Paeds · neurology-neurodisability-and-neuromuscular
Duchenne and Becker muscular dystrophy
Also known as Duchenne muscular dystrophy · DMD · Becker muscular dystrophy · BMD · Dystrophinopathy · Pseudohypertrophic muscular dystrophy
Fellowship guide to Duchenne and Becker muscular dystrophy. Covers the X-linked recessive dystrophinopathies from the loss of dystrophin and the reading-frame hypothesis, through the boy with delayed motor milestones, calf pseudohypertrophy, Gowers sign, and a creatine kinase above 10,000, to the genetic confirmation by multiplex ligation-dependent probe amplification, the glucocorticoid backbone of prednisolone 0.75 mg per kg per day or deflazacort 0.9 mg per kg per day started at the motor plateau, the cardiac surveillance with angiotensin-converting-enzyme inhibition and eplerenone, the respiratory surveillance with spirometry and non-invasive ventilation, and the precision therapies of exon-skipping and AAV micro-dystrophin gene therapy.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
Overview & Definition
A boy who cannot run as fast as his friends, who walks on his toes and cannot jump, may carry a single gene that slowly unmakes his muscle. Duchenne muscular dystrophy is the most common and the most severe inherited muscular dystrophy of childhood, an X-linked recessive disorder in which loss-of-function mutations in the DMD gene abolish the sarcolemmal protein dystrophin. The muscle fibre loses its mechanical anchor, tears itself apart with every contraction, and is replaced by fat and scar. The boy walks later than his peers, weakens through early childhood, loses the ability to walk in the first decade, and dies of the heart and the lungs unless the disease is modified. [4]
Becker muscular dystrophy is the allelic and milder partner, born of the same gene but of mutations that leave a shortened, partly functional dystrophin rather than none at all. The two are dystrophinopathies, and they sit on a continuous spectrum of severity that tracks with how much dystrophin survives. The Becker boy walks into adulthood, and his heart, not his legs, may be the organ that threatens him first. [11]
Three ideas make this topic central to the paediatric exam. The first is recognition, because the boy is often labelled clumsy or lazy for years before someone measures a creatine kinase that is off the scale. The second is the glucocorticoid backbone, because prednisolone or deflazacort, started at the right time, changes the trajectory of the disease. The third is the systems care, because the heart and the lungs now decide the outcome, and a boy who is watched and treated can live into his fourth decade. The modern care considerations of Birnkrant and colleagues, building on the Bushby guidelines, set these principles out and remain the single most testable source. [4][5][6]
Classification
The dystrophinopathies sort themselves by how much dystrophin the mutation leaves behind, and that amount sets the severity. Duchenne muscular dystrophy sits at the severe end, in which an out-of-frame mutation shifts the reading of the gene so that translation halts early and no functional dystrophin reaches the membrane. Becker muscular dystrophy sits at the mild end, in which an in-frame mutation keeps the gene readable, so a shorter but partly working protein is produced. Between the two lies an intermediate group, and the clinical severity tracks the residual dystrophin almost as a dial. [2]
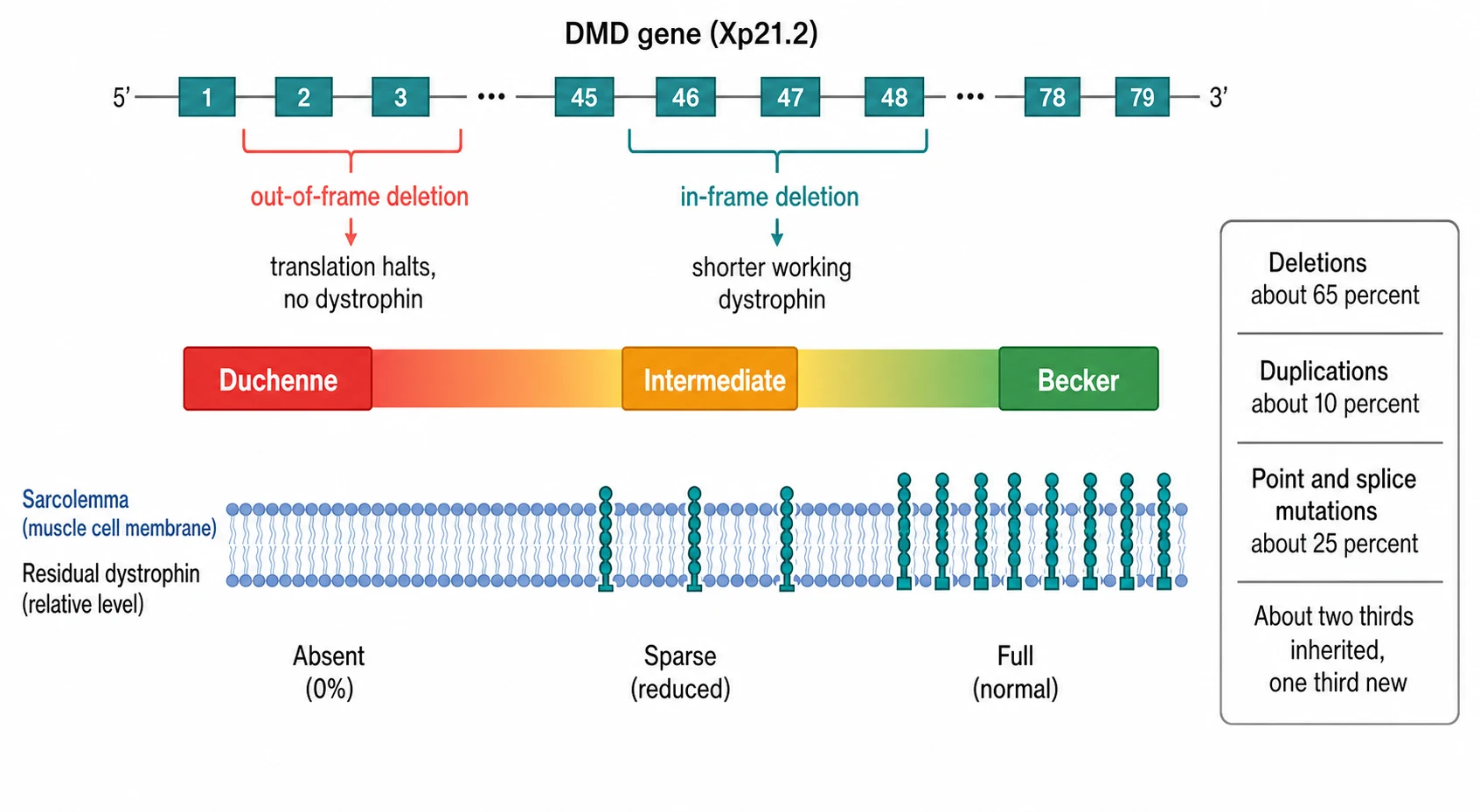
The molecular lesion comes in three shapes, and the geneticist searches for each in turn. A deletion of one or more exons is the commonest, found in roughly two thirds of boys, and it tends to cluster in two hot-spots of the giant gene. A duplication of exons accounts for about 10 percent, and a point mutation, a small insertion or deletion, or a splice change makes up the remainder. The shape of the mutation matters because it decides which precision therapy, if any, the boy is eligible for. [3]
