Paeds · neurology-neurodisability-and-neuromuscular
Developmental regression and neurodegeneration
Also known as Developmental regression · Loss of milestones · Neurodegeneration in childhood · Progressive encephalopathy · Childhood dementia
A fellowship approach to the child who loses previously acquired developmental milestones. Recognise regression as a red flag, distinguish true progressive neurodegeneration from plateau and static loss, read the tempo and bedside pattern to generate a structured differential (epileptic encephalopathies, genetic and syndromic neurodegeneration, leukodystrophies, neurodegeneration with brain iron accumulation, and the acquired and treatable mimics - autoimmune encephalitis, subacute sclerosing panencephalitis, autistic regression, Rett syndrome), and drive a tiered neuro-investigation anchored on the developmental history, brain MRI, EEG and genomic sequencing - never accepting a degenerative label until the treatable causes have been actively excluded.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
A toddler who was speaking in two-word phrases stops talking and begins to stumble, and a teenager with no past history arrives incoherent and twitching over a weekend. In both rooms the unifying question is the same: is this loss of skills a progressive disorder, and is there a therapy whose window is closing right now? The fellowship task is to convert that bedside worry into a structured, time-aware workup that does not forfeit a treatable cause to a comforting but wrong label such as autism, cerebral palsy, or behavioural change. [1] [9]
R · E · G · R · E · S · S
Overview & Definition
The clinician's first act is to confirm what kind of developmental problem this is, because the word "regression" is used loosely and the distinction changes everything. Developmental delay means milestones are not being met on time. A plateau means the child has stopped gaining new skills but has not lost old ones. Neurological regression - the entity this page owns - means the child has lost previously acquired skills. Only regression carries the weight of a presumed progressive process, and only regression mandates the search for a neurodegenerative or otherwise treatable cause at speed. [1]
Why does losing skills carry so much more weight than simply being slow? Because a child whose trajectory was once higher and is now falling has something actively injuring the brain, whereas a child who is slow from the start or who has simply stopped climbing may have a static encephalopathy. The injury may be a structural neurodegenerative disease, an epileptic network disrupting cortical function, a synaptic autoimmune process, or a treatable metabolic defect - and the tempo of the fall is itself diagnostic. Acute or subacute regression, over days to weeks, raises the treatable and time-critical causes such as autoimmune encephalitis and metabolic decompensation; insidious regression, over months to years, raises the leukodystrophies, the storage disorders, and the neurodegenerations with brain iron accumulation. [1] [8]
What makes this group worth knowing in depth is that a meaningful and growing subset is treatable, and that treatment works best - or only - when it is begun before the brain injury becomes irreversible. The diagnostic label matters: calling a treatable autoimmune encephalitis a degenerative disorder forfeits the only chance of recovery, while calling a static unrecognised cerebral palsy a progressive regression subjects a family to a frightening and unnecessary workup. The history, not the single examination, settles which it is. [1]
Classification
The causes of regression are best grouped by the bedside pattern and the mechanism, because the pattern predicts the first test, the imaging, and whether a disease-modifying therapy exists. The figure below lays out four neurodegenerative pattern groups alongside a lower band of the acquired and treatable mimics that must be excluded at the same time - because the mimics are the ones a general paediatrician can actually reverse. [1] [8]

The four groups behave differently at the bedside. Epileptic encephalopathies - infantile spasms, Landau-Kleffner syndrome, and the syndrome of continuous spikes and waves during slow sleep (CSWS) - cause regression through persistent interictal epileptiform activity that disrupts cortical function, so the seizure history and the sleep EEG are the keys. Genetic and syndromic neurodegeneration brings the storage disorders, the gangliosidoses, Rett syndrome, and the hereditary ataxias, where a named gene produces progressive neuronal loss. Leukodystrophies and white-matter disorders - metachromatic leukodystrophy, Krabbe, X-linked adrenoleukodystrophy, Pelizaeus-Merzbacher - injure myelin and declare themselves as regression with spasticity, ataxia, and characteristic white-matter imaging. Neurodegeneration with brain iron accumulation (NBIA), classically pantothenate kinase-associated neurodegeneration (PKAN), produces progressive dystonia with iron deposition in the basal ganglia. [5] [6]
The lower band is where the reversible causes live, and it is the band the general paediatrician must never miss. Autoimmune encephalitis, dominated by NMDA-receptor antibody encephalitis, produces subacute psychiatric and cognitive regression that responds to immunotherapy. Subacute sclerosing panencephalitis (SSPE), a late complication of measles, produces progressive myoclonic regression. Autistic regression and Rett syndrome are pattern diagnoses that mimic neurodegeneration but follow a recognisable course and need no transplant or immunotherapy - they are excluded by their phenotype and confirmed genetically when appropriate. Holding this band in mind prevents the most consequential error in the field: labelling a treatable autoimmune encephalitis as a degenerative disorder. [2] [8]
Pattern groups at a glance - pattern, key test, treatment lever
- Epileptic encephalopathy (infantile spasms, Landau-Kleffner, CSWS): regression driven by epileptiform EEG activity, often with seizures. Key test: sleep EEG (electrical status for CSWS, hypsarrhythmia for infantile spasms). Treatment: hormonal therapy or vigabatrin for infantile spasms; steroids and antiseizure medication for Landau-Kleffner and CSWS.
- Genetic and syndromic neurodegeneration (Rett, gangliosidoses, hereditary ataxias, storage disorders): progressive neuronal loss with a named gene. Key test: trio exome sequencing plus targeted metabolic assays. Treatment: disease-modifying for selected storage disorders; supportive and rehabilitative for many.
