Paeds · neurology-neurodisability-and-neuromuscular
Congenital myopathies and muscular dystrophies
Also known as Congenital muscular dystrophy · Congenital myopathy · Merosin-deficient congenital muscular dystrophy · Collagen VI-related dystrophy · Dystroglycanopathy · Central core disease · Nemaline myopathy · Centronuclear myopathy
Fellowship guide to congenital myopathies and muscular dystrophies in children. Covers the division into the dystrophic congenital muscular dystrophies and the structurally disordered congenital myopathies, the merosin-deficient LAMA2 form with its white matter changes and raised creatine kinase, the collagen VI spectrum of Ullrich and Bethlem with contractures and distal hyperlaxity, the dystroglycanopathies of Walker-Warburg and muscle-eye-brain disease with cobblestone lissencephaly and eye malformations, the LMNA and SEPN1 and titin forms, the congenital myopathies of central core disease with ryanodine receptor RYR1 mutations and malignant hyperthermia risk, nemaline myopathy with its rod bodies, and centronuclear myotubular myopathy with neonatal respiratory failure, the creatine kinase as the first fork, the brain and eye examination, the muscle biopsy findings of cores and rods and central nuclei, the multigene panel replacing biopsy, and the multidisciplinary management of respiratory support, nutrition, contractures, scoliosis, and rehabilitation.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
Overview & Definition
A baby born floppy, with a weak cry and a poor suck and limbs that slip through the hands, presents one of the defining diagnostic challenges of paediatric neurology, and congenital muscle disease is the explanation a clinician must not miss. Congenital myopathies and congenital muscular dystrophies are a group of inherited disorders of the structural and contractile proteins of skeletal muscle that present from birth or early infancy with hypotonia, weakness, and reduced or absent tendon reflexes, and they share a bedside picture that the paediatrician recognises as the floppy infant of neuromuscular origin. They are genetically diverse but clinically unified, and the task at the bedside is to separate the child whose creatine kinase is high and whose brain and eyes are involved from the child whose creatine kinase is normal and whose problem is confined to the muscle fibre. [1][5]
The single distinction that organises the whole field is the one between the muscular dystrophies and the myopathies, and it rests on what the muscle looks like under the microscope. In a congenital muscular dystrophy the muscle is dystrophic, meaning it shows active degeneration and regeneration with fibrosis and fatty replacement, because the protein that anchors the fibre to its surrounding basement membrane is defective and the fibre tears with every contraction. In a congenital myopathy the muscle is structurally disordered but not actively dying, and it shows a stable and characteristic abnormality such as central cores, nemaline rods, or centrally placed nuclei, because the proteins that build and organise the contractile apparatus are faulty. This distinction predicts the creatine kinase, the prognosis, and the genes, and it is the first fork in the diagnostic road. [1][4]
Why these diseases matter above all is that they can stop a child breathing even while the child is still walking, and they can hide a treatable or lethal complication such as malignant hyperthermia or cardiac arrhythmia behind a stable-looking weakness. The disease-modifying landscape is shifting, with emerging antisense and gene therapies for several forms, but the backbone of care remains the multidisciplinary management of respiratory failure, feeding difficulty, contractures, scoliosis, and rehabilitation. The paediatrician who recognises the floppy infant, sends the creatine kinase, and refers early to a neuromuscular service is the one who changes the trajectory. [2][3]
Classification
The practical classification runs on two questions that the clinician can answer at the bedside and the microscope, and both change the gene panel and the prognosis. The first question is whether the muscle is dystrophic or merely disordered, which separates the congenital muscular dystrophies from the congenital myopathies and predicts the creatine kinase. The second question is whether the brain, the eyes, or the heart are involved, which separates the merosin-deficient and the dystroglycanopathy forms of muscular dystrophy from the collagen VI and LMNA forms and from the pure-muscle myopathies. [1]
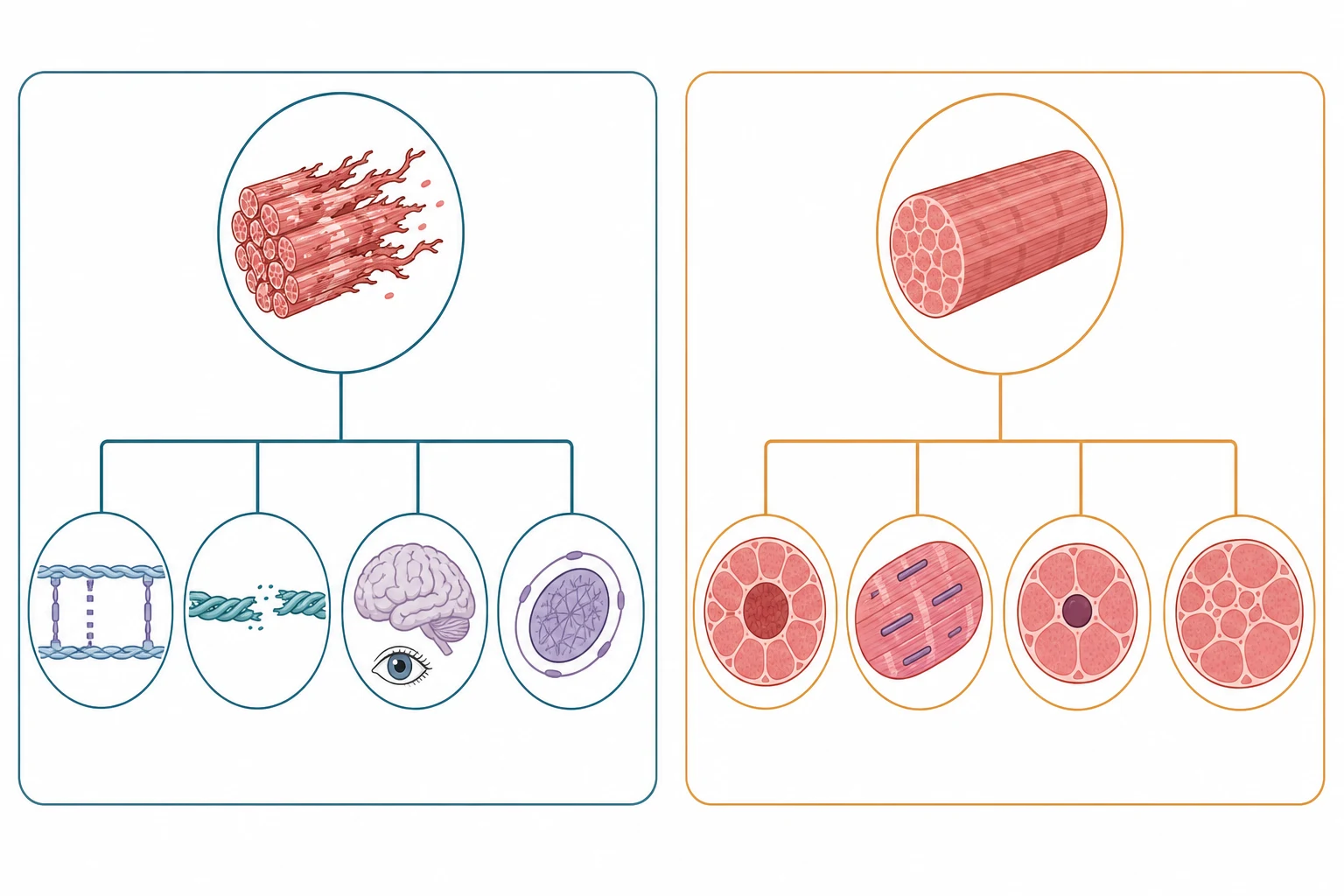
The congenital muscular dystrophies divide by the protein that is defective. Merosin-deficient congenital muscular dystrophy, caused by mutations in the LAMA2 gene that encodes the laminin-alpha-2 chain of the basement membrane, presents with severe congenital hypotonia and weakness, a markedly raised creatine kinase, diffuse white matter change on brain magnetic resonance imaging, and epilepsy, and it is the commonest form in Northern European populations. The collagen VI-related dystrophies form a spectrum from the severe Ullrich congenital muscular dystrophy, with proximal contractures and distal joint hyperlaxity and follicular hyperkeratosis, to the milder Bethlem myopathy, and they carry a normal or mildly raised creatine kinase and a distinctive restrictive respiratory failure. [1][10]
The dystroglycanopathies are the forms with brain and eye involvement, and they arise from defective glycosylation of alpha-dystroglycan, a protein that links the muscle fibre to the extracellular matrix and guides brain development. Walker-Warburg syndrome is the most severe, with cobblestone lissencephaly, hydrocephalus, eye malformations such as microphthalmia and retinal dysplasia, and severe congenital weakness, and muscle-eye-brain disease and Fukuyama congenital muscular dystrophy sit at milder points on the same spectrum. The LMNA-related congenital muscular dystrophy is the form with early axial and upper-limb weakness, a dropped head, spine rigidity, and a high risk of cardiac conduction disease, and it carries a normal or mildly raised creatine kinase. SEPN1-related myopathy brings a rigid spine and early respiratory failure, and congenital titinopathy from TTN mutations is among the most severe of all, with congenital contractures and respiratory failure at birth. [1][9][6]
