Paeds · neurology-neurodisability-and-neuromuscular
Guillain-Barré syndrome and acute flaccid paralysis
Also known as Acute inflammatory demyelinating polyradiculoneuropathy · AIDP · Acute motor axonal neuropathy · AMAN · Miller Fisher syndrome · Acute flaccid paralysis
Fellowship guide to Guillain-Barre syndrome and acute flaccid paralysis in children. Covers the Brighton and Asbury Cornblath diagnostic criteria with the albuminocytological dissociation in the cerebrospinal fluid, the subtypes AIDP, AMAN, AMSAN, the Miller Fisher syndrome, and Bickerstaff brainstem encephalitis with their anti-ganglioside antibody associations, the molecular mimicry by which Campylobacter jejuni lipo-oligosaccharide provokes cross-reactive antibodies against GM1, GD1a, and GQ1b with macrophage-mediated demyelination and axonal injury, the ascending symmetrical areflexic presentation and the paediatric differences of pain and refusal to walk, the acute flaccid paralysis differential including transverse myelitis, acute flaccid myelitis, and tick paralysis, the bedside respiratory monitoring with forced vital capacity thresholds under twenty millilitres per kilogram for intensive care, the first-line intravenous immunoglobulin two grams per kilogram over two to five days and plasma exchange as alternative, the lack of benefit of corticosteroids, the Erasmus scores for ventilation and outcome, and the paediatric recovery course.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
Overview & Definition
A previously well child who stops walking over a few days, with legs that feel weak and floppy and reflexes that have quietly disappeared, is presenting one of the most treatable and most time-critical neurological emergencies of childhood. Guillain-Barré syndrome is an acute, monophasic, immune-mediated attack on the peripheral nerves and nerve roots that produces rapidly progressive, symmetrical, ascending flaccid weakness with areflexia, reaching its worst within four weeks. It is the commonest cause of acute flaccid paralysis in children in the post-polio era. [1]
The reason this disease matters above all is that it can stop breathing. The same process that weakens the legs ascends to the respiratory muscles and the cranial nerves, so the child who is still walking in the morning can be in ventilatory failure by nightfall. The single most important habit at the bedside is to measure the forced vital capacity, repeatedly, and to escalate to intensive care before the child tires. Disease-modifying treatment with intravenous immunoglobulin speeds recovery, and the early and systematic supportive care of ventilation, autonomic instability, and pain is what carries the child through. [1]
The framing concept for the paediatrician is acute flaccid paralysis, the public-health and clinical term for any child under fifteen years with sudden-onset flaccid weakness. Guillain-Barré syndrome is its leading non-polio cause, but the same presentation hides transverse myelitis, acute flaccid myelitis, spinal cord compression, tick paralysis, and botulism, each demanding different treatment. The job is to recognise the syndrome, to treat it early, and to exclude the mimics that change everything. [2]