A second, clinical classification follows the boy through his life and guides the surveillance. He moves from the early ambulatory phase, in which he walks but cannot run or jump normally, through the late ambulatory phase, into the early and then the late non-ambulatory phases. The transition between phases triggers a change in the priorities of care, from the muscles and the steroids in the ambulatory years to the heart, the lungs, and the spine in the wheelchair years. [4]
Epidemiology & Risk Factors
Duchenne muscular dystrophy is the most frequent childhood muscular dystrophy, with an incidence of about 1 in 3,500 to 1 in 5,000 live male births, and it is found in every population. Becker muscular dystrophy is roughly a third as common. Because the disease is X-linked recessive, it affects boys almost exclusively, and the girls who carry one altered copy are usually well but at risk of the cardiomyopathy. [3]
The inheritance explains the family risk that the fellow must trace at the bedside. About two thirds of affected boys inherit the mutation from a carrier mother, who may have no idea she carries it, and about one third carry a new mutation that arose in the egg or in early embryonic life. A carrier mother has a one in two chance of passing the altered X to each son, in which case he will be affected, and a one in two chance of passing it to each daughter, in which case she will be a carrier. This pattern is the reason the sisters, the mother, and the maternal aunts of an affected boy are offered genetic testing and cardiac surveillance. [3]
The strongest risk factor for severity, beyond the Duchenne-versus-Becker split, is the completeness of the multidisciplinary care. A boy who is diagnosed late, who never receives glucocorticoids, and who has no cardiac or respiratory surveillance will lose ambulation earlier and die of the heart and the lungs in his teens. The same boy, watched and treated in a neuromuscular centre, can walk into his second decade and live into his fourth, which is why the gap in access, rather than the gene alone, now shapes the outcome. [6]
Pathophysiology
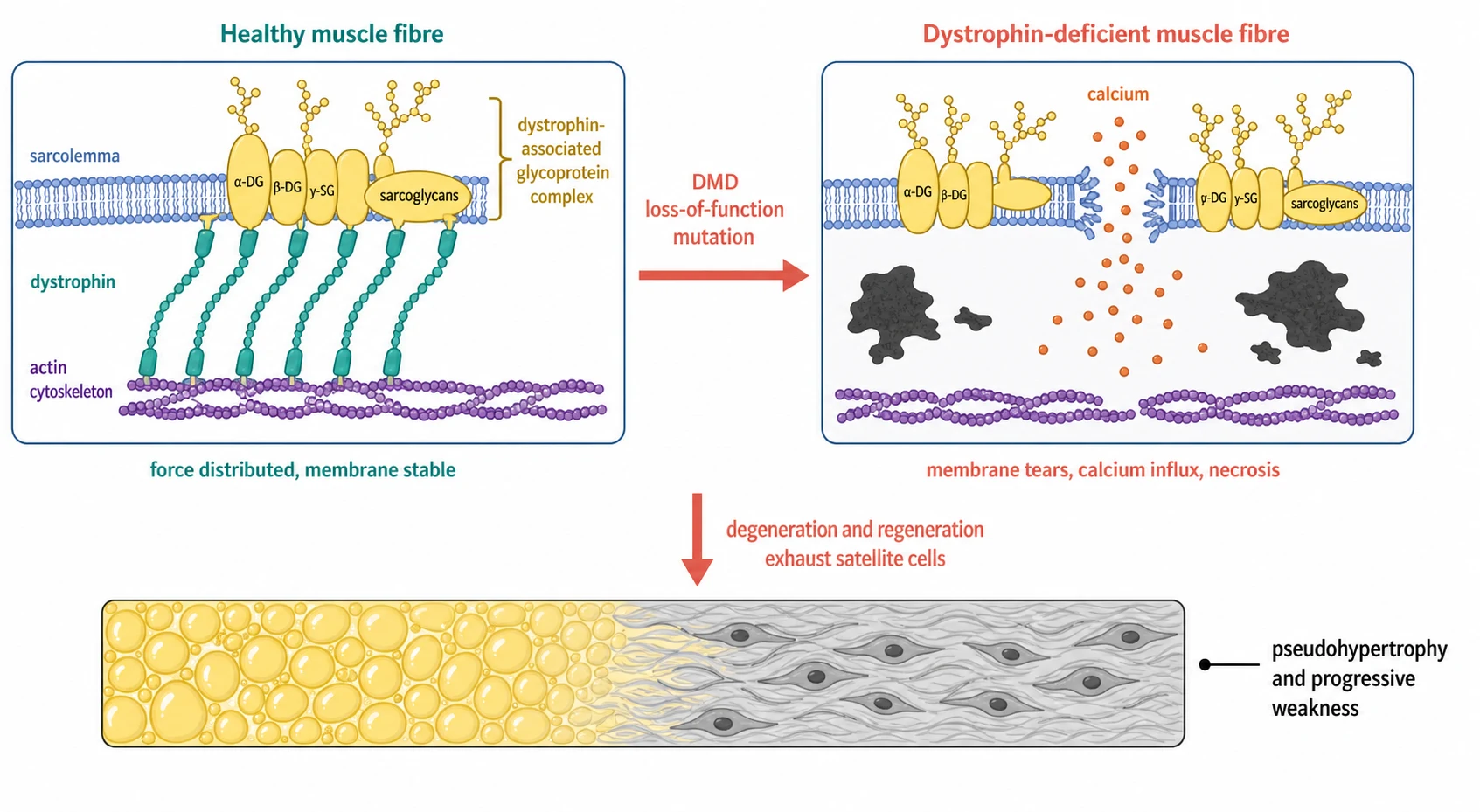
The protein at the centre of the disease is dystrophin, identified by Hoffman, Brown, and Kunkel in 1987 as the missing product of the Duchenne locus. Dystrophin sits just beneath the sarcolemma, the membrane of the muscle fibre, and it forms a mechanical link between the actin cytoskeleton inside the fibre and the dystrophin-associated glycoprotein complex that spans the membrane to anchor the cell to the surrounding matrix. This link is the shock absorber that distributes the force of contraction across the membrane, and when it is gone the membrane takes the strain alone. [1]
The mechanism of the weakness is mechanical failure followed by self-destruction. In Duchenne the membrane loses its anchor, and the repeated shearing of contraction tears the sarcolemma, admits a flood of calcium, and sets off necrosis and inflammation. The fibre tries to regenerate through its satellite cells, but the cycles of degeneration and regeneration exhaust the reserve, and the muscle is gradually replaced by fat and fibrous tissue. The fat is the calf pseudohypertrophy that the fellow feels, and the fibrosis is the firm, woody texture that replaces the muscle. [1]