- Leukodystrophy and white matter (MLD, Krabbe, X-ALD, Pelizaeus-Merzbacher): regression with spasticity, ataxia, white-matter change. Key test: brain MRI, very-long-chain fatty acids, arylsulfatase A. Treatment: haematopoietic stem cell transplant for early cerebral X-ALD and selected early leukodystrophies.
- Basal-ganglia iron and movement (NBIA) (PKAN, PLAN, MPAN, BPAN): progressive dystonia with iron in the globus pallidus. Key test: iron-sensitive brain MRI, molecular testing. Treatment: symptom management; cofactors for a few responsive subtypes.
- Acquired and treatable mimics (autoimmune encephalitis, SSPE, autistic regression, Rett): subacute or patterned regression. Key test: autoimmune and infective panels, sleep EEG, MECP2. Treatment: immunotherapy for autoimmune encephalitis; syndrome-specific for the rest. [5] [8]
Epidemiology & Risk Factors
Each major cause of regression is individually uncommon, but the probability that a child with true regression has a definable - and often treatable - cause is high enough to justify a systematic workup in every case. Trio exome sequencing now identifies a diagnostic variant in a substantial fraction of children with unexplained developmental regression, and a defined minority of these are treatable. The general paediatrician's job is not to pre-select who deserves a workup, because the phenotype alone rarely excludes a treatable cause. [1]
The strongest risk factors for the genetic neurodegenerative disorders are genetic. Consanguinity, a previously affected sibling, and a family history of unexplained childhood death or developmental failure all raise the probability of an autosomal recessive disorder and should lower the threshold for genomic testing in any child with a compatible phenotype. Sex-linked patterns reshape the counselling: Rett syndrome affects girls (MECP2 is X-linked, usually lethal in boys), while X-linked adrenoleukodystrophy and Pelizaeus-Merzbacher disease affect boys. Founder effects concentrate specific disorders in particular populations, so a careful three-generation pedigree is part of the workup, not an optional add-on. [2] [5]
The acquired and treatable mimics have their own epidemiology that changes the differential. Autoimmune encephalitis has a bimodal age distribution, with a peak in young children and a larger peak in adolescents and young adults, where NMDA-receptor encephalitis may be paraneoplastic (ovarian teratoma in adolescent girls). SSPE follows wild-type measles infection, typically six to ten years later, and remains common wherever measles immunisation coverage is incomplete - a reminder that the differential is region-dependent. A recent viral illness, a new tumour, or a recent vaccination may all reshape the pre-test probability of an acquired cause. [8] [9]
Pathophysiology
A single upstream cause becomes a whole-brain disease because the central nervous system is exquisitely dependent on the pathways each disorder disrupts: tight energy supply, clean substrate handling, intact myelin, controlled metal traffic, and orderly synaptic transmission. The cascade runs from a gene variant or an acquired insult to a specific mechanism of neuronal injury - storage engorgement, energy failure, demyelination, synaptic autoimmunity, iron accumulation, or an epileptic network - and that mechanism is what defines the clinical pattern and the tempo. The mechanism, not the gene, is what the clinician reads at the bedside. [1]
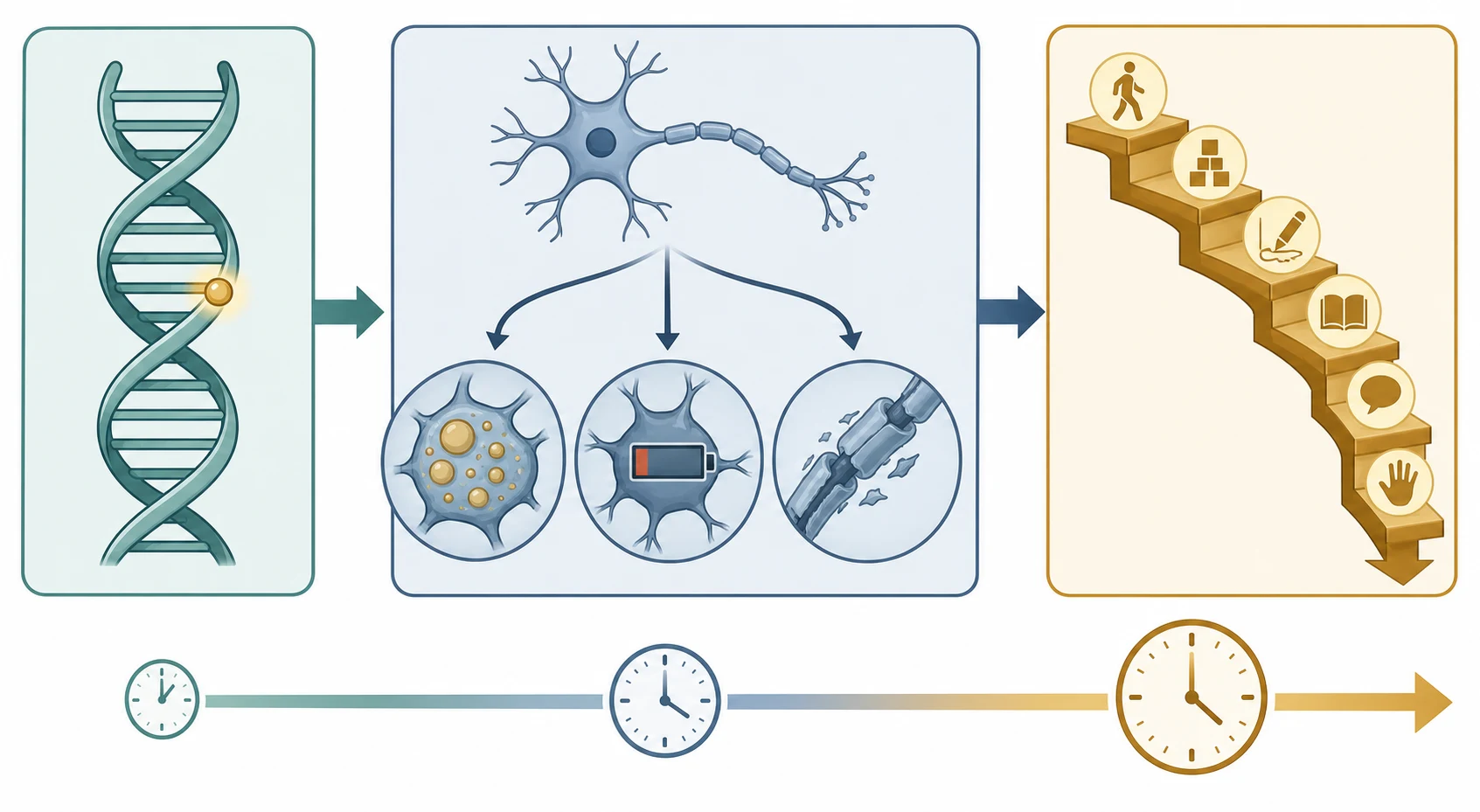
The mechanism of neuronal injury differs by group and explains the imaging and clinical patterns. Storage and lysosomal disorders engorge neurons with undegraded substrate, triggering demyelination, neuronal swelling, and gliosis, and they often declare themselves with organomegaly or a cherry-red spot. Leukodystrophies disrupt myelin synthesis or maintenance, producing symmetric white-matter change with spasticity and ataxia. NBIA deposits iron in the basal ganglia, producing a progressive movement disorder. Epileptic encephalopathies do not primarily destroy neurons - they disrupt cortical function through persistent epileptiform activity, which is why the regression can plateau or partially reverse when the EEG is controlled. [5] [6]
The autoimmune and infectious mimics have a distinct and treatable mechanism. In NMDA-receptor antibody encephalitis, antibodies bind the GluN1 subunit of the NMDA receptor on the neuronal surface, causing receptor internalisation and a reversible fall in synaptic NMDA-receptor signalling; the clinical picture - psychiatric onset, seizures, movement disorder, dysautonomia - maps onto the limbic and basal-ganglia distribution of these receptors, and recovery follows when the antibody titre falls with immunotherapy. This reversibility is the biological basis for the urgency of treatment: a degenerative label here forfeits a recoverable brain. [7] [8]