The congenital myopathies divide by the structural abnormality on the biopsy. Central core disease, the commonest congenital myopathy, is caused most often by mutations in the ryanodine receptor gene RYR1 and shows central areas of absent oxidative enzyme activity called cores, and it carries the life-threatening association of malignant hyperthermia susceptibility. Nemaline myopathy shows rod-shaped bodies on the Gomori trichrome stain and is caused most often by mutations in the nebulin NEB or the skeletal muscle actin ACTA1 genes, and it presents with facial weakness and a slender build across a severity spectrum. Centronuclear myopathy shows centrally placed nuclei and includes the severe X-linked myotubular myopathy from MTM1 mutations, which causes neonatal respiratory failure, and the milder autosomal dominant DNM2 form. Congenital fibre-type disproportion and core-rod myopathy complete the group. [4][11]
Epidemiology & Risk Factors
Congenital muscle disease is individually rare but collectively common enough that every general paediatrician will meet a case, and the combined incidence of the congenital muscular dystrophies sits around one in twenty thousand live births, with the congenital myopathies at a similar order. Merosin-deficient congenital muscular dystrophy is the commonest form in Northern Europe, the dystroglycanopathies and Fukuyama disease are commoner in populations with founder mutations, and collagen VI-related disease is among the commonest worldwide. Central core disease from RYR1 is the single commonest congenital myopathy, and the severe X-linked myotubular myopathy affects boys with an incidence of around one in fifty thousand male births. [1][5]
The risk factor for every form is a pathogenic variant in the relevant gene, and the inheritance pattern is the second piece of family information the clinician gathers. Merosin-deficient disease and nemaline myopathy are mostly autosomal recessive, the collagen VI spectrum is autosomal recessive for Ullrich and autosomal dominant for Bethlem, the LMNA and DNM2 forms are autosomal dominant, and the myotubular myopathy is X-linked recessive. A family history of a floppy infant who died, of consanguinity, or of male siblings affected with neonatal respiratory failure raises the prior probability and directs the panel. [1][4]
Pathophysiology
The mechanism that unites these diseases is the failure of a single structural or contractile protein of the muscle fibre, and the consequence depends on whether that protein holds the fibre to its matrix or builds the contractile machinery inside it. In the congenital muscular dystrophies the defective protein sits in the basement membrane or the dystrophin-glycoprotein complex that anchors the fibre to the extracellular matrix, so every contraction tears the membrane, allowing calcium influx and fibre necrosis, and the muscle shows cycles of degeneration, regeneration, fibrosis, and fatty replacement. In the congenital myopathies the defective protein builds or regulates the contractile apparatus or the calcium release channel, so the fibre survives but is structurally disordered, and the biopsy shows the stable cores, rods, or central nuclei that name the disease. [5][4]
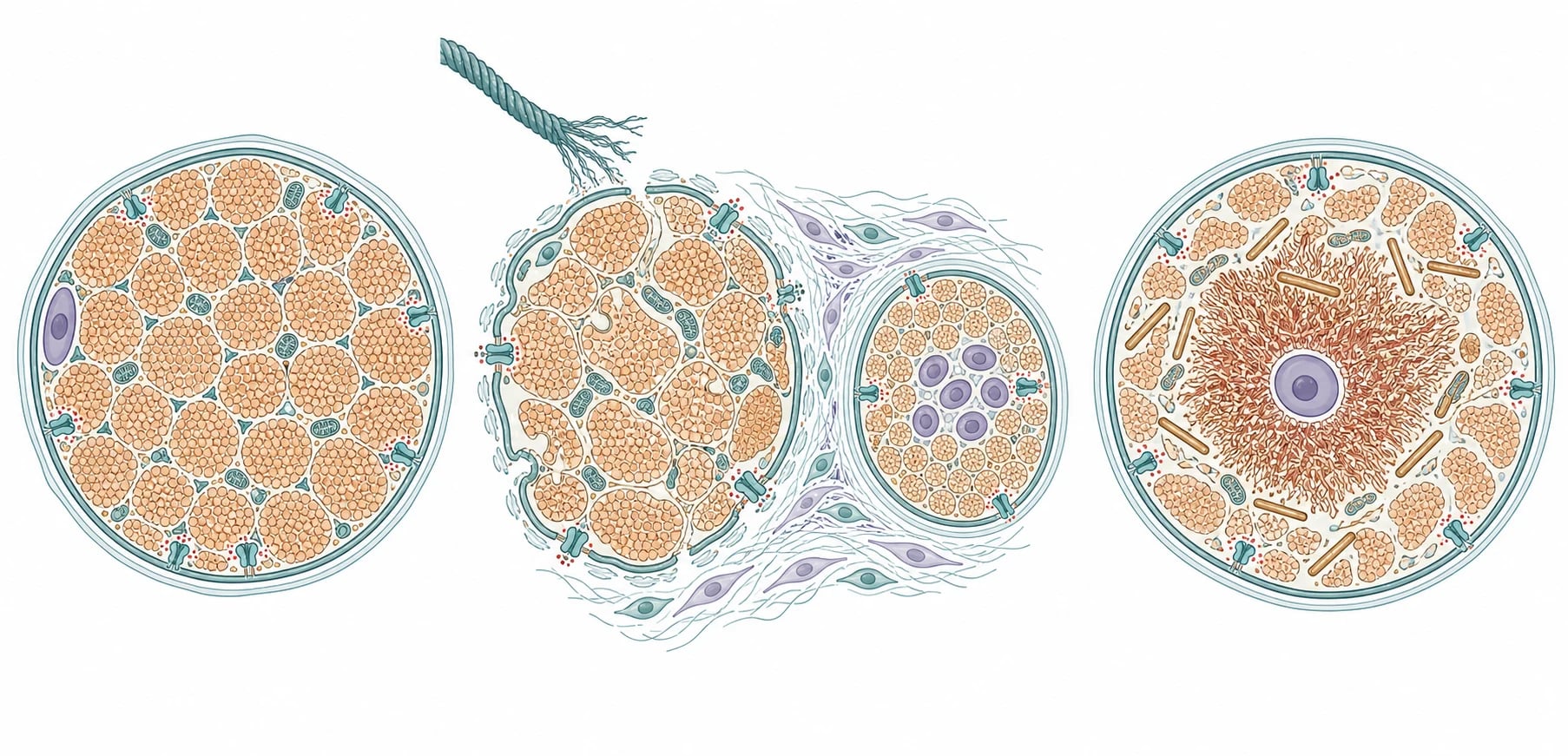
The merosin-deficient form shows this tearing mechanism in its purest form. The laminin-alpha-2 chain, encoded by LAMA2, is a key strand of the basement membrane that laminin uses to anchor the muscle fibre, and its absence strips the membrane of its anchor, so the fibre necroses and the creatine kinase leaks out in large quantities. The same laminin-alpha-2 chain is expressed in the white matter of the brain and in the peripheral nerve, which explains the diffuse white matter change on imaging, the demyelinating peripheral neuropathy, and the epilepsy that accompany the muscle disease, while the cognition is typically spared. [10]
The dystroglycanopathies add a developmental dimension, because alpha-dystroglycan is not only a muscle anchor but also a guide for the migration of neurons in the developing brain. When glycosylation of alpha-dystroglycan fails, the neurons over-migrate past the cortical surface and form the cobblestone lissencephaly that defines Walker-Warburg syndrome, and the same glycosylation failure disrupts the development of the eye, producing the microphthalmia, cataract, glaucoma, and retinal dysplasia that accompany the brain and muscle disease. The brain malformation, not the muscle weakness, sets the prognosis in these children. [9]
The collagen VI mechanism carries its own therapeutic story. Collagen VI is secreted by the muscle fibre and the surrounding fibroblasts and forms a beaded microfibril network that protects the fibre from mechanical stress, and its absence in Ullrich disease triggers mitochondrial dysfunction and apoptosis through the opening of the permeability transition pore. The Angelin 2007 study showed that cyclosporin and its non-immunosuppressive analogue Debio-025 close the pore and rescue the mitochondrial dysfunction in cells from patients, which opened the first mechanistic therapy for a congenital muscular dystrophy. The ryanodine receptor mechanism in central core disease is the link to malignant hyperthermia, because the same calcium release channel that forms the cores also leaks uncontrolled calcium into the muscle in response to volatile anaesthetics and succinylcholine, triggering the hypermetabolic crisis. [8][11]
Clinical Presentation