Classification
Guillain-Barré syndrome is sorted in two ways that both change what you do at the bedside. The first sort is by how sure you are of the diagnosis, which is captured by the Brighton criteria and sets when you treat. The second sort is by which part of the nerve is injured, which predicts recovery and the antibody pattern. [2]
The certainty sort is the validated Brighton criteria of Fokke 2014, which tier the diagnosis into three levels. Level one needs the typical clinical picture plus at least one supportive feature and an electrophysiology pattern plus a raised cerebrospinal fluid protein with a low cell count. Level two drops one of the ancillary supports. Level three is the clinical picture alone with no better explanation. The point of the tiering is practical: a Brighton level three child with ascending areflexic weakness is treated now, not when the lumbar puncture returns, because the cerebrospinal fluid can be normal in the first week. [2]
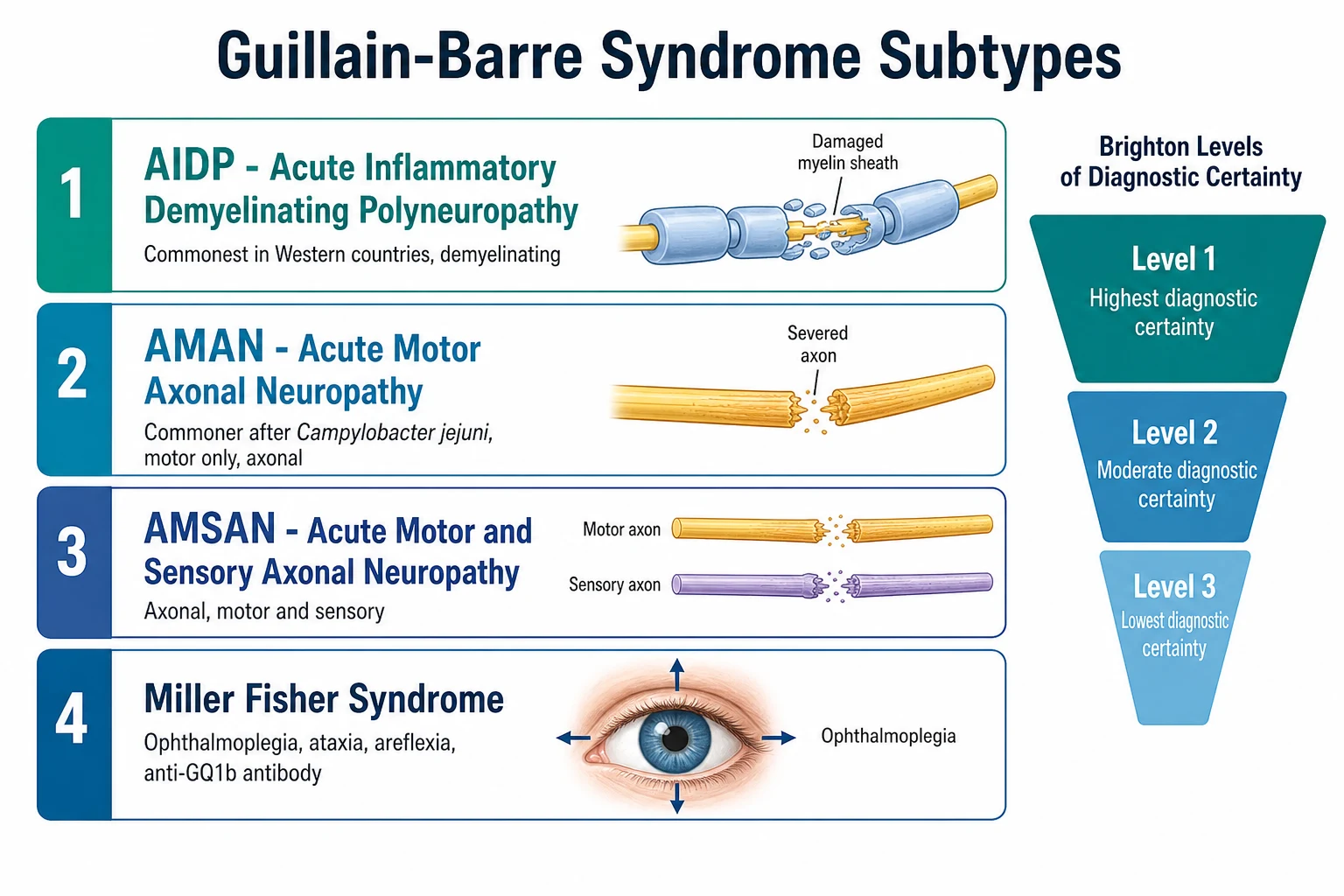
The injury sort divides the disease into the demyelinating and the axonal families. Acute inflammatory demyelinating polyradiculoneuropathy, or AIDP, is the dominant form in Western children and damages the myelin sheath, so it usually recovers well as myelin regrows. Acute motor axonal neuropathy, or AMAN, and acute motor and sensory axonal neuropathy, or AMSAN, sever the axons themselves and are commoner after Campylobacter jejuni and in East Asian children, with a recovery that depends on axonal regrowth over months. The Miller Fisher syndrome adds ophthalmoplegia, ataxia, and areflexia and carries the anti-GQ1b antibody, and it overlaps with Bickerstaff brainstem encephalitis, which adds drowsiness. [1]
Epidemiology & Risk Factors
Guillain-Barré syndrome can occur at any age, but its incidence rises through life. In children the rate sits around one to two per one hundred thousand per year, and boys are affected slightly more often than girls. In the prospective Korinthenberg multicentre study of childhood disease, the median age was around six to seven years, and recovery was faster and more complete than in adults. [11]
Around two thirds of children have an infection in the one to three weeks before the weakness begins, and the antecedent illness is the single most useful clue to the mechanism and the subtype. Campylobacter jejuni gastroenteritis is the strongest and commonest antecedent, and it is linked specifically to the axonal AMAN and AMSAN subtypes and to anti-ganglioside antibodies. Cytomegalovirus, Epstein-Barr virus, Mycoplasma pneumoniae, influenza, and upper respiratory viruses follow, and Zika virus triggered well-described outbreaks in adults. Rarely, the disease follows surgery or a vaccine, but modern influenza vaccines carry only a tiny excess risk. [1]
Pathophysiology
The mechanism is molecular mimicry, and the single sentence to hold is that the germ wears the same coat as the nerve, so the immune attack on the germ turns on the nerve. [1]