The reading-frame hypothesis of Koenig and colleagues is the key that links the mutation to the severity, and it is one of the most testable molecular ideas in paediatrics. The DMD gene is read in groups of three bases, and a deletion that removes a whole number of triplets keeps the reading frame intact, so translation continues past the gap and produces a shorter but folded protein. This is the in-frame lesion of Becker. A deletion that throws the reading frame out of step introduces a premature stop, the ribosome falls off, and almost no protein is made. This is the out-of-frame lesion of Duchenne. The same logic, restored in reverse, is the very basis of exon-skipping therapy, which snips the gene back into frame. [2]
Clinical Presentation
The boy with Duchenne presents to the paediatrician not with weakness that anyone has measured, but with the slow failure of a child to do what his peers do. He walks late, often after eighteen months, and he never runs normally. He cannot jump with both feet, he falls often, and he climbs stairs only by pulling himself up the rail. His calves look large and firm, the pseudohypertrophy of fat and fibrosis, and his gait waddles and pitches him onto his toes. When he is asked to rise from the floor he turns, braces his hands on his knees, and climbs his own thighs, the Gowers sign, and the time he takes to do it is a measure of his disease. [4]
The weakness is proximal and symmetrical, and it spares the face and the eyes. The pelvic girdle fails before the shoulder girdle, so the legs go first, and the deep tendon reflexes fade as the muscle is lost. The boy also carries a cognitive burden that is independent of his muscle, with a mean intelligence quotient shifted about one standard deviation below the mean and a higher rate of autism, attention deficit, and learning difficulty, because dystrophin is expressed in the brain as well as the muscle. None of this is progressive, but it shapes the classroom as much as the playground. [4]
The natural history of Duchenne muscular dystrophy
The Becker boy writes a slower story. He walks at a normal age, and his weakness declares itself later, between the ages of five and fifteen, and it advances over decades rather than years. He keeps walking well into adulthood, and the calf pseudohypertrophy and the Gowers sign may be the only signs for years. The danger for the Becker patient is that his heart, rather than his legs, may fail him first, and a dilated cardiomyopathy can appear before any weakness is noticed, which is why every unexplained cardiomyopathy in a male deserves a creatine kinase. [11]
Differential Diagnosis
The boy with a waddling gait and a high creatine kinase has a dystrophic process until proven otherwise, but the fellow must separate the dystrophinopathies from the other chronic neuromuscular causes of proximal weakness. The creatine kinase is the first fork in the road, because it is markedly elevated in the dystrophinopathies and only mildly raised or normal in the spinal and junctional causes. The pattern of the weakness and the reflexes then separates the muscle disease from the nerve, the junction, and the anterior horn cell. [4]
Duchenne muscular dystrophy
highest creatine kinase
- X-linked recessive, absent dystrophin, onset 2 to 5 years
- Creatine kinase above 10,000, pseudohypertrophy, Gowers sign
- Loss of ambulation in the first decade
- Confirmed by DMD genetic testing
Becker muscular dystrophy
allelic and milder
- Same gene, partly functional dystrophin, onset 5 to 15 years
- Ambulation preserved into adulthood
- Cardiomyopathy may precede weakness
- Creatine kinase markedly but variably elevated
Spinal muscular atrophy
anterior horn cell
- Symmetric proximal weakness with areflexia
- Creatine kinase normal or only mildly raised
- Fasciculations of the tongue in the infant
- SMN1 deletion on genetic testing
Limb-girdle muscular dystrophy
phenocopy
- Proximal weakness that mimics dystrophinopathy
- Creatine kinase markedly elevated