The bedside picture is the floppy infant, and the features that point to a neuromuscular rather than a central cause are the distribution and the reflexes. The hypotonia is the presenting sign, and in a neuromuscular cause it is accompanied by weakness, so the limbs move little against gravity and the baby feels limp and slips through the hands. The tendon reflexes are reduced or absent, which is the single most useful sign in separating a neuromuscular from a central cause of floppiness, because a central cause usually preserves or exaggerates the reflexes. The cry is weak, the suck is poor, and the baby has feeding difficulty and slow weight gain, and there may be joint contractures from reduced fetal movement, the so-called arthrogryposis. [1][4]
The face and the eyes narrow the diagnosis further. Facial weakness with a long myopathic face, a tented upper lip, and ptosis points to a congenital myopathy, especially the nemaline and centronuclear forms, and ophthalmoplegia with the eyes held still is characteristic of the myotubular and the centronuclear myopathies. A high arched palate, a thin build, and respiratory difficulty from birth point to the severe congenital myopathies. In the muscular dystrophies the face is often less affected, and the clues lie instead in the contractures, the skin, and the brain. [4]
The extramuscular features are the ones that target the gene. White matter change on the brain magnetic resonance imaging with epilepsy and a markedly raised creatine kinase points to merosin-deficient disease. Cobblestone lissencephaly with eye malformations and severe weakness points to a dystroglycanopathy such as Walker-Warburg syndrome, and that combination carries the grimmest prognosis in the field. Proximal contractures with distal joint hyperlaxity and follicular hyperkeratosis of the skin points to collagen VI-related Ullrich disease, and the same child will develop restrictive respiratory failure even while still walking. A dropped head with spine rigidity and upper-limb weakness points to LMNA disease and its cardiac risk. A rigid spine with early respiratory failure and preserved walking points to SEPN1 disease. [1][7]
FLOPPY
The tempo and the severity span a wide spectrum within each gene, and the clinician must not assume the worst or the best from the label alone. A child with collagen VI-related disease may walk independently and yet need nocturnal ventilation by the teenage years, while a child with congenital titinopathy or myotubular myopathy may need ventilation and a tracheostomy from the neonatal period. The motor trajectory is usually static or slowly progressive in the myopathies and more variable in the muscular dystrophies, and the cognition is usually spared except in the dystroglycanopathies, where the brain malformation dominates. [2][6]
Differential Diagnosis
The differential of the floppy infant is the practical frame, and the first fork is between a central and a peripheral cause. A central cause, such as hypoxic-ischaemic encephalopathy, chromosomal disorder, or metabolic encephalopathy, usually presents with altered alertness, seizures, dysmorphic features, and preserved or brisk reflexes, and the floppiness is a hypotonia without proportionate weakness. A peripheral neuromuscular cause presents with a baby who is alert and socially engaged but weak, with reduced or absent reflexes, and the congenital muscle diseases sit in this group alongside spinal muscular atrophy, the congenital myasthenic syndromes, and the hereditary motor and sensory neuropathies. [1]
The next fork separates the muscle diseases from their neuromuscular mimics, and the creatine kinase and the neurophysiology do the work. Spinal muscular atrophy, caused by survival motor neuron gene deletion, presents with severe symmetric proximal weakness and tongue fasciculations and a normal creatine kinase, and it is the single most important mimic to exclude, because it has a specific disease-modifying therapy. The congenital myasthenic syndromes present with fatigable weakness involving the eyes and the face and a normal creatine kinase, and they respond to specific drug therapy. A markedly raised creatine kinase at presentation raises the muscular dystrophies above the myopathies and the mimics, and a neurophysiology showing a myopathic pattern confirms the muscle origin. [1][5]
The remaining mimics each carry a specific discriminator. Prader-Willi syndrome presents with severe neonatal hypotonia and feeding difficulty but with preserved reflexes and distinctive dysmorphism and later hyperphagia. Metabolic myopathies such as the glycogen and fatty-acid oxidation disorders present with exercise intolerance or episodic collapse rather than static congenital weakness. The congenital myopathies must also be distinguished among themselves, because the malignant hyperthermia risk of RYR1 disease and the cardiac risk of LMNA disease change the management even when the weakness looks similar. The clinching combination is the alert but weak baby with reduced reflexes, the creatine kinase level, and the brain and eye pattern, confirmed by the gene panel. [4][11]
Clinical & Bedside Assessment
The bedside assessment begins with the general observation of the alert but weak baby, because the combination of a socially engaged infant with hypotonia and weakness is the signature of a neuromuscular cause. Assess the tone by the traction test, the ventral suspension, and the scarf sign, and assess the power by the spontaneous movement against gravity and the anti-gravity posture. Examine the tendon reflexes at the biceps, the knees, and the ankles, because their reduction or absence in a weak limb is the single sign that moves the diagnosis from central to peripheral. Watch the respiratory effort, the intercostal recession, and the paradoxical abdominal breathing that signal diaphragmatic weakness. [1]
