical story of acute cerebellar ataxia is a previously well preschool child who develops an unsteady gait over a few hours to a day or two, one to three weeks after a viral illness, and is brought to the emergency department because the parents fear a serious neurological event. The child is alert, conversant, and afebrile, but walks with a wide-based, staggering, reeling gait and may fall when turning. The ataxia is predominantly truncal and gait-based, the speech may be mildly slurred, and there may be nystagmus, but there is no weakness, no altered conscious state, and no focal deficit. The examination between the ataxia is otherwise normal, and that normality is the central reassuring feature. [1] [2]
The child with a posterior fossa tumour tells a different story, and recognising it is the high-stakes skill of the topic. The ataxia is subacute or progressive over days to weeks rather than hours, and it is accompanied by the features of raised intracranial pressure from obstruction of the fourth ventricle and the cerebral aqueduct. The child has a headache that is worse in the early morning or on waking, vomiting that is characteristically effortless and early-morning, a head tilt as the child compensates for a cranial nerve palsy, a new squint from a sixth nerve palsy, papilloedema on fundoscopy, and a declining school performance or change in behaviour. Any one of these, in a child with ataxia, converts the consultation from reassurance to urgent imaging. [3] [4]
| Clinical picture | What it implies | Act |
|---|
Friedreich ataxia presents in an older child or adolescent with a slowly progressive ataxia that has been present for months to a few years before presentation. The onset is typically between five and fifteen years of age, always before twenty-five, and the gait and limb ataxia is accompanied by the cardinal neurological signs of absent lower limb tendon reflexes, an extensor plantar response, and loss of position and vibration sense in the lower limbs. The skeletal features of pes cavus and scoliosis are often present, and the systemic features of hypertrophic cardiomyopathy and, less often, diabetes mellitus are part of the multisystem disease and the principal determinants of survival. The clinical task is to recognise the constellation and to confirm it with a targeted genetic test. [5] [10]
Opsoclonus-myoclonus-ataxia syndrome presents with a combination that is unmistakable once seen. The child has acute or subacute ataxia with marked, often extreme, irritability and sleep disturbance, the chaotic multidirectional saccades of opsoclonus that are present in all directions of gaze and persist in sleep, and myoclonus of the trunk and limbs that gives the condition its name of dancing eyes and dancing feet. The clinical task is to recognise the syndrome, because it is under-recognised and frequently misdiagnosed as acute cerebellar ataxia, and to initiate the search for neuroblastoma with urine catecholamines and imaging of the chest and abdomen. [8]
Differential Diagnosis
The first differential question is whether the unsteadiness is cerebellar ataxia at all, because weakness, vertigo, and sensory disturbance all produce an unsteady gait that the family and sometimes the clinician label as ataxia. A child with an acute limp or limb weakness from a transient synovitis, a fracture, or a Guillain-Barre syndrome walks unsteadily but has a motor rather than a coordination problem, and the examination of tone, power, and reflexes separates them. Vertigo from vestibular neuritis or otitis media produces a reeling gait with prominent nausea and a subjective sense of spinning, and sensory ataxia from a neuropathy or a dorsal column lesion produces an unsteady gait that is worse in the dark and with eyes closed. [1]