- Mostly autosomal recessive, affects girls equally
- Dystrophin normal on biopsy, panel of genes diagnostic
Spinal muscular atrophy is the chief mimic, because it also produces a symmetric, proximal weakness in a young child, but its creatine kinase is normal and its hallmark is the areflexia and the tongue fasciculations of anterior horn cell loss, and it yields to an SMN1 deletion rather than a DMD variant. The limb-girdle muscular dystrophies are the other close phenocopies, with a proximal weakness and a high creatine kinase, but they are mostly autosomal recessive, they affect girls as often as boys, and they need a gene panel or a muscle biopsy to separate them from the dystrophinopathies. [4]
The inflammatory myopathies, dermatomyositis and polymyositis, can raise the creatine kinase and weaken the proximal muscles, but their course is faster, often over weeks, and dermatomyositis carries its skin signs. Congenital myopathies and metabolic myopathies are considered when the creatine kinase is lower or the history points to an infant-onset hypotonia. The message for the exam is that a markedly elevated creatine kinase in a weak boy points to a dystrophic process, and the first and most important test after the creatine kinase is the DMD genetic test. [4]
Clinical & Bedside Assessment
The assessment begins with the history, and the two questions that matter most are when the boy walked and whether any male relative was weak or died young. A boy who walked after eighteen months, who never ran or jumped, and whose maternal uncle uses a wheelchair or died of heart failure in his twenties is the boy the fellow must not send home. The history also gathers the cognitive and behavioural picture, the school performance, and the swallowing and breathing concerns, because these shape the care as much as the muscle. [4]
The examination looks for the signs that the muscle is failing. The gait is observed first, and the waddle of pelvic-girdle weakness, the toe-walking of calf tightness, and the arched back of lumbar lordosis are sought. The boy is asked to rise from the floor, and the Gowers sign, the hands climbing the thighs, is noted and timed. The calves are palpated for the firm pseudohypertrophy, the deep tendon reflexes are tested for their loss, and the joints are checked for contractures of the Achilles, the hamstrings, and the iliotibial bands. The spine is examined for scoliosis once the boy is in a wheelchair. [4]
The bedside and functional assessment of a boy with a dystrophinopathy
Observe the gait for the waddle, the toe-walking, and the lumbar lordosis of pelvic-girdle weakness
Ask the boy to rise from the floor and time it, looking for the Gowers sign and the hands climbing the thighs
Palpate the calves for the firm pseudohypertrophy of fat and fibrosis and test the deep tendon reflexes for their loss
Run the timed function tests, the time to stand, the ten-metre run, and the climb of four stairs, to grade and track the motor function
Check the joints for contractures, the spine for scoliosis, and the heart for the gallop or murmur of early cardiomyopathy
Confirm the weight and plot the growth, because the glucocorticoid dose is weight-based and the steroids alter both
Assess the development and the cognition, because dystrophin is expressed in the brain and the learning profile shapes the plan
The timed function tests turn the bedside observation into numbers that track the disease and the response to treatment. The time to stand from the floor, the ten-metre run, the time to climb four stairs, and the six-minute walk are measured at each visit, because the trend over time, more than any single value, reveals the plateau that should trigger the glucocorticoid decision and the decline that heralds the loss of ambulation. The cardiac and respiratory findings are sought at every visit, because a gallop or a raised respiratory rate may be the first sign that the heart or the lungs have joined the muscle in failing. [4]