The focused examination then looks for the features that target the gene. Examine the face for the long myopathic face, the tented lip, the ptosis, and the ophthalmoplegia that point to the myopathies. Examine the joints for the proximal contractures and the distal hyperlaxity of collagen VI disease, and examine the skin for the follicular hyperkeratosis that accompanies it. Palpate the skull and the anterior fontanelle for the hydrocephalus of Walker-Warburg syndrome, and examine the eyes with the red reflex and the ophthalmoscope for the microphthalmia and the cataract. Examine the spine for the rigidity and the scoliosis of LMNA and SEPN1 disease, and examine the heart for the rhythm disturbance that signals the cardiac risk. [1][7]
The respiratory assessment is the part that saves lives, and it is the single most examined bedside skill in the topic, because a child can walk yet still develop diaphragmatic respiratory failure. Measure the forced vital capacity seated and supine in any cooperative child, and arrange an overnight sleep study, because the supine fall and the nocturnal hypoventilation appear before the daytime symptoms. Assess the cough strength and the bulbar function, because a weak cough and pooled secretions predict aspiration and chest infection. Palpate the abdomen and assess the swallow and the feeding, because feeding difficulty and failure to thrive are nearly universal and drive the decision for gastrostomy. [2][3]
In ANZ practice, the diagnosis and the care of congenital muscle disease are centralised in the tertiary paediatric neuromuscular services, and the general paediatrician who meets a floppy infant sends the creatine kinase and the survival motor neuron gene test and refers early. Retrieval networks coordinate the transfer of any child with respiratory failure, and the multidisciplinary clinics bring together neurology, respiratory, gastroenterology, orthopaedics, and rehabilitation. The gene panel and the muscle biopsy are performed in the specialist centre, and the family is offered genetic counselling and prenatal diagnosis. The threshold to refer and to transfer is low, because early respiratory and nutritional support change the trajectory. [2]
Investigations
The investigations confirm the muscle origin, fork the diagnosis, and target the gene, and the creatine kinase is the test that does the first two at once. A markedly raised creatine kinase, in the thousands of units per litre, points to a congenital muscular dystrophy, especially the merosin-deficient and the dystroglycanopathy forms, and it raises the muscular dystrophies above the myopathies and the mimics. A normal or mildly raised creatine kinase points to a congenital myopathy or a mimic such as spinal muscular atrophy, and it directs the workup toward the survival motor neuron gene and the muscle biopsy. The creatine kinase is sent on every floppy infant with reduced reflexes, and it is the single highest-yield first test. [1]
The brain magnetic resonance imaging and the eye examination fork the muscular dystrophies among themselves. Diffuse white matter abnormality with a structurally normal brain and epilepsy points to merosin-deficient disease, while cobblestone lissencephaly with hydrocephalus or encephalocoele and eye malformations points to a dystroglycanopathy such as Walker-Warburg syndrome. A normal brain with normal eyes points to the collagen VI, LMNA, SEPN1, and titin forms of muscular dystrophy and to the congenital myopathies, and it narrows the panel. The nerve conduction studies add the demyelinating peripheral neuropathy of merosin-deficient disease and the myopathic pattern of the muscle diseases. [1][9]
The muscle biopsy was once the diagnostic standard and it remains definitive when the gene panel is negative, and it shows the cores, the rods, and the central nuclei that name the congenital myopathies and the degeneration, the fibrosis, and the fatty replacement that define the congenital muscular dystrophies. Immunohistochemistry for merosin, collagen VI, and alpha-dystroglycan adds the protein pattern. In modern practice, however, the next-generation sequencing muscle panel has moved to first-line, because it is faster, it is less invasive, and it identifies the gene in the majority of children, and the biopsy is reserved for the panel-negative case or when the histological pattern is itself the question. The gene panel covers the major genes LAMA2, the collagen VI genes COL6A1 to COL6A3, the dystroglycanopathy genes, LMNA, SEPN1, TTN, RYR1, NEB, ACTA1, MTM1, and DNM2. [1][4]
[2] [3]The supporting investigations complete the assessment. The echocardiogram and the electrocardiogram screen for the cardiomyopathy and the conduction disease that complicate the LMNA and some dystroglycanopathy forms, and the Holter monitor is added when the conduction risk is high. The overnight sleep study screens for the nocturnal hypoventilation that drives the decision to start non-invasive ventilation, and the respiratory function tests track the forced vital capacity over time. The swallowing assessment and the dietetic review guide the feeding plan, and the audiology and the ophthalmology review complete the baseline. The diagnostic workup is coordinated with the genetic counsellor, because the gene result sets the inheritance, the prognosis, and the reproductive options. [2][7]
Management — Resuscitation
Resuscitation in the neonate or infant with congenital muscle disease secures the airway and the breathing and the feeding while the diagnostic workup runs. Any baby with respiratory distress, a weak cough, or ventilatory failure is admitted to a neonatal or paediatric intensive care unit, and the airway is secured and the breathing supported with non-invasive or invasive ventilation. The threshold to ventilate is driven by the work of breathing and the gas exchange, and the child with a congenital titinopathy or a myotubular myopathy may need prolonged ventilation from birth, while the child with a milder myopathy may need only transient support for a chest infection. [2][6]