The second and dangerous question is which acute ataxia this is, and the red-flag screen and the tempo answer it. A purely acute onset with a preceding viral illness, a normal conscious state, and a negative red-flag screen points to post-infectious acute cerebellar ataxia. An acute onset with encephalopathy, fever, or meningism points to central nervous system infection or acute disseminated encephalomyelitis and demands a lumbar puncture and imaging. An acute onset in a toddler with access to medications points to drug ingestion, and the toxicology history and screen are the diagnostic tools. A subacute or progressive course with headache, vomiting, or focal signs points to a posterior fossa tumour or cerebellitis and demands urgent imaging. [1] [3]
Cerebellar ataxia
- Wide-based staggering gait, present eyes open or closed
- Dysmetria, intention tremor, dysdiadochokinesia
- Slurred scanning speech, nystagmus
- Romberg negative
- Localises to the cerebellum or its connections
Sensory ataxia
- Unsteady gait, worse in the dark or with eyes closed
- Loss of position and vibration sense
- Romberg positive
- Absent reflexes if a neuropathy
- Localises to the dorsal columns or peripheral nerve
Vestibular ataxia
- Reeling gait with vertigo and nausea
- Prefers to lie still, worse with head movement
- No limb dysmetria
- Nystagmus may be present
- Vestibular neuritis or otitis media
Weakness mimicking ataxia
- Unsteady because of limb or trunk weakness
- Reduced tone, power, or reflexes
- Guillain-Barre, transient synovitis, fracture
- Not a coordination problem
- Examine tone and power first
The differential of chronic progressive ataxia is the genetic and metabolic panel. Friedreich ataxia is distinguished by the autosomal recessive pattern, the early onset, the absent lower limb reflexes with extensor plantars, and the cardiomyopathy. Ataxia telangiectasia is distinguished by the earlier onset in a toddler, the oculocutaneous telangiectasias, the recurrent sinopulmonary infections, and the raised serum alpha-fetoprotein. Ataxia with vitamin E deficiency and abetalipoproteinemia mimic Friedreich ataxia but are distinguished by a low serum vitamin E and, for abetalipoproteinemia, acanthocytes and a low lipid profile, and they are treatable with vitamin E supplementation. The episodic ataxias present with attacks of ataxia lasting minutes to hours rather than a progressive course, and they respond to acetazolamide. [5] [9]
Clinical & Bedside Assessment
The assessment of the ataxic child begins with the tempo and the conscious state, because these two features alone triage the urgency. Establish whether the ataxia came on over hours, days, or months, whether it is improving, static, or worsening, and whether the child is alert and interactive or drowsy and encephalopathic. A child with an altered conscious state is not a candidate for a diagnosis of acute cerebellar ataxia, and the search shifts immediately to encephalitis, meningitis, intoxication, and stroke. Ask about a preceding viral illness, about access to medications and substances, and about headache, vomiting, visual change, and behavioural or school decline. [1]
The gait is the single most informative sign in ataxia, and observing the child walk is non-negotiable. A wide-based, staggering, reeling gait that is present whether the eyes are open or closed is the hallmark of cerebellar ataxia. Ask the child to walk heel-to-toe along a line, to turn sharply, and to stand and walk in the dark or with the eyes closed. A child whose gait deteriorates markedly with eye closure has a positive Romberg sign and a sensory ataxia, which redirects the workup to the dorsal columns and the peripheral nerves. A child who refuses to walk, or who develops a head tilt or a new squint, has a posterior fossa lesion until proven otherwise. [1]
The coordination examination of the limbs confirms and localises the cerebellar lesion. Perform the finger-to-nose test, looking for dysmetria (past-pointing) and intention tremor that worsens as the finger approaches the nose, and the heel-to-shin test, looking for a wavering, unsteady movement as the heel slides down the opposite shin. Test rapid alternating movement with dysdiadochokinesia, asking the child to pronate and supinate the hands rapidly, and look for slurred, scanning speech and for nystagmus, particularly on lateral gaze. Examine the fundi for papilloedema, the eye movements for a cranial nerve palsy, and the reflexes and plantar responses, because the pattern of reflex change and the presence of an extensor plantar are the clues that separate Friedreich ataxia from a pure cerebellar lesion. [1]