Investigations
The creatine kinase is the single most powerful screening test, and it is the test that should come to mind the moment a boy is suspected of a muscle disease. In Duchenne the creatine kinase is markedly elevated, typically above 10,000 units per litre and often between 10,000 and 100,000, which is ten to a hundred times the normal value, and it is raised from birth, before any weakness appears, which is the basis of newborn screening. The liver enzymes, the alanine and aspartate aminotransferase, are also raised, because they leak from the same damaged muscle, and the fellow who orders them for a suspected liver problem must remember that a high transaminase in a weak boy is muscle, not liver. [3]
The diagnosis is now confirmed by genetic testing, and the genetic test has largely replaced the muscle biopsy as the first investigation. Multiplex ligation-dependent probe amplification is the first-line method, because it detects the exon deletions and duplications that account for the majority of cases. If the multiplex test is normal but the creatine kinase is high, the next step is sequencing of the whole DMD gene to find the point mutations, the small insertions, and the splice changes. A muscle biopsy with dystrophin immunostaining and western blot is reserved for the small number of boys whose genetics are inconclusive, and it shows the absent or reduced protein directly. [3]
Prednisolone or prednisone for Duchenne muscular dystrophy
Dose
0.75 mg per kg per day, started at the motor plateau and continued through loss of ambulation
Deflazacort for Duchenne muscular dystrophy
Dose
0.9 mg per kg per day, started at the motor plateau
The systems surveillance runs in parallel with the genetics, and it begins at diagnosis. A baseline electrocardiogram and echocardiogram are done when the diagnosis is made, and they are repeated at least annually, because the cardiomyopathy is progressive and often silent in the young boy. Spirometry is added from the age of five or six, or once the boy is non-ambulatory, and the forced vital capacity is tracked as the bellwether of the respiratory decline. Bone density, the eyes, and the swallowing are assessed as the disease and the steroids demand, and a developmental and cognitive assessment completes the picture. [5]
Management — Resuscitation
Duchenne muscular dystrophy is a chronic disease, but it carries a small set of acute, time-critical dangers that the fellow must meet with the same discipline as a resuscitation. The first and the most feared is the anaesthetic crisis. A boy with a dystrophinopathy who receives suxamethonium or a volatile anaesthetic agent can suffer a catastrophic, anaesthesia-induced rhabdomyolysis with a runaway hyperkalaemia and a cardiac arrest, and the safe path is a total intravenous technique that avoids both. Every boy with a confirmed or suspected dystrophinopathy who presents for surgery is flagged to the anaesthetist before the day. [6]
The second danger hides in the glucocorticoid itself. A boy on chronic prednisolone or deflazacort has a suppressed adrenal axis, and the stress of a febrile illness, a fracture, or an operation can tip him into an adrenal crisis with hypotension and collapse. The rule is to double or triple his usual steroid dose for a moderate illness and to give parenteral hydrocortisone for a severe illness or a major operation, and the family is taught this sick-day rule as surely as a family with congenital adrenal hyperplasia is taught it. [6]