Tempo - the dimension that triages urgency
The tempo is itself diagnostic and triages urgency. Acute or subacute regression demands same-day biochemistry, an autoimmune and infective screen, and an EEG, because the time-critical causes - autoimmune encephalitis and metabolic decompensation - can be reversed if caught. Insidious regression demands a deliberate, tiered workup, but with the same governing principle: name the disorder before a therapy window closes. A child who regressed overnight is not the same problem as a child who has been quietly slipping for a year, and the urgency of the first shapes the pace of the second. [1] [9]
Clinical Presentation
The presenting complaint is always the same in words - a child has lost skills - but the pattern of what is lost, what accompanies it, and how fast it happens is what points to the cause. The first pattern is regression with epilepsy. The seizure type and the EEG do much of the localising work here: infantile spasms in clusters with hypsarrhythmia point to West syndrome; an acquired loss of language comprehension in a child aged three to eight, with sleep-activated epileptiform activity, points to Landau-Kleffner syndrome; and global regression with electrical status epilepticus of sleep - a spike-wave index occupying most of non-rapid-eye-movement sleep - points to CSWS. The teaching point is that an epileptic encephalopathy that is not behaving as expected should prompt a sleep EEG and a metabolic workup rather than a fourth anticonvulsant. [4]
The second pattern is regression with a movement disorder. Progressive dystonia, parkinsonism, chorea, or ataxia in a child who is losing skills points to a basal-ganglia or cerebellar disorder: PKAN and the wider NBIA group produce dystonia with iron in the globus pallidus; Wilson disease produces a movement disorder with psychiatric change and hepatic disease in the older child; Niemann-Pick type C produces a vertical supranuclear gaze palsy with cognitive decline; and the hereditary ataxias, including ataxia-telangiectasia, produce progressive ataxia with their own systemic signatures. A movement disorder that progresses is a neuroimaging and metabolic decision, not a watch-and-wait. [6]
The third pattern is the autistic and Rett-syndrome regression. Autistic regression produces loss of language and social skills, usually before age three and without motor or systemic signs, and without the organomegaly, imaging change, or biochemistry of a neurodegenerative disease. Rett syndrome affects girls, classically after six to eighteen months of apparently normal development, and follows a four-stage course through stagnation, rapid regression with loss of purposeful hand use and the emergence of hand stereotypies, a pseudo-stationary plateau, and late motor deterioration. Deceleration of head growth is a hallmark. Rett is distinguished from autistic regression by the hand stereotypies, the gait dyspraxia, and the MECP2 test, and it is one of the pattern diagnoses that must sit alongside the search for a treatable cause. [2] [3]
The fourth pattern is the acquired, subacute encephalopathy that mimics degeneration - and this is the one to catch. A child or adolescent who develops psychiatric change, new seizures, a movement disorder such as orofacial dyskinesia, altered sleep, autonomic instability, and a fluctuating or reduced level of consciousness over days to weeks has autoimmune encephalitis until proved otherwise. This presentation is regularly mislabelled as a primary psychiatric disorder, drug ingestion, or a degenerative process in its early phase, and the cost of the delay is measurable in lost recovery. The Graus diagnostic framework - possible, probable, and confirmed autoimmune encephalitis - exists precisely to let the clinician act on the phenotype before the antibody panel returns. [8] [9]
Differential Diagnosis
The differential of regression is broad, and several treatable non-neurodegenerative causes must be actively excluded because each changes the trajectory if missed. Autoimmune encephalitis heads the list of treatable mimics, because it is commoner than many of the degenerative disorders and is fully reversible with early treatment. SSPE, though ultimately irreversible, must be recognised for its counselling implications. Landau-Kleffner syndrome and CSWS are treatable epileptic encephalopathies hidden behind a normal waking EEG. Hypothyroidism, coeliac disease, nutritional deficiency (including vitamin B12 and folate), chronic illness, and medication effects can all mimic regression and are excluded by a targeted screen. Hearing and vision loss can masquerade as developmental loss, and a child who appears to have stopped engaging may simply not hear or see well. [1] [8]