Campylobacter jejuni carries on its surface a lipo-oligosaccharide whose terminal sugars are structurally almost identical to the gangliosides GM1, GD1a, and GT1a that stud the surface of peripheral nerve axons and Schwann cells. The host makes IgG antibodies against the bacterial lipo-oligosaccharide, and those antibodies cross-react with the neural gangliosides. The antibodies bind, fix complement, and form the membrane attack complex at the nodes of Ranvier, and macrophages are recruited and strip and sever the myelin or the axon. In AIDP the dominant injury is macrophage-mediated demyelination, which produces conduction block. In AMAN and AMSAN the macrophages invade the periaxonal space and cut the axons, which produces a purely motor or a motor and sensory axonal picture. [1]
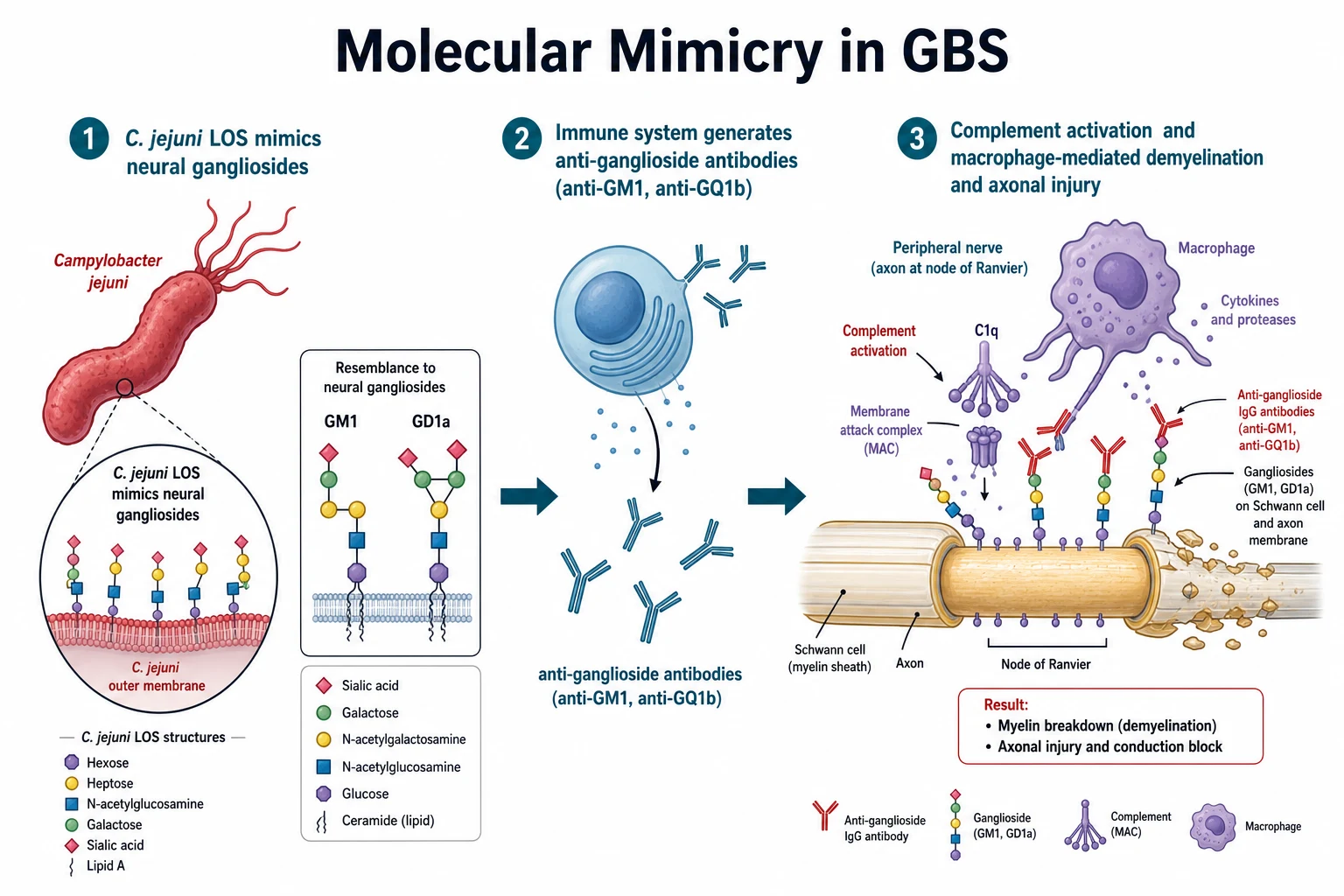
The Miller Fisher syndrome works through the same logic but at a different target. The antibodies bind GQ1b and GT1a gangliosides, which are concentrated in the oculomotor nerves and the muscle spindles, and that distribution explains the ophthalmoplegia, the ataxia, and the areflexia that define the syndrome. The reason the illness is monophasic and self-limiting is that the antibody response resolves over weeks once the antigen is cleared, which is why the disease reaches a nadir and then recovers rather than relapsing. [9]
Clinical Presentation
The child is almost always previously well, and the story is one of progressive weakness over days. The weakness is symmetrical and ascending, beginning in the legs and climbing to the arms, the trunk, and the cranial nerves. A child who walked on Monday is unsteady on Tuesday, refuses to stand on Wednesday, and is paralysed from the neck down by Friday. The deep tendon reflexes disappear early, often before the weakness is severe, and their loss in a weak limb is one of the most valuable bedside signs in paediatrics. [11]
The features that surround the weakness matter because they change the immediate management. Pain in the back and legs is common in children and may be the first complaint, sometimes before overt weakness, and it is easily mistaken for a viral myalgia. Cranial nerve involvement brings facial diplegia, a weak suck and swallow, pooled secretions, and a weak cough, and it predicts aspiration and ventilatory failure. Sensory loss is usually mild and distal, and autonomic dysfunction brings a fluctuating blood pressure, a fast or slow heart rate, and bladder and bowel disturbance that can be mistaken for a cord lesion. [11]
The age of the child bends the presentation, and this is where the diagnosis is missed. A young child often cannot report weakness and instead presents with pain, irritability, reluctance to walk, or regression in walking, so a low threshold for examining the reflexes and for spirometry is essential. The adolescent may present with a Miller Fisher or bulbar variant, and the post-infectious story is clearer. The tempo is subacute over days to a maximum of four weeks to the clinical nadir, and a faster onset to nadir predicts a more severe course and a higher chance of ventilation. [12]
ASCEND