[6]The third danger is the respiratory failure that creeps in once the boy is in a wheelchair. The respiratory muscles weaken, the cough loses its force, and the boy hypoventilates at night before he struggles by day. A boy who presents with new breathlessness, orthopnoea, morning headaches, or a faltering concentration is tested at once with a forced vital capacity, a peak cough flow, and an overnight oximetry or capnography, and non-invasive ventilation is started when the night-time breathing fails. The fourth is the cardiac decompensation, a sudden heart failure from the dilated cardiomyopathy, which is managed with the standard heart-failure drugs and an urgent cardiology review. [5]
Management — Definitive & Stepwise
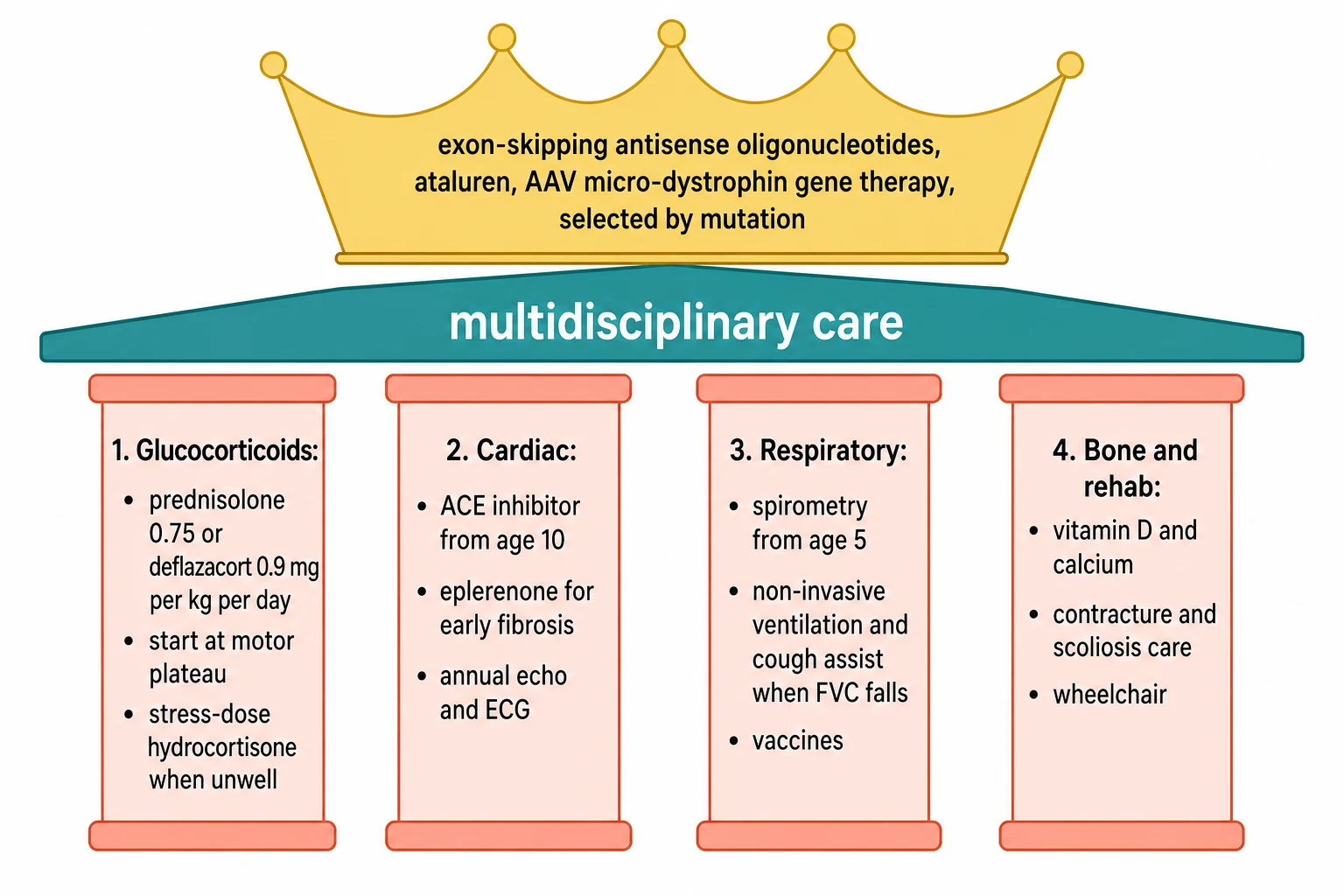
The definitive care of a boy with a dystrophinopathy is a lifelong, multidisciplinary programme built on four pillars and crowned by the precision therapies. The first pillar is the glucocorticoid, and it is the single most important disease-modifying treatment in the disease. The care considerations say to start the steroid when the boy reaches his motor plateau, the point at which he stops gaining new skills, typically between four and seven years, and to continue it through the loss of ambulation and beyond. The FOR-DMD trial of Griggs and colleagues settled the choice of agent and the dose, showing that prednisone 0.75 mg per kg per day, prednisolone at the equivalent dose, and deflazacort 0.9 mg per kg per day all improve motor function over placebo, and that deflazacort carries less weight gain. [7][4]
[4] [5]The second pillar is the cardiac care. The dilated cardiomyopathy is progressive and often silent in the young boy, so an angiotensin-converting-enzyme inhibitor or an angiotensin receptor blocker is started prophylactically by the age of ten in the boy with Duchenne, even before symptoms, because the early suppression of the harmful remodelling delays the heart failure. The eplerenone trial of Raman and colleagues showed that the mineralocorticoid antagonist slows the fibrosis that precedes the pump failure in boys with early cardiac changes on magnetic resonance imaging. An echocardiogram and an electrocardiogram are done at diagnosis and at least yearly, and the manifesting carriers and the Becker patients are watched just as closely, because their hearts can fail before their legs. [9][5]
The third pillar is the respiratory care. The respiratory function is tracked with spirometry from the age of five or six, and the forced vital capacity is the bellwether of decline. When the forced vital capacity falls below half of the predicted value, or the peak cough flow drops, the boy is assessed for nocturnal hypoventilation, and non-invasive ventilation is started for the overnight failure, with a cough-assist device to clear the secretions and the routine immunisations of influenza and pneumococcus to prevent the infections that tip the weak lungs into failure. The fourth pillar is the bone and the rehabilitation, with calcium and vitamin D to blunt the steroid osteoporosis, bisphosphonates for the fractures, physiotherapy and orthoses for the contractures, and the timely provision of the wheelchair and the scoliosis surgery. [5]
FOR-DMD — deflazacort versus prednisone versus placebo (Griggs et al, Neurology 2016)
Key finding
In 196 ambulatory boys aged 4 to 7 years with Duchenne muscular dystrophy, both deflazacort 0.9 mg per kg per day and prednisone 0.75 mg per kg per day improved the muscle strength score over placebo over 52 weeks, with a comparable gain in motor function, but deflazacort produced significantly less weight gain than prednisone. Prednisolone 0.75 mg per kg per day and deflazacort 0.9 mg per kg per day are thus the equivalent, evidence-based glucocorticoid doses, and deflazacort is preferred where weight gain is the dominant concern.