Why does a normal initial screen not exclude a neurodegenerative disorder? Because several disorders are intermittent or tissue-specific. Lactate may be normal between mitochondrial flares; urine organic acids may be normal outside a decompensation; a single waking EEG may miss the electrical status of CSWS, which appears only in sleep; and a disorder that is not on the newborn-bloodspot panel will not be caught by the panel. When the clinical phenotype is highly suggestive, a normal screen lowers the probability but does not close the case, and the next step is targeted assays, a sleep EEG, and trio exome sequencing. [1]
Clinical & Bedside Assessment
The single most informative bedside act is to chart the developmental trajectory against specific earlier milestones. Ask what the child could do at six months, at twelve months, at eighteen months, and now - could they sit, babble, use a pincer grasp, walk, speak in phrases, follow a two-step instruction. A trajectory that rises then falls is regression; a line that flattens is a plateau; a line that was always low is delay. Anchor each milestone to an age and a witness, because the trajectory is the diagnosis in a way that no single examination finding can be, and a caregiver's account of a skill the child demonstrably had six months ago is the evidence that mandates the workup. [1]
Three questions frame every consultation. First, is the central nervous system involved and how fast - acute, subacute, or insidious? Second, which systems are involved and how severely - is there organomegaly, skin change, hepatic dysfunction, immunodeficiency? Third, is there consanguinity, a previously affected sibling, a family history of unexplained childhood death, or a relevant exposure such as recent measles or a new tumour? These three questions convert a vague worry into a structured hypothesis about the pattern group and the urgency, and they shape which test is sent first. [1] [9]
The examination is then directed and recorded. Measure growth and head circumference and plot them against earlier values - deceleration of head growth suggests Rett or a storage disorder; macrocephaly suggests a leukodystrophy or a storage disorder. Examine the skin for café-au-lait patches, hypopigmentation, and angiokeratomata. Examine the eyes for oculocutaneous telangiectasia (ataxia-telangiectasia), a Kayser-Fleischer ring (Wilson), and the fundus for a cherry-red spot, optic atrophy, and retinitis pigmentosa. Assess tone and movement for dystonia, parkinsonism, chorea, and ataxia. Palpate the liver and spleen. The discriminating neuro-ophthalmic and movement signs localise the disorder before any test is sent, which is why a focused neurological examination is the second pillar of the workup after the developmental history. [2] [6]
Investigations
The investigation strategy is tiered, and each tier either names the disorder or justifies the next. The first tier is the broad screen, sent on every child with unexplained regression: a full blood count, electrolytes, liver and thyroid function, a creatine kinase, a venous lactate drawn free-flowing, ammonia on ice, plasma amino acids, an acylcarnitine profile, urine organic acids, total homocysteine, and an autoimmune and infective screen to exclude the mimics. A hearing and vision assessment is part of the first tier, not an afterthought, because sensory loss masquerades as regression. [1]
Brain magnetic resonance imaging is central at this stage because the pattern of white-matter, basal-ganglia, cortical, or atrophic change points to a group. Symmetric confluent white-matter change raises a leukodystrophy; a contrast-enhancing parieto-occipital lesion in a boy raises cerebral X-linked adrenoleukodystrophy; iron deposition in the globus pallidus with an eye-of-the-tiger sign raises PKAN; and symmetric basal-ganglia or brainstem necrosis with a raised lactate raises Leigh syndrome. Magnetic resonance spectroscopy adds a lactate peak and a metabolic dimension. The EEG is the key investigation for the epileptic encephalopathies, and a sleep recording is essential - the electrical status epilepticus of CSWS and the epileptiform activity of Landau-Kleffner are missed on a routine waking trace. [4] [5]
The tiered workup
- Recognise regression - chart the trajectory against earlier milestones; confirm loss of previously acquired skills with a witness.
- First-line screen - bloods, metabolic markers (lactate, ammonia, amino acids, acylcarnitines, organic acids, homocysteine, creatine kinase), autoimmune and infective panels, hearing and vision, plus brain MRI and a sleep EEG.
- Targeted assays by pattern - storage (urine glycosaminoglycans and enzyme panel), leukodystrophy (very-long-chain fatty acids, arylsulfatase A), NBIA and Wilson (copper, caeruloplasmin, iron-sensitive MRI), autoimmune (cell-based antibody panel), CSF analysis where indicated.
- Chromosomal microarray - the first-tier genetic test for any child with a developmental disorder, to detect copy-number variants.