The feeding and the airway protection are addressed in parallel, because the weak suck and the bulbar weakness cause aspiration and failure to thrive. A nasogastric tube is placed for feeding, and the swallow is assessed, and the decision for a gastrostomy is made early in the severe forms, because it secures the nutrition and the medication route for the long term. The fluid and the electrolyte balance and the temperature are maintained, and the baby is monitored for the seizures of merosin-deficient disease and for the cardiac arrhythmia of LMNA disease. The diagnostic workup, including the creatine kinase, the gene panel, and the brain imaging, is sent in parallel, and the paediatric neurology and the neuromuscular service are involved from the outset. [2][3]
The workup is coordinated with the intensive care and the surgical teams, because the child with congenital muscle disease is at risk of complications under anaesthesia. Any child with a suspected or confirmed RYR1 mutation is flagged for malignant hyperthermia risk before any anaesthetic, and the volatile anaesthetics and succinylcholine are avoided, and a trigger-free technique is planned with the anaesthetist. The family is given a medic-alert and counselled on the risk, because the malignant hyperthermia crisis is preventable with avoidance and treatable with dantrolene if it occurs. [3][11]
Management — Definitive & Stepwise
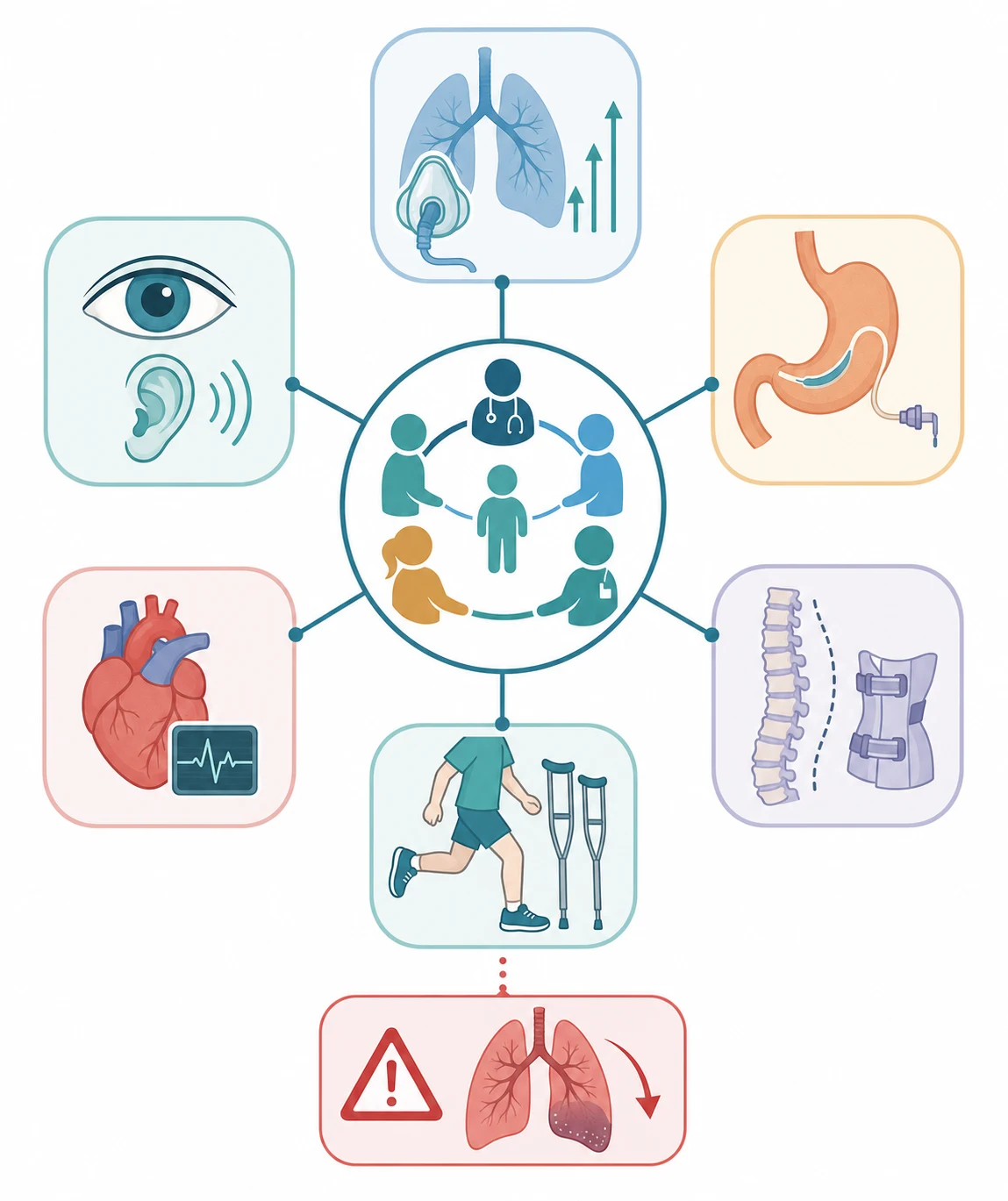
The definitive management is multidisciplinary supportive care, because most congenital muscle diseases have no curative therapy, and the disease-modifying landscape is still emerging for most genes. The care is built around the prevention and the management of the four complications that dominate the course, which are the respiratory failure, the feeding difficulty and the malnutrition, the contractures and the scoliosis, and the cardiac involvement. A coordinated neuromuscular clinic that brings together the neurology, the respiratory, the gastroenterology, the orthopaedic, and the rehabilitation teams is the standard of care set out in the international consensus statements. [2][3]
The respiratory management is the part that changes the survival, and it is built on the early detection and the treatment of the ventilatory failure. The forced vital capacity and the overnight sleep study are tracked at least annually in any child with a respiratory-risk form such as collagen VI, SEPN1, myotubular, or merosin-deficient disease. Non-invasive ventilation, most often bilevel positive airway pressure delivered overnight, is started for the symptomatic nocturnal hypoventilation or the objective sleep-study finding, and it is the intervention that most extends the life. The cough assistance, with the mechanical insufflation-exsufflation device, and the airway clearance are added to prevent the chest infections that drive the acute deterioration, and a minority of children, especially those with myotubular myopathy, need a tracheostomy and long-term invasive ventilation. [2]
Respiratory and nutritional support in congenital muscle disease (paediatric)
The nutritional and the orthopaedic management run alongside the respiratory care. The gastrostomy secures the nutrition and the medication route in any child with feeding difficulty or failure to thrive, and it is performed early in the severe forms, because the malnutrition worsens the weakness and the respiratory reserve. The contractures are managed with passive stretching, splinting, and serial casting, and the scoliosis, which is driven by the truncal weakness, is monitored with the spinal radiograph and managed with the seating and the bracing, with the spinal fusion reserved for the progressive and severe curve that threatens the respiratory function. The physiotherapy, the occupational therapy, and the standing frames maintain the range of movement and the bone density and the function, and the rehabilitation is a lifelong process. [2]
The management ladder, from diagnosis to transition
At diagnosis: Confirm the gene with the next-generation sequencing panel; send the baseline creatine kinase, brain MRI, echocardiogram, and sleep study; refer to the multidisciplinary neuromuscular clinic
Infancy: Secure the feeding with nasogastric or gastrostomy; support the breathing as needed; start physiotherapy and stretching for the contractures; flag any RYR1 case for malignant hyperthermia
Early childhood: Track the forced vital capacity and the overnight sleep study annually in the respiratory-risk forms; start nocturnal non-invasive ventilation for the objective hypoventilation; add cough assistance and airway clearance
School age: Manage the scoliosis with seating, bracing, and the spinal fusion for the severe curve; maintain the nutrition and the bone density; support the education and the social development
Adolescence: Continue the respiratory and the nutritional surveillance; screen the cardiac function annually in LMNA and the dystroglycanopathy forms; plan the transition to the adult neuromuscular service
Throughout: Genetic counselling and prenatal diagnosis for the family; psychological and social support; coordination with the school and the community services
The cardiac and the specific gene surveillance complete the management. The echocardiogram and the electrocardiogram, and often the Holter monitor, screen for the cardiomyopathy and the conduction disease that complicate the LMNA and some dystroglycanopathy forms, and the LMNA form carries a risk of sudden cardiac death even with a preserved ejection fraction, so the cardiac surveillance is mandatory from diagnosis and the pacemaker or the defibrillator is considered when the conduction abnormality appears. The ophthalmology review screens for the eye malformations and the visual impairment of the dystroglycanopathies, and the audiology review completes the baseline. The family is offered the genetic counselling and the prenatal or the preimplantation diagnosis, because the gene result sets the recurrence risk and the reproductive options. [7][9]
Specific Subtypes & Scenarios