Crowning the four pillars is the precision therapy, and the choice rests entirely on the mutation. The exon-skipping antisense oligonucleotides bind the messenger RNA and skip a neighbouring exon to restore the reading frame, so a shorter but functional dystrophin is produced, and each drug targets a specific exon, eteplirsen for exon 51, golodirsen and viltolarsen for exon 53, and casimersen for exon 45. The proof that a skipped exon makes dystrophin came from the eteplirsen study of Mendell and colleagues. For the boys with a nonsense mutation, ataluren promotes read-through of the premature stop codon. For boys with a confirmed mutation and suitable age, the adeno-associated-virus gene therapy of delandistrogene moxeparvovec delivers a micro-dystrophin, and the EMBARK phase 3 trial of Mendell and colleagues tested its effect on motor function. [8][10]
Specific Subtypes & Scenarios
Becker muscular dystrophy is the subtype that rewards careful surveillance more than aggressive muscle therapy. The Becker patient walks into adulthood, his creatine kinase is high but his weakness is mild, and his chief threat is the heart. A dilated cardiomyopathy can declare itself before any weakness, and the fellow who meets a young man with an unexplained cardiomyopathy orders a creatine kinase and a DMD analysis, because the diagnosis changes the surveillance of his heart and the hearts of his female relatives. [11]
The manifesting carrier is the girl who carries one altered copy and shows signs, and she is the scenario that the exam tests through the family. Skewed X-inactivation leaves enough diseased fibres to raise the creatine kinase, to weaken her proximal muscles, and, most importantly, to risk a dilated cardiomyopathy that can strike even a carrier who has no weakness at all. Every confirmed or potential carrier is offered a baseline cardiac assessment and lifelong cardiac surveillance, because the carrier heart fails without warning. [3]
The intermediate dystrophinopathy is the boy who sits between Duchenne and Becker, ambulating into his mid-teens but with a more severe course than Becker, and his management follows the Duchenne pathway. The exon-skipping-eligible boy is a scenario of its own, because his deletion must be tested against the drugs, and the small fraction who are amenable to exon 51, 53, or 45 skipping are identified only by a careful reading of his genetic report. The newborn-screened infant is the newest scenario, in which a markedly elevated creatine kinase on a heel-prick card brings a well baby to the clinic months before any weakness, and the family is counselled, the diagnosis is confirmed, and the surveillance begins before the disease declares itself. [4]
Complications & Pitfalls
The complications of the dystrophinopathies are the complications of the muscle, the heart, the lungs, and the bone. The progressive weakness costs the boy his walking, then his arms, and finally his breathing, and the contractures and the scoliosis fix his posture as the muscle is lost. The dilated cardiomyopathy is now the leading cause of death in the well-treated boy, because the steroids and the ventilation have pushed the respiratory failure back, and the heart is the organ that the care must now protect. The respiratory failure, the nocturnal hypoventilation, and the weak cough that cannot clear a chest infection are the other end of the disease. [5]
The complications of the glucocorticoid are the price of the benefit, and the fellow must anticipate each. The weight gain and the growth retardation change the body, the behavioural change and the mood swings test the family, the delayed puberty compounds the short stature, and the cataracts and the glucose intolerance are watched for at each visit. The osteoporosis is the gravest of the steroid harms, because a vertebral crush fracture or a fractured femur can tip an ambulatory boy out of his walking for good, and the calcium, the vitamin D, the bisphosphonates, and the fall precautions are the countermeasures. [4]
The pitfalls are the errors that cost the boy years. The first is the delayed diagnosis, the boy sent away as clumsy or lazy until the calf pseudohypertrophy and the high creatine kinase are finally noticed, by which time the window for the early steroid has narrowed. The second is the failure to look at the heart, because a silent cardiomyopathy is missed without the annual echocardiogram, and a carrier mother or sister can die of an unwatched heart. The third is the anaesthetic death, the boy given suxamethonium for a routine procedure by a team that did not know the diagnosis. The fourth is the untreated respiratory failure, the boy who struggles at night but whose overnight oximetry was never checked. [6]
Prognosis & Disposition
The prognosis of Duchenne has been rewritten by the modern care, and the fellow must give the family a prognosis that reflects the present, not the past. Untreated, the boy loses ambulation between the ages of nine and eleven, and he dies of the heart and the lungs in his late teens. With the glucocorticoids, the cardiac and the respiratory surveillance, and the timely ventilation, the boy walks into his second decade, and his survival now extends into his twenties and thirties, and for some into the fourth decade. The Becker patient, with his milder course, walks into middle age, but his life is still bounded by his heart. [4]
The determinants of the outcome are the completeness of the care and the age at which it begins. The boy who is diagnosed early, who receives the glucocorticoid at the motor plateau, whose heart is treated before it fails, and whose lungs are ventilated before they collapse, is the boy who lives longest. The determinants also include the mutation, because the precision therapy now opens a different door for the boy whose deletion is amenable to an exon-skip or a gene transfer. [10]
The disposition is to a neuromuscular multidisciplinary centre, and this is where the modern care is delivered. The neurologist, the cardiologist, the respiratory physician, the physiotherapist, the occupational therapist, the orthotist, the endocrinologist, the genetic counsellor, the psychologist, and the social worker meet the boy and the family together, because the fragmentation of the care is itself a threat to the outcome. As the boy grows, the plan moves with him, from the playground and the school, through the wheelchair and the spine, to the transition to the adult neuromuscular service, where the heart, the lungs, and the independence are the new frontiers. [6]
Special Populations