- Trio exome or genome sequencing - the single highest-yield test for unexplained neuroregression when targeted testing is unrevealing, analysing the child and both parents together. [1]
The third tier is genomic, and it has transformed the field. Chromosomal microarray is the first-line genetic test for any child with a developmental disorder. When the microarray and the targeted assays are unrevealing, trio exome or genome sequencing is the single highest-yield test for unexplained neuroregression, identifying a diagnostic variant in a substantial fraction of cases and often surfacing a treatable disorder that targeted testing had missed. A lumbar puncture is reserved for the phenotype that demands it - suspected GLUT1 deficiency shows a low cerebrospinal-fluid glucose with a low CSF-to-plasma ratio; suspected cerebral folate deficiency shows a low CSF 5-methyltetrahydrofolate; and suspected autoimmune or infective encephalitis shows the cells, protein, and oligoclonal bands of an inflammatory process. A molecular diagnosis enables carrier testing, prenatal diagnosis, and preimplantation genetic testing for the wider family, and it carries prognosis. [1] [12]
Management — Resuscitation
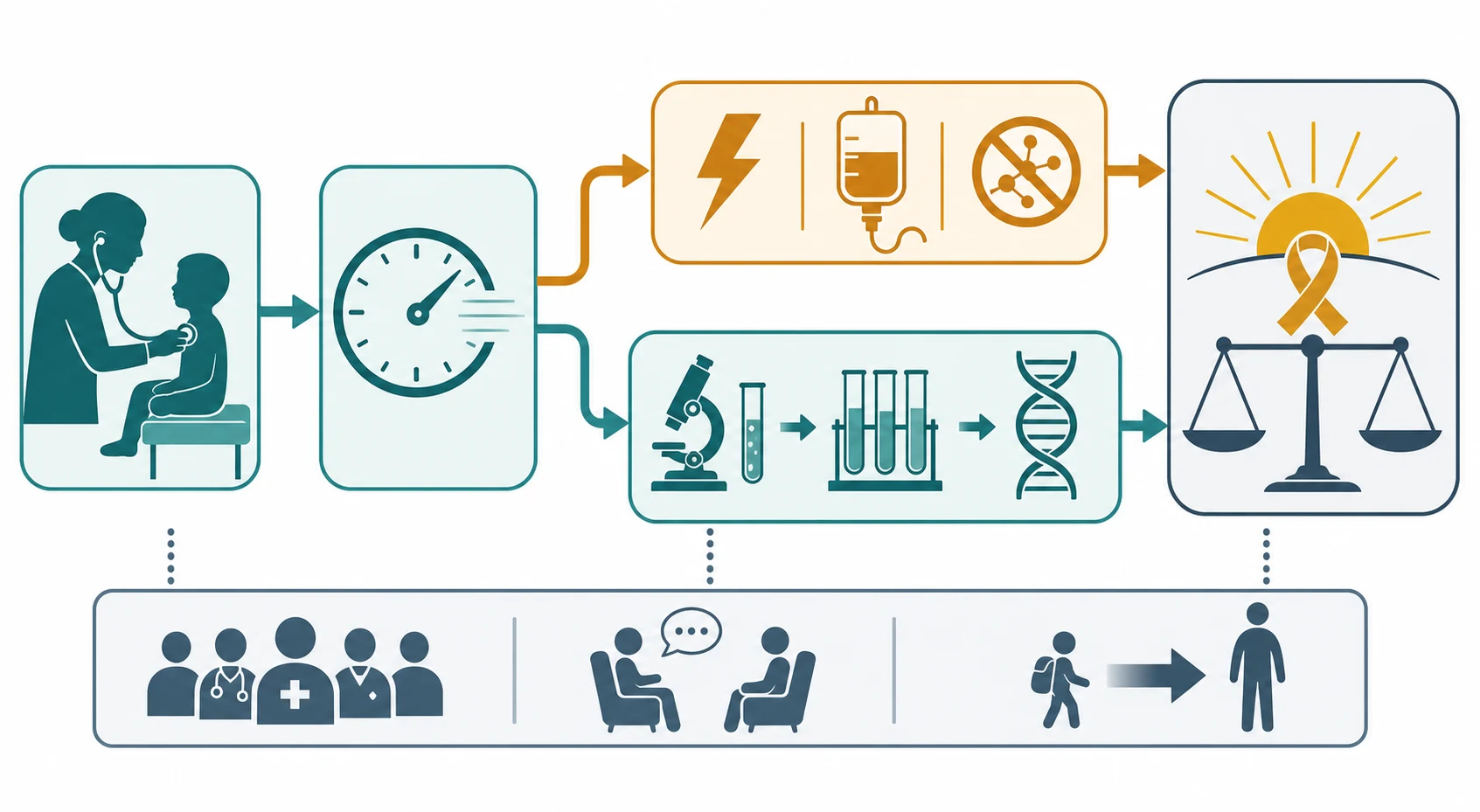
When regression presents as acute or subacute encephalopathy - altered consciousness, seizures, a movement disorder, or metabolic derangement - the response is resuscitation first and the diagnostic workup in parallel. Secure the airway, breathing, and circulation; treat seizures early; and move immediately to exclude and treat the time-critical causes. The two resuscitation-pace decisions are autoimmune encephalitis and metabolic decompensation, because both are reversible if caught and both worsen if they are watched. [1] [9]
For suspected autoimmune encephalitis, empirical immunotherapy is warranted while the antibody panel returns, because the panel can take weeks and the outcome is better the earlier treatment begins. First-line therapy is high-dose corticosteroids (methylprednisolone), intravenous immunoglobulin, plasma exchange, or a combination, guided by the paediatric neurology and neuroimmunology service. The paediatric consensus framework structures first- and second-line therapy - adding rituximab or cyclophosphamide when the response is poor - and the principle is that the phenotype justifies treatment before the antibody result confirms it. [9] [10]
For suspected metabolic decompensation, the emergency protocol aims to halt catabolism and clear the toxin while the workup proceeds. Stop all protein and any potential toxin intake, give intravenous dextrose at maintenance-plus rate to suppress catabolism and provide an alternative fuel, treat acidosis with bicarbonate if severe, and manage hyperammonaemia with nitrogen-scavenger drugs and, if severe, haemofiltration, because ammonia itself is neurotoxic. Status epilepticus is managed in parallel with a first-line benzodiazepine followed by a second-line agent, chosen to avoid worsening an underlying metabolic defect. These resuscitation decisions are owned by the general paediatrician in the first hours, with the specialist service taking over as the diagnosis declares itself. [1]
Management — Definitive & Stepwise
Definitive management means naming the disorder and matching it to the disease-modifying or syndrome-specific therapy that fits its mechanism. For autoimmune encephalitis, the lever is immunotherapy, with the staged regimen and the prognostic weight of early treatment now well characterised; most children treated early make a good recovery, and a poor early response is the signal to escalate to second-line therapy. For GLUT1 deficiency, the ketogenic diet provides ketones as an alternative brain fuel and can control seizures, improve the movement disorder, and stabilise cognition - a genuinely treatable cause of epileptic encephalopathy and regression that is missed when the epilepsy is treated without a metabolic workup. [7] [12]
For the epileptic encephalopathies, the antiseizure strategy is tailored to the syndrome. Infantile spasms respond to hormonal therapy (adrenocorticotrophic hormone or high-dose prednisolone) or vigabatrin, the latter preferred when the cause is tuberous sclerosis. Landau-Kleffner syndrome and CSWS respond to corticosteroids alongside antiseizure medication such as ethosuximide, levetiracetam, or clobazam, with the goal of suppressing the electrical status that is driving the cognitive regression. For selected storage and leukodystrophy disorders, substrate reduction and pharmacological chaperones, enzyme replacement for non-neuronopathic viscera, and haematopoietic stem cell transplant for early cerebral adrenoleukodystrophy and selected neuronopathic disease are the disease-modifying levers, and the window for transplant closes once the injury becomes fixed - which is why early accurate diagnosis is more valuable now than ever. [1] [5]