[1] [3]Investigations
The guiding principle for the child who meets the clinical picture of post-infectious acute cerebellar ataxia, with a normal conscious state and a negative red-flag screen, is that investigation is minimal and imaging is not routine. The diagnosis is clinical, the child is observed, and no blood test, lumbar puncture, or scan is required when the pattern is classic. Over-investigation exposes the child to radiation and to incidental findings that raise anxiety, and it delays the reassurance and observation that constitute the treatment. The exceptions that warrant targeted testing are the toxicology screen when ingestion is suspected, and the inflammatory markers and lumbar puncture when central nervous system infection or acute disseminated encephalomyelitis is in the differential. [1] [2]
Neuroimaging is reserved for the child with a red flag, a focal deficit, an altered conscious state, or a progressive or unexplained course, and the modality is magnetic resonance imaging whenever possible. Computed tomography is reserved for the acute emergency where magnetic resonance imaging is unavailable or where the question is an acute haemorrhage or hydrocephalus that needs an immediate answer, because it delivers less detail of the posterior fossa and a radiation dose. The magnetic resonance imaging of a child with a posterior fossa tumour shows the mass, its effect on the fourth ventricle and the degree of hydrocephalus, and its imaging characteristics, which guide the neurosurgical and oncological management. [4]
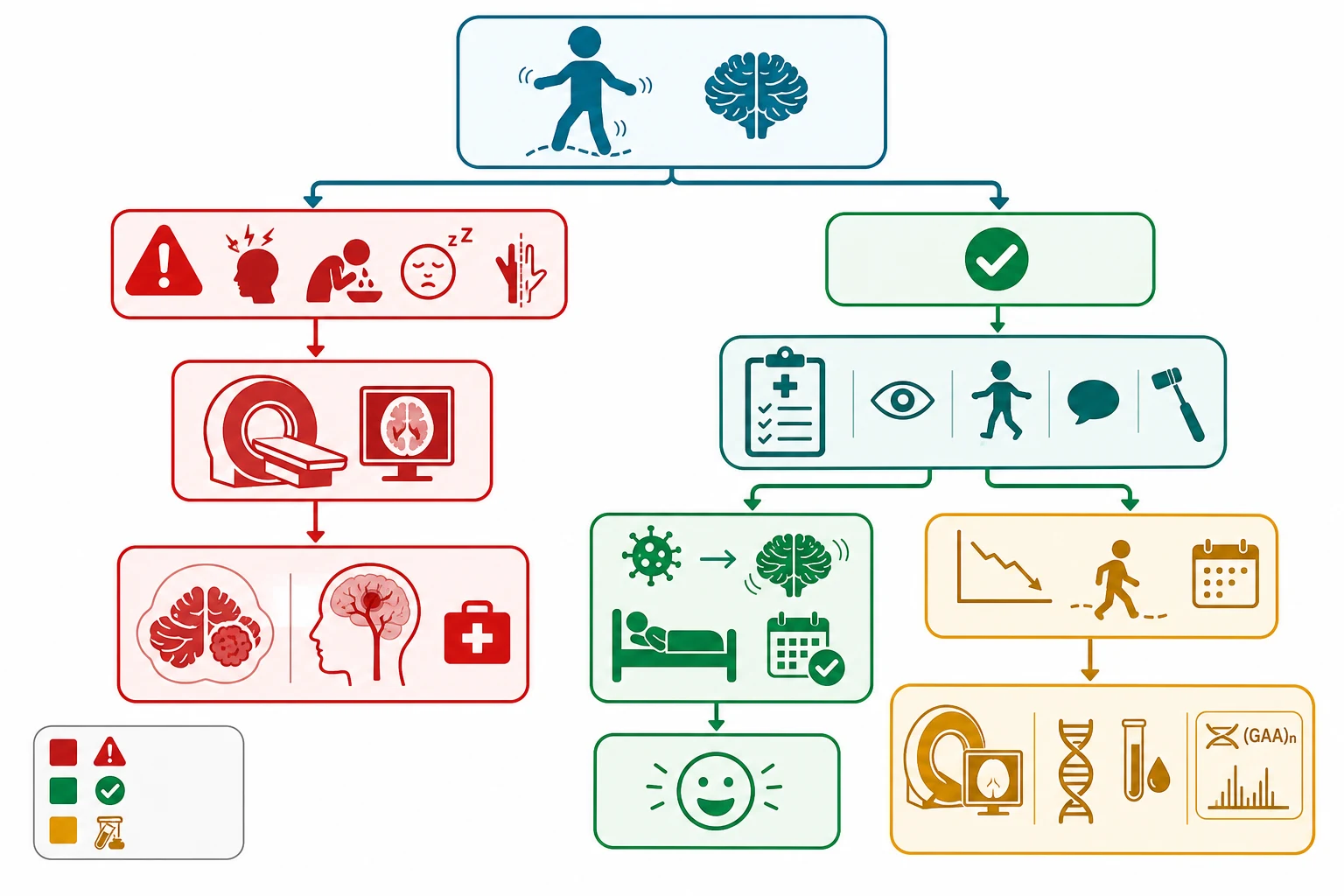
The investigation of chronic progressive ataxia is magnetic resonance imaging followed by a directed genetic and metabolic evaluation. The magnetic resonance imaging looks for cerebellar atrophy, a structural lesion, and the signal changes of a metabolic or neurodegenerative disorder, and it guides the differential before the genetic panel is sent. The genetic testing for Friedreich ataxia is the targeted test for the GAA trinucleotide repeat expansion in the FXN gene, and it is the first and definitive test when the clinical picture fits. A broader ataxia gene panel, a serum vitamin E level to exclude ataxia with vitamin E deficiency and abetalipoproteinemia, and a serum alpha-fetoprotein and immunoglobulin level to screen for ataxia telangiectasia are added when the clinical picture is less specific. [5] [9]
The child with suspected opsoclonus-myoclonus-ataxia syndrome has a dedicated workup for neuroblastoma, because around half harbour an occult neural crest tumour. This workup includes a urine collection for catecholamine metabolites, the homovanillic and vanillylmandelic acids, and magnetic resonance imaging or computed tomography of the chest, abdomen, and pelvis, with an iodine-123 metaiodobenzylguanidine scan when a primary lesion is not found on cross-sectional imaging. A normal imaging panel does not exclude the syndrome, which is treated on its clinical merits with immunotherapy. [8]
Management — Resuscitation
The resuscitation priority in the ataxic child is to secure the airway, breathing, and circulation and to treat the treatable before the diagnostic refinement. A child with an altered conscious state, whatever the underlying cause, is managed with airway protection and a structured approach to the comatose child, and the treatable causes of an encephalopathy, hypoglycaemia, meningitis, encephalitis, and drug ingestion, are sought and reversed. A child with a posterior fossa tumour and obstructive hydrocephalus who is deteriorating needs urgent neurosurgical decompression, and the recognition of raised intracranial pressure and the early involvement of the neurosurgical and retrieval teams is the resuscitation act that saves the life. [3] [4]