Merosin-deficient congenital muscular dystrophy, caused by complete deficiency of the laminin-alpha-2 chain encoded by LAMA2, presents with severe congenital hypotonia and weakness, a markedly raised creatine kinase, diffuse white matter change on the brain imaging, and epilepsy in around a third of the children, while the cognition is usually spared. The children rarely achieve independent walking, and they develop the contractures and the restrictive respiratory failure that dominate the course. The diagnosis rests on the gene panel and on the immunohistochemistry or the western blot for merosin on the muscle biopsy, and the management is the multidisciplinary supportive care with the respiratory and the orthopaedic focus. A milder, partial-deficiency form and a limb-girdle muscular dystrophy phenotype broaden the LAMA2 spectrum. [1][10]
The collagen VI-related dystrophies form the spectrum from Ullrich congenital muscular dystrophy to Bethlem myopathy, and they carry the distinctive combination of proximal joint contractures with distal joint hyperlaxity, the follicular hyperkeratosis of the skin, and a normal or mildly raised creatine kinase. Ullrich disease presents at birth with the contractures and the weakness, and the child develops the restrictive respiratory failure, often in the first or the second decade, even while still walking, which makes the sleep-study surveillance essential. Bethlem myopathy is the milder dominant form, with the finger contractures and the later-onset weakness. The Angelin 2007 study of the mitochondrial permeability transition pore opened the mechanistic therapy, and the cyclosporin analogue is under investigation. [1][8]
The dystroglycanopathies are the forms with the brain and the eye, and they arise from the defective glycosylation of the alpha-dystroglycan. Walker-Warburg syndrome is the most severe, with the cobblestone lissencephaly, the hydrocephalus or the encephalocoele, the severe eye malformations, and the profound weakness, and the prognosis is grim, with most children dying in infancy. Muscle-eye-brain disease and Fukuyama congenital muscular dystrophy sit at milder points on the spectrum, with the less severe brain malformation and the longer survival. The brain malformation, not the muscle weakness, sets the prognosis, and the management is supportive with the feeding, the seizure control, and the comfort. [9]
The congenital myopathies each carry their own scenario. Central core disease from RYR1 is the commonest, and it presents with the congenital hypotonia, the delayed motor milestones, the congenital hip dislocation, and the later scoliosis, and it carries the malignant hyperthermia risk that demands the anaesthetic precautions. Nemaline myopathy from the nebulin NEB or the actin ACTA1 presents with the facial weakness, the slender build, and the respiratory involvement across a severity spectrum from the severe congenital form to the adult-onset form. The centronuclear myopathies show the centrally placed nuclei, and the severe X-linked myotubular myopathy from MTM1 presents with the neonatal respiratory failure, the ophthalmoplegia, and the macrocephaly in a boy, and it often needs the tracheostomy and the long-term ventilation, while the DNM2 form is the milder autosomal dominant variant. Congenital titinopathy from TTN is among the most severe of all, with the congenital contractures and the respiratory failure at birth. [4][6][11]
The LMNA-related congenital muscular dystrophy deserves its own scenario, because it carries the cardiac risk that can kill. The Ben Yaou 2021 natural history study showed that the LMNA form presents with the early axial and the upper-limb weakness, the dropped head, and the spine rigidity, and that the cardiac conduction disease and the arrhythmia can appear even with a preserved ejection fraction and even before the weakness is severe. The cardiac surveillance with the echocardiogram, the electrocardiogram, and the Holter monitor is mandatory from the diagnosis, and the pacemaker or the implantable defibrillator is considered when the conduction abnormality appears, because the sudden cardiac death is the threat that the management must prevent. [7]
Complications & Pitfalls
The life-threatening complications are the respiratory failure, the cardiac arrhythmia, and the malignant hyperthermia, and each is preventable with the right surveillance. The respiratory failure from the diaphragmatic and the intercostal weakness is the commonest cause of death in the severe forms, and it is managed with the non-invasive ventilation and the cough assistance, but it is missed when the clinician waits for the daytime symptoms instead of screening with the sleep study. The cardiac conduction disease and the sudden death of the LMNA form are missed when the echocardiogram alone is relied on and the Holter monitor is omitted, because the arrhythmia can precede the contractile failure. The malignant hyperthermia crisis of the RYR1 form is missed when the family is not flagged before the anaesthetic. [2][7]
The classic pitfalls are the ones the examiner rewards a candidate for naming. The first is attributing the floppy infant to a central or a benign cause and missing the reduced reflexes that point to the neuromuscular origin. The second is sending the survival motor neuron gene and the brain imaging but omitting the creatine kinase, the single highest-yield first test. The third is relying on the daytime symptoms and the walking ability to screen for the respiratory failure, instead of the sleep study and the forced vital capacity. The fourth is failing to flag the RYR1 case for the malignant hyperthermia risk before the anaesthetic. The fifth is omitting the cardiac surveillance in the LMNA form and missing the arrhythmia. The sixth is under-treating the feeding difficulty and the malnutrition that worsen the weakness. [1][11]