The care plan is multidisciplinary and lifelong, built around the specialist neurology, metabolic, and genetics service. Developmental and educational support, allied health (physiotherapy, occupational therapy, speech and language), orthopaedics and respiratory for complications, and palliative care involved early in the most severe disorders all coexist with disease-modifying treatment rather than replacing it. Once a molecular diagnosis is made, genetic counselling, carrier testing, prenatal diagnosis, and preimplantation genetic testing reshape the reproductive risk for the whole family, and this is part of management, not an afterthought. [1]
Specific Subtypes & Scenarios
Rett syndrome is the prototypic syndromic regression and every candidate must know its course. A girl who develops normally for the first six to eighteen months then enters a phase of rapid regression, losing purposeful hand use and spoken language and acquiring characteristic midline hand stereotypies - wringing, washing, and clapping - alongside gait dyspraxia and deceleration of head growth, has Rett syndrome until proved otherwise. The revised 2010 diagnostic criteria anchor the diagnosis on a period of regression followed by loss of purposeful hand skills and the emergence of stereotypies, with molecular confirmation through MECP2 testing. After the regression phase the disease enters a plateau that can last years before the late motor-deterioration stage, and management is supportive, rehabilitative, and surveillance-based. [2]
Landau-Kleffner syndrome and CSWS are the treatable epileptic encephalopathies that hide behind a normal waking EEG. Landau-Kleffner presents in a child aged three to eight with an acquired verbal auditory agnosia - a loss of language comprehension that progresses to loss of expressive language - alongside seizures and, on EEG, epileptiform activity that is markedly activated in sleep. CSWS extends this to global regression with electrical status epilepticus of sleep, defined by a spike-wave index occupying most of non-rapid-eye-movement sleep. Both are treatable: corticosteroids and antiseizure medication can suppress the electrical status and recover function, and the principle is that any child with acquired loss of language or global regression must have a sleep EEG before the regression is labelled degenerative. [4]
Autoimmune (NMDA-receptor) encephalitis is the treatable mimic that every general paediatrician must recognise. A child or adolescent presents subacutely with psychiatric symptoms - agitation, hallucinations, behavioural change - followed by seizures, an orofacial or limb movement disorder, autonomic instability, altered sleep, and a decreasing level of consciousness that may progress to catatonia and intensive-care support. The phenotype justifies empirical immunotherapy before the antibody panel confirms it, and the paediatric consensus framework structures the staged regimen. Most children treated early make a good recovery; a poor early response is the signal to escalate, and the presence of an ovarian teratoma in an adolescent girl must be sought and removed. [9] [10]
The leukodystrophies and the NBIA group are the insidious neurodegenerations that imaging names. X-linked adrenoleukodystrophy affects boys and may declare itself in mid-childhood with behavioural change, school decline, new seizures, and adrenal insufficiency, with a characteristic contrast-enhancing parieto-occipital white-matter lesion - once the cerebral inflammatory form begins, progression is rapid and the transplant window is short. PKAN and the wider NBIA group present with progressive dystonia and cognitive decline, with iron deposition in the globus pallidus (the eye-of-the-tiger sign in classic PKAN); a few NBIA subtypes are cofactor-responsive, which is why a diagnosis of NBIA is the start of a search for a treatable subset, not the end of it. [5] [6]
Complications & Pitfalls
The most consequential complications are the consequences of delay: a forfeited transplant or enzyme window, irreversible brain injury, and the lost opportunity to offer the family accurate genetic counselling. Every day matters for the disorders whose therapy works only early - cerebral X-ALD, the neuronopathic storage disorders, the cofactor-responsive epileptic encephalopathies - and the single biggest contributor to delay is mislabelling. Calling a treatable autoimmune encephalitis a primary psychiatric disorder, or an early epileptic encephalopathy a degenerative process, closes the diagnostic search and redirects the family away from the treatment that would have worked. [7] [9]
Over-reliance on a normal initial screen is a second pitfall. Lactate can be normal between mitochondrial flares, urine organic acids can be normal outside a decompensation, a single waking EEG can miss the electrical status of CSWS, and a disorder that is not on the newborn-bloodspot panel will not be caught by the panel. When the phenotype is suggestive, the answer is a sleep EEG, targeted assays, and trio exome - not reassurance. A third pitfall is underestimating anaesthetic and fasting risk in a child with an undiagnosed neurodegenerative or metabolic disorder: prolonged fasting before a procedure, or certain anaesthetic agents, can trigger a metabolic decompensation that becomes the first presentation of the disorder, so any child under investigation needs a peri-procedure metabolic safety plan. [1]
Prognosis & Disposition