Drug ingestion, once identified, is managed with supportive care in a safe setting, with attention to airway protection, cardiovascular monitoring, and the specific antidotes where they apply. The toxicology history, the agents to which the child had access, and the toxicology screen guide the supportive plan, and the child is observed until the ataxia resolves. The principle is that most childhood ingestions resolve with observation and supportive care, and the role of the clinician is to ensure a safe setting, to identify the agent, and to recognise the small minority that need an antidote or escalation. [1]
[1] [3]Management — Definitive & Stepwise
The definitive management of post-infectious acute cerebellar ataxia is reassurance and observation, because the condition is self-limiting and the majority of children recover fully within weeks to months without specific treatment. The family is counselled on the expected course, the safety of a normal gait over the recovery period, and the warning signs that should prompt return, and the child is reviewed in clinic until the ataxia resolves. The child with acute cerebellitis who has cerebellar swelling on imaging, or signs of brainstem compression, is managed with neurosurgical and intensive care involvement, and occasionally with corticosteroids or decompression, but these are the rare severe end of the spectrum. [1] [2]
The definitive management of opsoclonus-myoclonus-ataxia syndrome is immunotherapy and the treatment of any underlying neuroblastoma, because the syndrome is autoimmune or paraneoplastic rather than a self-limiting post-infectious process. The international consensus perspective described a stepwise approach built on corticosteroids or adrenocorticotropic hormone, intravenous immunoglobulin, and, for refractory or relapsing disease, rituximab and cyclophosphamide, and the neuroblastoma, when found, is managed by the paediatric oncology and surgical teams. The motor symptoms often improve with immunotherapy, but the cognitive and behavioural sequelae frequently persist, which is why long-term developmental surveillance is part of the plan. [8]
FRIEDREICH: the bedside signature of Friedreich ataxia
The definitive management of Friedreich ataxia is multidisciplinary and now includes the first disease-modifying drug. Omaveloxolone was shown in the MOXIe trial to improve the modified Friedreich Ataxia Rating Scale score compared with placebo, and the delayed-start analysis of the extension showed that patients who began the drug later improved but did not fully catch up with those who started earlier, which supports early treatment. The clinical management guidelines by Corben and colleagues framed the broader care around the neurological, cardiac, endocrine, and rehabilitative needs, with surveillance for the cardiomyopathy that is the leading cause of death, for diabetes, for scoliosis, and for the disability that the multidisciplinary team addresses. [7] [12] [10]
Specific Subtypes & Scenarios
Acute cerebellitis is the severe end of the acute cerebellar ataxia spectrum, in which the cerebellum is swollen and inflamed, sometimes with effacement of the fourth ventricle and early hydrocephalus. The child may have more pronounced ataxia, headache, and drowsiness, and the magnetic resonance imaging shows cerebellar swelling and signal change, occasionally with herniation. The management shifts from observation to neurosurgical and intensive care involvement, with corticosteroids to reduce oedema and, in the severe case with brainstem compression, suboccipital decompression, because the swollen cerebellum can compress the brainstem and be life-threatening. [1]