The long-term complications dominate the lived burden of the disease. The contractures, the scoliosis, and the reduced mobility limit the function and the independence, and the respiratory dependence on the non-invasive or the invasive ventilation reshapes the daily life and the family. The learning difficulty and the visual impairment complicate the dystroglycanopathies, and the psychological burden of a chronic and progressive disease falls on the child and the family. The coordination of the multidisciplinary care, the school support, and the community services is part of the treatment, not an afterthought, and the transition to the adult neuromuscular service is planned from the early teenage years. [2]
Prognosis & Disposition
The prognosis is set by the specific gene and the pattern of the involvement, and it spans the full spectrum from the death in infancy of Walker-Warburg syndrome to the near-normal life span of the mild congenital myopathies. The merosin-deficient form carries a survival into adolescence and adulthood with the respiratory support, and the collagen VI and the LMNA forms carry a survival into adulthood with the multidisciplinary care. The severe congenital myopathies, the congenital titinopathy, and the myotubular myopathy carry a high early mortality from the respiratory failure, but the survival has improved with the non-invasive ventilation, and many of these children now reach adulthood with the technology-dependent care. [1][6]
The predictors of the outcome are the levers worth knowing, because they set the prognosis and the surveillance. The brain malformation of the dystroglycanopathies is the single strongest predictor of the poor outcome, and the eye malformation accompanies it. The respiratory involvement, measured by the forced vital capacity and the sleep study, is the predictor of the respiratory failure and the ventilation need, and it is tracked at least annually in the respiratory-risk forms. The cardiac involvement of the LMNA form is the predictor of the sudden death, and the conduction abnormality is the trigger for the pacemaker or the defibrillator. The achievement of the independent walking is the functional milestone that sets the motor trajectory. [2][7]
disease trajectory
Moderate to severe: survival into adolescence and adulthood with the respiratory and the orthopaedic support
The disposition follows the respiratory and the nutritional status and the specific gene. Any neonate or infant with the respiratory distress or the ventilatory failure is admitted to the neonatal or the paediatric intensive care unit, and the child with the severe myopathy or the titinopathy may need the prolonged ventilation. The child with the stable respiratory status is managed in the outpatient neuromuscular clinic with the annual surveillance, and the child with the acute deterioration from the chest infection is admitted for the respiratory support and the airway clearance. The transition to the adult neuromuscular service is planned from the early teenage years, because the multidisciplinary care and the technology dependence continue across the life span. [2]
Special Populations
The neonate with the severe congenital myopathy or the titinopathy presents with the respiratory failure at birth, and the management is the intensive care with the ventilation and the feeding, and the diagnostic workup is sent in parallel, because the gene result sets the prognosis and the long-term plan. The decision to continue the long-term ventilation in a child with the severe titinopathy or the myotubular myopathy is a complex one that involves the family, the intensive care, the neuromuscular, and the ethics teams, and it is informed by the gene result and the natural history, and the family is supported through the process. [6]
The child from a consanguineous family or a population with a founder mutation carries a higher prior probability of an autosomal recessive form, and the family history and the ancestry direct the gene panel. The child from a refugee or a migrant family needs an early interpreter and a broad workup, and the genetic counselling is adapted to the language and the culture. The Aboriginal and Torres Strait Islander child and the child from a remote setting may present late, and the retrieval pathways and the telehealth link to the tertiary neuromuscular service are central to the equity of the care. The girl with the floppy infant is not exempt from the X-linked myotubular myopathy, because the manifesting carrier does occur, and the skewed X-inactivation is the explanation. [1]
Evidence, Guidelines & Regional Differences
The diagnostic framework rests on the Bönnemann 2014 diagnostic approach to the congenital muscular dystrophies, which set out the creatine-kinine and the brain and the eye and the biopsy logic that still organises the field, and on the Muntoni and Voit 2004 century-of-progress review, which framed the modern classification. The Nance 2012 update and the Amburgey 2013 RYR1 genotype-phenotype study refined the congenital myopathies and their genotype-phenotype correlations, and the Coppens 2025 congenital titinopathy study defined the most severe end of the spectrum. [1][5][4][11][6]
The management evidence rests on the two international consensus statements, the Wang 2010 consensus on the standard of care for the congenital muscular dystrophies and the Wang 2012 consensus on the standard of care for the congenital myopathies, which set out the multidisciplinary framework of the respiratory, the nutritional, the orthopaedic, and the rehabilitation care that remains the standard today. The Ben Yaou 2021 LMNA natural history study and the Angelin 2007 cyclosporin study defined the cardiac risk and the mechanistic therapy, and the Magri 2020 LAMA2 study and the Mercuri 2006 FKRP study defined the brain and the eye patterns. [2][3][7][8][10][9]
Angelin 2007 — cyclosporin in Ullrich congenital muscular dystrophy
A study of the muscle cells and the cultured myotubes from patients with collagen VI-related Ullrich congenital muscular dystrophy, testing the effect of cyclosporin and its non-immunosuppressive analogue on the mitochondrial permeability transition pore
Key finding
Cyclosporin and the analogue closed the permeability transition pore and rescued the mitochondrial dysfunction and the apoptosis in the patient cells.