Prognosis is determined by the specific disorder, the age and tempo of onset, the genotype, and - critically - the timing of treatment. The early-treated treatable causes have a transformed prognosis: a child with autoimmune encephalitis treated early may recover fully; a child with GLUT1 deficiency started on a ketogenic diet early may have near-normal cognition; a boy with cerebral X-ALD transplanted early may halt the disease. The untreatable neuronopathic disorders - the severe leukodystrophies, the gangliosidoses, infantile neuronal ceroid lipofuscinosis - have a guarded outlook measured in years, and the task shifts to symptom control, developmental support, and palliative care. [7] [12]
Disposition is structured around the specialist neurology, metabolic, and genetics service, with lifelong surveillance and a named coordinator. The general paediatrician holds the whole child, coordinates the multidisciplinary team, and owns the emergency sick-day plan and the peri-procedure metabolic safety plan. As the child grows, structured transition to adult neurology and disability services begins in early adolescence, with explicit handover of the diagnosis, the surveillance plan, and the reproductive counselling. Palliative care is involved early in the most severe disorders, coexisting with disease-modifying treatment rather than replacing it: symptom control for seizures, dystonia, feeding difficulty, and respiratory compromise improves the child's quality of life and the family's capacity to cope. [1]
The conversation about goals of care is iterative, revisited as the disease declares its trajectory, and it is held with honesty and without abandoning the search for a treatable subset. Even when a disorder is not yet treatable, an accurate molecular diagnosis brings prognosis, family planning, and the possibility of future trial eligibility, which is why the workup is completed even when the immediate treatment options are limited. [1]
Special Populations
The approach differs across the age span. Infants and toddlers present with the acute encephalopathies, infantile spasms, and the early storage disorders, where the tempo is fast and a sleep EEG and an emergency metabolic protocol are early decisions. School-age children and adolescents present with autoimmune encephalitis, cerebral adrenoleukodystrophy, Wilson disease, the leukodystrophies, and PKAN, declared through psychiatric change, school decline, a movement disorder, or new seizures. Recognising the age-typical presentation sharpens the differential and the first test. [2] [9]
Consanguineous and founder populations carry a higher burden of autosomal recessive neurodegenerative disorders, and the consultation reframes reproductive risk for the whole family: carrier testing, prenatal diagnosis, and preimplantation genetic testing become central to management once a molecular diagnosis is made. For Indigenous, migrant, refugee, and remote populations, equitable and culturally safe access to specialist neurology and genomic services is a real challenge, concentrated as those services are in tertiary centres. The principles are constant - recognise regression early, deploy the tiered workup, exclude the treatable causes - but the pathway must be adapted to geography, language, and cultural safety, often with telehealth and a local coordinator. [1]
The technology-dependent and complex-chronic child, and the transition-age adolescent, need a plan that adapts across the lifespan. The adolescent who has lived with a neurodegenerative diagnosis since childhood faces new questions - adherence to therapy, transfer to adult services, reproductive decisions, and the risk of decompensation with new freedoms - and a structured transition, begun in early adolescence and completed before the eighteenth birthday, is the safeguard. The SSPE-prone population, where measles immunisation coverage is incomplete, is a reminder that prevention sits upstream of the workup. [11]
Evidence, Guidelines & Regional Differences
The conceptual backbone of the field is a set of consensus frameworks that let the clinician act on the phenotype before the confirmatory test returns. The revised Rett syndrome diagnostic criteria of Neul and colleagues standardised the recognition of the prototypic syndromic regression. The Graus diagnostic framework for autoimmune encephalitis - possible, probable, and confirmed categories built on the clinical phenotype, the EEG, the CSF, and the imaging - exists so that empirical immunotherapy is justified before the antibody panel confirms the diagnosis, and the paediatric adaptation and the international treatment consensus structure the staged regimen. The leukodystrophy consensus and the NBIA reviews converge across regions on the same imaging-led and genotype-led workup. [2] [8] [9]
In Australia and Aotearoa New Zealand, the newborn-bloodspot panel, funded access to exome, genome, enzyme, and gene therapies, and transplant services are governed by jurisdictional programmes that evolve over time. Specialist neurology, metabolic, and genetics services are concentrated in tertiary paediatric centres, with outreach to regional and remote areas via telehealth and retrieval networks. The autoimmune-encephalitis antibody panel may have a turnaround of weeks in centres without on-site neuroimmunology, which is precisely why the empirical-immunotherapy principle matters. State the local panel and the local funded pathway rather than assuming a universal list, and involve the regional service early. [1] [9]