Opsoclonus-myoclonus-ataxia syndrome deserves its own scenario because it is the most under-recognised of the acute ataxias and because its long-term prognosis is driven by the cognitive and behavioural outcome rather than by the motor recovery. The child presents with the dancing eyes of opsoclonus, myoclonus, ataxia, and extreme irritability, and around half have an occult neuroblastoma that is found on urine catecholamines and cross-sectional or metaiodobenzylguanidine imaging. The treatment is immunotherapy and the management of the tumour, and the family is counselled that the motor signs often resolve while the cognitive and behavioural problems frequently persist and require long-term developmental and educational support. [8]
Acute cerebellar ataxia
the safe common one
- Acute, post-infectious, preschool child
- Normal conscious state, no red flags
- Observation and reassurance
- Full recovery in most over weeks to months
- No specific treatment
Acute cerebellitis
the severe end
- Cerebellar swelling on imaging
- Headache, drowsiness, early hydrocephalus
- Neurosurgical and intensive care involvement
- Corticosteroids and occasionally decompression
- Brainstem compression can be life-threatening
Opsoclonus-myoclonus
the under-recognised one
- Dancing eyes, myoclonus, extreme irritability
- Occult neuroblastoma in around half
- Immunotherapy, not observation
- Motor recovery but persistent cognitive sequelae
- Long-term developmental surveillance
Ataxia telangiectasia presents with a progressive cerebellar ataxia in a toddler or preschool child, and the oculocutaneous telangictasias appear later, typically on the bulbar conjunctiva and the pinnae, which is why the early presentation may be mistaken for a chronic cerebellar problem of unknown cause. The condition is caused by biallelic mutations in the ATM gene, and it is associated with immunodeficiency and recurrent sinopulmonary infection, with a predisposition to malignancy, particularly leukaemia and lymphoma, and with a characteristically raised serum alpha-fetoprotein that supports the diagnosis. The management is supportive and multidisciplinary, with surveillance for infection and malignancy and the avoidance of radiation. [9]
Complications & Pitfalls
The complications of ataxia in children fall into the complications of the underlying disease and the complications of a missed or delayed diagnosis. A child with Friedreich ataxia develops the complications of a progressive multisystem disease, with cardiomyopathy that is the leading cause of death, diabetes, progressive scoliosis, and loss of mobility that typically requires a wheelchair within ten to fifteen years of onset. A child with opsoclonus-myoclonus syndrome develops the persistent cognitive and behavioural sequelae that are the principal long-term burden. A child with ataxia telangiectasia develops recurrent infection and malignancy. [5] [8] [9]
The avoidable pitfalls are several and recur in every examination. The first is the catastrophic error of discharging a posterior fossa tumour as a post-infectious acute cerebellar ataxia, which happens when the red-flag screen is omitted or when a headache, a head tilt, or a papilloedema is attributed to a viral illness. The second is the under-recognition of opsoclonus-myoclonus syndrome, which is mistaken for acute cerebellar ataxia and denied the immunotherapy and the neuroblastoma workup that change the outcome. The third is the misattribution of a sensory ataxia or a weakness to a cerebellar lesion, which misdirects the workup, and the fourth is the over-investigation of a classic post-infectious acute cerebellar ataxia with unnecessary imaging and blood tests that raise anxiety without benefit. [1] [3]
Missed tumour
- Discharging a posterior fossa tumour as post-infectious ataxia
- Omitting the red-flag screen
- Attributing headache, head tilt, or papilloedema to a virus
- The catastrophic error
- Urgent imaging for any red flag
Missed OMAS
- Opsoclonus mistaken for nystagmus
- Irritability dismissed as a viral prodrome
- No neuroblastoma workup
- No immunotherapy
- Persistent cognitive sequelae
Mislocalised lesion
- Sensory ataxia or weakness called cerebellar
- Workup misdirected to the cerebellum
- Romberg sign and reflexes separate them
- Examine tone and power first