Practice change
The study identified the mitochondrial permeability transition pore as a drug target in collagen VI disease and opened the first mechanistic therapy for a congenital muscular dystrophy, with the analogue under clinical investigation.
The treatment landscape is shifting toward the disease-modifying therapy. The next-generation sequencing panel has moved to first-line and replaced the muscle biopsy in the majority, and the genetic confirmation enables the emerging antisense and the gene therapy trials that are now underway for several forms. The cyclosporin analogue for the collagen VI disease, the exon-skipping and the gene-replacement approaches for the merosin-deficient disease, and the myotubularin gene therapy for the MTM1 myopathy are the active frontiers, and the paediatrician who refers early and confirms the gene is the one who opens the trial option for the family. [1]
Regional differences are small in principle because the framework is international, but the access and the organisation of the care differ. The tertiary paediatric neuromuscular services in ANZ, the UK, and North America provide the gene panel, the multidisciplinary clinic, and the respiratory and the cardiac surveillance promptly, but the retrieval from the remote settings can stretch to days, and the telehealth link to the specialist service is central to the equity. The threshold to refer and to transfer is low, and the choice of the first-line investigation is broadly consistent across the RACP, the RCPCH, the ABP, and the RCPSC contexts, because the creatine kinase fork and the multidisciplinary framework are international. [2]
Exam Pearls
The floppy infant with reduced or absent reflexes and a socially engaged manner has a neuromuscular cause until proven otherwise, and the creatine kinase is the first fork, with a markedly raised kinase pointing to a congenital muscular dystrophy and a normal or mildly raised kinase pointing to a congenital myopathy or a mimic. The congenital muscular dystrophies are dystrophic, with the active degeneration and the fibrosis and the raised kinase, and they include the merosin-deficient LAMA2, the collagen VI, the dystroglycanopathy, the LMNA, the SEPN1, and the titin forms. The congenital myopathies are structurally disordered but not dystrophic, with the cores, the rods, and the central nuclei, and they include the central core RYR1, the nemaline NEB, and the centronuclear MTM1 forms. [1]
The brain and the eye fork the muscular dystrophies. The diffuse white matter change with the epilepsy and the spared cognition points to the merosin-deficient LAMA2 form, and the cobblestone lissencephaly with the eye malformation points to a dystroglycanopathy such as the Walker-Warburg syndrome, which carries the grimmest prognosis in the field. The collagen VI Ullrich form carries the proximal contractures and the distal hyperlaxity and the follicular hyperkeratosis, and the LMNA form carries the dropped head and the spine rigidity and the cardiac conduction risk. The next-generation sequencing muscle panel has replaced the biopsy as the first-line diagnostic test in the majority. [9][1]
The management is the multidisciplinary supportive care, because most forms have no curative therapy, and the respiratory failure, the feeding difficulty, the contractures, the scoliosis, and the cardiac involvement are the complications that the care is built around. The nocturnal non-invasive ventilation is the single intervention that most extends the survival in the respiratory-risk forms, and a walking child can still be in the respiratory failure, so the sleep study and the forced vital capacity, not the walking ability, drive the decision to ventilate. The RYR1 central core disease carries the malignant hyperthermia risk that demands the anaesthetic precautions, and the LMNA form carries the sudden cardiac death risk that demands the Holter surveillance and the pacemaker consideration. [2][11][7]
The prognosis is set by the specific gene and the pattern of the involvement, and it spans from the death in infancy of the Walker-Warburg syndrome and the congenital titinopathy to the near-normal life span of the mild congenital myopathies. The single most useful habit at the bedside is to send the creatine kinase on every floppy infant with the reduced reflexes, to examine the brain and the eyes, and to refer early to the neuromuscular service, because the gene result sets the prognosis and opens the emerging trial option. The two rules that protect the child are to screen the respiratory and the cardiac function with the objective tests and to flag the RYR1 case for the malignant hyperthermia before the anaesthetic. [1][3]
References
- [1]Bönnemann CG, Wang CH, Quijano-Roy S, et al Diagnostic approach to the congenital muscular dystrophies. Neuromuscul Disord, 2014.PMID 24581957
- [2]Wang CH, Bonnemann CG, Rutkowski A, et al Consensus statement on standard of care for congenital muscular dystrophies. J Child Neurol, 2010.PMID 21078917
- [3]Wang CH, Dowling JJ, North K, et al Consensus statement on standard of care for congenital myopathies. J Child Neurol, 2012.PMID 22431881
- [4]Nance JR, Dowling JJ, Gibbs EM, et al Congenital myopathies: an update. Curr Neurol Neurosci Rep, 2012.PMID 22392505
- [5]Muntoni F, Voit T The congenital muscular dystrophies in 2004: a century of exciting progress. Neuromuscul Disord, 2004.PMID 15351421
- [6]Coppens S, Deconinck N, Sullivan P, et al Congenital Titinopathy: Comprehensive Characterization of the Most Severe End of the Disease Spectrum. Ann Neurol, 2025.PMID 39853809
- [7]Ben Yaou R, Yun P, Dabaj I, et al International retrospective natural history study of LMNA-related congenital muscular dystrophy. Brain Commun, 2021.PMID 34240052
- [8]Angelin A, Tiepolo T, Sabatelli P, et al Mitochondrial dysfunction in the pathogenesis of Ullrich congenital muscular dystrophy and prospective therapy with cyclosporins. Proc Natl Acad Sci U S A, 2007.PMID 17215366
- [9]Mercuri E, Topaloglu H, Brockington M, et al Spectrum of brain changes in patients with congenital muscular dystrophy and FKRP gene mutations. Arch Neurol, 2006.PMID 16476814
- [10]Magri F, Brusa R, Bello L, et al Limb girdle muscular dystrophy due to LAMA2 gene mutations: new mutations expand the clinical spectrum of a still challenging diagnosis. Acta Myol, 2020.PMID 32904964
- [11]Amburgey K, Bailey A, Hwang JH, et al Genotype-phenotype correlations in recessive RYR1-related myopathies. Orphanet J Rare Dis, 2013.PMID 23919265