The strength of evidence varies across the field. The evidence for early immunotherapy in autoimmune encephalitis and for early haematopoietic stem cell transplant in cerebral X-ALD is mature enough to justify the urgency of diagnosis. The evidence for the ketogenic diet in GLUT1 deficiency and for hormonal therapy in infantile spasms is strong. The evidence for the long-term outcomes of gene therapy and for the natural history of ultra-rare disorders is still maturing, and SSPE management remains limited to disease-modulation rather than cure. Uncertainty changes counselling: a family should be told what is known, what is likely, and what is genuinely unknown, so that decisions rest on real information rather than false reassurance or undue pessimism. [7] [11]
Exam Pearls
Remember the golden rule that frames every consultation: never label a regressing child degenerative or palliative until the treatable causes have been actively excluded. The treatable subset spans every pattern - immunotherapy for autoimmune encephalitis, the ketogenic diet for GLUT1 deficiency, steroids for the epileptic encephalopathies, haematopoietic stem cell transplant for early cerebral adrenoleukodystrophy, and cofactors for the responsive metabolic disorders. The diagnostic rate-limiting step is naming the disorder early, and trio exome is now the single highest-yield test for unexplained neuroregression when targeted testing is unrevealing. [1] [7]
Self-test: a fourteen-year-old with subacute regression
A previously well fourteen-year-old presents over ten days with insomnia, agitation and hallucinations, then a generalised tonic-clonic seizure, and on examination has orofacial dyskinesia and a fluctuating conscious level. Basic bloods, a first brain MRI, and a waking EEG are unrevealing. What is the most likely diagnosis, what framework justifies acting now, and what is the first step? [8] [9]
Answer: This is anti-NMDA-receptor antibody encephalitis until proved otherwise. The Graus diagnostic framework - which permits a diagnosis of probable autoimmune encephalitis on the phenotype (rapid onset of psychiatric symptoms, seizures, a movement disorder, and altered consciousness) with supportive EEG and CSF findings, before the antibody panel confirms it - justifies empirical immunotherapy now rather than waiting for the result. The first step is urgent paediatric neurology and neuroimmunology referral, CSF and serum for the autoimmune panel sent in parallel, a screen for an ovarian teratoma, and first-line immunotherapy with high-dose corticosteroids plus intravenous immunoglobulin or plasma exchange. [9] [10]
References
- [1]Moeschler JB, Shevell M, Committee on Genetics. Comprehensive evaluation of the child with intellectual disability or global developmental delays. Pediatrics, 2014.PMID 25157020
- [2]Neul JL, Kaufmann WE, Glaze DG, Christodoulou J, Clarke AJ, Bahi-Buisson N, et al. Rett syndrome: revised diagnostic criteria and nomenclature. Ann Neurol, 2010.PMID 21154482
- [3]Stefanatos GA. Regression in autistic spectrum disorders. Neuropsychol Rev, 2008.PMID 18956241
- [4]Stefanatos G. Changing perspectives on Landau-Kleffner syndrome. Clin Neuropsychol, 2011.PMID 21955111
- [5]Parikh S, Bernard G, Leventer RJ, van der Knaap MS, van Hove J, Pizzino A, et al. A clinical approach to the diagnosis of patients with leukodystrophies and genetic leukoencephelopathies. Mol Genet Metab, 2015.PMID 25655951
- [6]Schneider SA. Neurodegeneration with Brain Iron Accumulation. Curr Neurol Neurosci Rep, 2016.PMID 26739693
- [7]Titulaer MJ, McCracken L, Gabilondo I, Armangue T, Glaser C, Iizuka T, et al. Treatment and prognostic factors for long-term outcome in patients with anti-NMDA receptor encephalitis: an observational cohort study. Lancet Neurol, 2013.PMID 23290630
- [8]Graus F, Titulaer MJ, Balu R, Benseler S, Bien CG, Cellucci T, et al. A clinical approach to diagnosis of autoimmune encephalitis. Lancet Neurol, 2016.PMID 26906964
- [9]Cellucci T, Van Mater H, Graus F, Muscal E, Gallentine W, Klein-Gitelman MS, et al. Clinical approach to the diagnosis of autoimmune encephalitis in the pediatric patient. Neurol Neuroimmunol Neuroinflamm, 2020.PMID 31953309
- [10]Nosadini M, Thomas T, Eyre M, Anlar B, Armangue T, Benseler SM, et al. International Consensus Recommendations for the Treatment of Pediatric NMDAR Antibody Encephalitis. Neurol Neuroimmunol Neuroinflamm, 2021.PMID 34301820
- [11]Gascon GG, International Consortium on Subacute Sclerosing Panencephalitis. Randomized treatment study of inosiplex versus combined inosiplex and intraventricular interferon-alpha in subacute sclerosing panencephalitis (SSPE): international multicenter study. J Child Neurol, 2003.PMID 14736075
- [12]Klepper J, Akman C, Armeno M, Auvin S, Cervenka M, Cross HJ, et al. Glut1 Deficiency Syndrome (Glut1DS): State of the art in 2020 and recommendations of the international Glut1DS study group. Epilepsia Open, 2020.PMID 32913944