- Guillain-Barre and neuropathy are the mimics
Over-investigation
- Imaging a classic post-infectious ataxia
- Incidental findings and anxiety
- Radiation from unnecessary computed tomography
- Observation is the treatment
- Reserve imaging for red flags and progression
The complications of a delayed diagnosis of a posterior fossa tumour are acute and severe, because a tumour that obstructs the fourth ventricle produces obstructive hydrocephalus that can precipitate a rapid neurological decline, and a cerebellar swelling can compress the brainstem. The Wilne systematic review of childhood central nervous system tumours documented the long interval from symptom onset to diagnosis that characterises this group, and the principle that flows from it is that a persistent or progressive ataxia, alone, is a red flag and an indication for imaging regardless of the absence of other features. [3]
Prognosis & Disposition
The prognosis of post-infectious acute cerebellar ataxia is excellent and is the foundation of the family consultation. The classic study by Connolly and colleagues established that the large majority of children recover fully, with the ataxia resolving over weeks to months without specific treatment and without neurological sequelae. A small minority have a more prolonged course or minor residual signs, and the rare child with acute cerebellitis and cerebellar swelling may have a more guarded outlook, but the rule for the classic case is reassurance and full recovery. The child is discharged with safety netting advice and reviewed in clinic until the gait is normal. [2] [1]
The prognosis of a posterior fossa tumour depends on the tumour type, the extent of resection, and the response to adjuvant therapy, and it is the domain of the paediatric neuro-oncology team rather than the generalist. The disposition is urgent referral for magnetic resonance imaging, neurosurgical assessment, and the staging and molecular classification that now drive the treatment and the prognosis. The role of the generalist and the emergency clinician is to recognise the red flags, to image, and to refer, and the early recognition shortens the interval from symptom onset to treatment that the Wilne review identified as the avoidable delay. [3] [4]
The prognosis of Friedreich ataxia is that of a progressive degenerative disease, with loss of ambulation typically within ten to fifteen years of onset and a reduced life expectancy driven by the cardiomyopathy. The natural history is heterogeneous, as the Rummey study documented, and the recent arrival of omaveloxolone as a disease-modifying therapy offers the prospect of slowing the neurological decline, particularly when started early. The disposition is a multidisciplinary clinic with cardiology, endocrinology, rehabilitation, and genetic counselling, and the family is supported through the progressive loss of function with attention to mobility, communication, cardiac health, and the psychosocial impact of a chronic diagnosis. [6] [11]
Special Populations
The child with disability and neurodiversity may struggle to articulate the quality of the unsteadiness or to cooperate with the coordination examination, so the behavioural signs of ataxia, a refusal to walk, a change in gait pattern, or a loss of a previously acquired motor skill, take greater diagnostic weight. The developmental history, the trajectory of motor milestones, and the carer's report of a change from the baseline are the high-yield elements, and the coordination examination is adapted to the developmental level. The threshold for imaging is lower when the examination is unreliable or when the history is one of regression rather than an acute post-infectious event. [1]