The manifesting carrier is the population that the fellow must not forget, because she sits in the family of every affected boy. The girl or woman who carries one altered DMD copy is usually well, but the skewed inactivation of her X chromosome can leave her with a raised creatine kinase, a mild proximal weakness, and, above all, a cardiomyopathy that is independent of her muscle strength. Every carrier is offered a baseline cardiac assessment and lifelong cardiac surveillance, because the carrier heart fails without the warning that the weakness gives the boy. [3]
The Aboriginal and Torres Strait Islander child and the child from a remote setting more often meets the disease late, after the window for the early steroid has narrowed, because the access to the creatine kinase and the genetic test is distant and the multidisciplinary centre is far. The retrieval and the telehealth connection to the neuromuscular centre are part of the care for these families, and the cultural safety of the counselling and the kinship mapping of the inheritance are part of the genetic work. [6]
The technology-dependent boy, the young man on the non-invasive or the invasive ventilation, is the population that the paediatrician and the adult physician share. The transition to the adult neuromuscular service is a deliberate, planned handover that begins in early adolescence, because the young man who is lost between the services is the young man whose heart and lungs go unwatched. The family that carries the gene, the sibling who is a carrier, and the affected boy who becomes a young man all move through a lifelong plan that the fellow helps to hold. [6]
Evidence, Guidelines & Regional Differences
The evidence base of the dystrophinopathies is anchored by the care considerations, which are the single most testable source. The Bushby guidelines of 2010 laid out the diagnosis and the pharmacological and psychosocial management, and the Birnkrant update of 2018 extended them across the three parts of neuromuscular and rehabilitation care, respiratory and cardiac and bone care, and primary care and emergency and transition care. These considerations give the glucocorticoid doses, the cardiac surveillance schedule, the respiratory thresholds, and the bone protection that define the modern standard. [3][4][5][6]
The trial evidence settles the two questions that the exam asks. The FOR-DMD trial of Griggs and colleagues established that deflazacort 0.9 mg per kg per day and prednisone 0.75 mg per kg per day are equivalent and superior to placebo, and that deflazacort spares the weight gain. The eplerenone trial of Raman and colleagues showed that the mineralocorticoid antagonist slows the early myocardial fibrosis, and the EMBARK phase 3 trial of Mendell and colleagues tested the adeno-associated-virus micro-dystrophin gene therapy, which is the newest of the precision therapies. [7][9][10]
The regional differences turn on the access to the multidisciplinary centre, the newborn screening, and the funding of the precision drugs. Australia funds the multidisciplinary care through the national disability scheme and the drug access through the pharmaceutical benefits scheme, the United Kingdom through the National Health Service and the National Institute for Health and Care Excellence, and the access in the rural and the remote and the low-resource settings is uneven. The newborn screening for Duchenne, by a creatine kinase on the heel-prick card, is offered in some regions and debated in others, because it brings the diagnosis to a well baby months before any weakness. [6]
Exam Pearls
The creatine kinase is the answer that opens the topic, and a value above 10,000 units per litre in a weak boy is a dystrophic process until the genetics prove otherwise. The disease is X-linked recessive, the boys are affected, the girls are carriers, and about a third of cases are new mutations. Dystrophin links the actin cytoskeleton to the dystrophin-associated glycoprotein complex, its loss tears the sarcolemma, and the reading-frame hypothesis explains why the in-frame deletion gives Becker and the out-of-frame deletion gives Duchenne. [2]
[4] [7]The glucocorticoid is the disease-modifying backbone, with prednisolone 0.75 mg per kg per day or deflazacort 0.9 mg per kg per day, and its harms are the weight, the growth, the behaviour, the bone, and the adrenal axis. The cardiac care turns on the angiotensin-converting-enzyme inhibitor by age 10 and the eplerenone for the early fibrosis, the respiratory care turns on the forced vital capacity and the non-invasive ventilation, and the bone care turns on the vitamin D and the bisphosphonates. The precision therapy is chosen by the mutation, with exon-skipping for the eligible deletions and the adeno-associated-virus micro-dystrophin for the suitable boy, and the two trials to name are FOR-DMD and EMBARK. [7][10]
References
- [1]Hoffman EP, Brown RH Jr, Kunkel LM Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell, 1987.PMID 3319190
- [2]Koenig M, Beggs AH, Moyer M The molecular basis for Duchenne versus Becker muscular dystrophy: correlation of severity with type of deletion. Am J Hum Genet, 1989.PMID 2491009
- [3]Bushby K, Finkel R, Birnkrant DJ Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and pharmacological and psychosocial management. Lancet Neurol, 2010.PMID 19945913
- [4]Birnkrant DJ, Bushby K, Bann CM Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and neuromuscular, rehabilitation, endocrine, and gastrointestinal and nutritional management. Lancet Neurol, 2018.PMID 29395989
- [5]Birnkrant DJ, Bushby K, Bann CM Diagnosis and management of Duchenne muscular dystrophy, part 2: respiratory, cardiac, bone health, and orthopaedic management. Lancet Neurol, 2018.PMID 29395990
- [6]Birnkrant DJ, Bushby K, Bann CM Diagnosis and management of Duchenne muscular dystrophy, part 3: primary care, emergency management, psychosocial care, and transitions of care across the lifespan. Lancet Neurol, 2018.PMID 29398641
- [7]Griggs RC, Miller JP, Greenberg CR Efficacy and safety of deflazacort vs prednisone and placebo for Duchenne muscular dystrophy. Neurology, 2016.PMID 27566742
- [8]Mendell JR, Rodino-Klapac LR, Sahenk Z Eteplirsen for the treatment of Duchenne muscular dystrophy. Ann Neurol, 2013.PMID 23907995
- [9]Raman SV, Hor KN, Mazur W Eplerenone for early cardiomyopathy in Duchenne muscular dystrophy: a randomised, double-blind, placebo-controlled trial. Lancet Neurol, 2015.PMID 25554404
- [10]Mendell JR, Muntoni F, McDonald CM AAV gene therapy for Duchenne muscular dystrophy: the EMBARK phase 3 randomized trial. Nat Med, 2025.PMID 39385046
- [11]Emery AE, Skinner R Clinical studies in benign (Becker type) X-linked muscular dystrophy. Clin Genet, 1976.PMID 975594