Aboriginal and Torres Strait Islander children and those in rural and remote settings may have reduced access to magnetic resonance imaging, paediatric neurology, and genetic testing, so the emphasis falls on the high-yield bedside screen and on the treatable and preventable causes. The delayed diagnosis of a posterior fossa tumour and of the hereditary ataxias is a recurring problem where access is limited, which is why a persistent or progressive ataxia is treated as a red flag and an indication for transfer and imaging regardless of the distance. The culturally safe consultation involves the family and the local health service in the decision to observe or to transfer. [3]
The adolescent with Friedreich ataxia faces the compounded burden of a progressive loss of mobility, the transition from paediatric to adult services, and the psychosocial impact of a chronic genetic diagnosis on identity, relationships, and future planning. The genetic counselling addresses the autosomal recessive inheritance, the carrier testing of siblings, and the reproductive options, and the transition of care is planned early with the adult neurology and cardiology services. The multidisciplinary team addresses the mobility, the communication, the cardiac and endocrine surveillance, and the mental health that the diagnosis affects. [10] [11]
Evidence, Guidelines & Regional Differences
The international evidence base for ataxia in children has converged on clear principles, though it rests more on cohort studies and consensus than on randomised trials for most causes. The classic study by Connolly and colleagues anchored the benign natural history of post-infectious acute cerebellar ataxia in 1994, and the consensus since has been to observe the classic case without routine imaging. The systematic review by Wilne and colleagues transformed the recognition of childhood central nervous system tumours by documenting the features at presentation and the long interval to diagnosis, and it underpins the red-flag screen that now governs the imaging decision. [2] [3]
The evidence base for Friedreich ataxia has advanced rapidly with the molecular understanding and the arrival of a disease-modifying therapy. The clinical features and the GAA repeat expansion in the FXN gene are settled, and the natural history study by Rummey and colleagues documented the heterogeneity of progression that shapes the prognosis and the trial design. The MOXIe trial by Lynch and colleagues showed that omaveloxolone improved the modified Friedreich Ataxia Rating Scale score, and the delayed-start analysis confirmed a durable benefit, which is the evidence that supports its use as the first disease-modifying therapy. The clinical management guidelines by Corben and colleagues framed the multidisciplinary care. [7] [12] [6] [10]
The evidence for opsoclonus-myoclonus-ataxia syndrome rests on the international consensus perspective by Rossor and colleagues, which described a standardised approach to diagnosis and treatment in the absence of randomised trials, and on the cohort studies that established the association with neuroblastoma and the response to immunotherapy. The evidence for ataxia telangiectasia rests on the comprehensive review by Rothblum-Oviatt and colleagues, which framed the multisystem manifestations and the surveillance for infection and malignancy. The 2025 Lancet Neurology review by Reetz and colleagues brought the Friedreich ataxia field up to date with the multisystem burden and the therapeutic horizon. [8] [9] [11]
The regional difference that matters is the selective versus the reflex use of neuroimaging. Across Australia and New Zealand, the United Kingdom, and Canada, the consensus is to observe a classic post-infectious acute cerebellar ataxia without routine imaging and to reserve magnetic resonance imaging for the child with a red flag or a progressive course. The temptation in settings with ready access to imaging is to scan for reassurance, but the consensus holds that over-imaging exposes the child to radiation and to incidental findings without benefit, and that the red-flag screen is the more discriminating and the safer gatekeeper. [1]
Exam Pearls
Diagnose post-infectious acute cerebellar ataxia from the clinical picture: a preschool child with an acute wide-based gait one to three weeks after a viral illness, a normal conscious state, and a negative red-flag screen. The prognosis is excellent, with the majority recovering fully over weeks to months, and the management is observation and reassurance without routine imaging. The single high-yield skill is the red-flag screen: headache, early-morning vomiting, an altered conscious state, papilloedema, a head tilt, a cranial nerve palsy, a progressive course, opsoclonus, and fever with meningism each demand urgent magnetic resonance imaging. [1] [2]
Recognise Friedreich ataxia from the constellation: a progressive ataxia with onset before twenty-five, absent lower limb reflexes, an extensor plantar response, loss of position and vibration sense, pes cavus, scoliosis, and hypertrophic cardiomyopathy, confirmed by the GAA repeat expansion in the FXN gene. The cardiomyopathy is the leading cause of death, and omaveloxolone is the first disease-modifying therapy, shown in the MOXIe trial to improve the modified Friedreich Ataxia Rating Scale score. Recognise opsoclonus-myoclonus-ataxia syndrome from the dancing eyes and extreme irritability, investigate for neuroblastoma, and treat with immunotherapy. Recognise ataxia telangiectasia from the progressive ataxia, the telangiectasias, the recurrent infection, and the raised serum alpha-fetoprotein. [5] [8] [9]
Localise the lesion at the bedside: a vermis lesion gives truncal and gait ataxia, a hemisphere lesion gives appendicular limb ataxia with dysmetria and dysdiadochokinesia, a dorsal column lesion gives sensory ataxia with a positive Romberg and loss of position and vibration sense, and a vestibular lesion gives vertigo and nausea. Separate cerebellar from sensory ataxia with the Romberg test, and separate ataxia from weakness with the examination of tone, power, and reflexes. The cardinal pitfall is the discharge of a posterior fossa tumour as a post-infectious ataxia, and the cardinal safeguard is the red-flag screen applied to every ataxic child. [1] [3]
References
- [1]Desai J, Mitchell WG Acute cerebellar ataxia, acute cerebellitis, and opsoclonus-myoclonus syndrome. J Child Neurol, 2012.PMID 22805251
- [2]Connolly AM, Dodson WE, Prensky AL, Rust RS Course and outcome of acute cerebellar ataxia. Ann Neurol, 1994.PMID 8210223
- [3]Wilne S, Collier J, Kennedy C, Koller K, Grundy R, Walker D Presentation of childhood CNS tumours: a systematic review and meta-analysis. Lancet Oncol, 2007.PMID 17644483
- [4]Brandão LA, Young Poussaint T Posterior Fossa Tumors. Neuroimaging Clin N Am, 2017.PMID 27889018
- [5]Cook A, Giunti P Friedreich's ataxia: clinical features, pathogenesis and management. Br Med Bull, 2017.PMID 29053830
- [6]Rummey C, Corben LA, Delatycki M, Wilmot G, Mathews K, Subramony SH, et al Natural History of Friedreich Ataxia: Heterogeneity of Neurologic Progression and Consequences for Clinical Trial Design. Neurology, 2022.PMID 35817567
- [7]Lynch DR, Chin MP, Delatycki MB, Subramony SH, Corti M, Hoyle JC, et al Safety and Efficacy of Omaveloxolone in Friedreich Ataxia (MOXIe Study). Ann Neurol, 2021.PMID 33068037
- [8]Rossor T, Yeh EA, Khakoo Y, Angelini P, Bellimapura S, Benseler S, et al Diagnosis and Management of Opsoclonus-Myoclonus-Ataxia Syndrome in Children: An International Perspective. Neurol Neuroimmunol Neuroinflamm, 2022.PMID 35260471
- [9]Rothblum-Oviatt C, Wright J, Lefton-Greif MA, McGrath-Morrow SA, Crawford TO, Lederman HM Ataxia telangiectasia: a review. Orphanet J Rare Dis, 2016.PMID 27884168
- [10]Corben LA, Collins V, Milne S, Farmer J, Florde J, Hynson L, et al Clinical management guidelines for Friedreich ataxia: best practice in rare diseases. Orphanet J Rare Dis, 2022.PMID 36371255
- [11]Reetz K, Lischewski SA, Dogan I, Didszun C, Roeh A, Romanzetti S, et al Friedreich's ataxia-a rare multisystem disease. Lancet Neurol, 2025.PMID 40541211
- [12]Lynch DR, Chin MP, Boesch S, Delatycki MB, Giunti P, Goldsberry A, et al Efficacy of Omaveloxolone in Friedreich's Ataxia: Delayed-Start Analysis of the MOXIe Extension. Mov Disord, 2023.PMID 36444905