Differential Diagnosis
The differential is the acute flaccid paralysis list, and the task is not to memorise it but to separate the mimics that change the treatment. The discriminating features are the symmetry, the reflex pattern, the presence of a sensory level, and the bowel and bladder function. [1]
The first fork is between Guillain-Barré syndrome and a spinal cord lesion. Transverse myelitis presents with weakness that is more often asymmetric and below a spinal cord level, with a clear sensory level, and with bladder and bowel dysfunction, and it needs an urgent spinal MRI. Acute flaccid myelitis, the polio-like anterior horn cell lesion, presents with focal, asymmetric, flaccid limb weakness, often after a viral respiratory illness, with an MRI showing longitudinally extensive anterior cord T2 hyperintensity. Spinal cord compression from tumour, abscess, or epidural haematoma produces back pain and a sensorimotor level and is a surgical emergency. The rule that protects all three is that any sensory level, any bladder or bowel dysfunction, or any asymmetric weakness triggers an urgent MRI before a confident diagnosis of Guillain-Barré syndrome is made. [1]
The remaining mimics each carry a specific pitfall. Tick paralysis produces an ascending areflexic weakness that can fool even an experienced clinician, and the cure is to find and remove the embedded tick, so examine the scalp and skin. Botulism descends rather than ascends, with cranial nerve palsies, dilated or sluggish pupils, and constipation, and it needs antitoxin. Periodic paralysis presents more acutely and is usually associated with a low potassium. Myasthenia gravis is fatigable rather than fixed. Hypokalaemic or hyperkalaemic paralysis and heavy-metal or toxin exposure complete the list, and the clinching combination is the symmetrical ascending areflexic weakness with albuminocytological dissociation. [1]
Clinical & Bedside Assessment
The bedside assessment runs in parallel with monitoring, because a child with Guillain-Barré syndrome can deteriorate quickly. Secure the airway and assess the bulbar function, the swallow, and the cough, because aspiration and ventilatory failure are the immediate threats. Place the child on cardiac and haemodynamic monitoring for autonomic instability, and measure the bedside spirometry in any child with progressive weakness. [12]
The respiratory monitoring is the part that saves lives, and it is the single most examined bedside skill in this topic. Measure the forced vital capacity, the maximum inspiratory pressure, and the maximum expiratory pressure every two to six hours in any child with progressive weakness or bulbar involvement. Escalate to intensive care for a forced vital capacity under twenty millilitres per kilogram, a fall of greater than thirty percent from baseline, a maximum inspiratory pressure under negative sixty centimetres of water, or bulbar weakness with aspiration risk. The child who is still talking can have a falling vital capacity, so the numbers, not the bedside appearance, drive the decision to intubate. [12]
The focused examination looks for the pattern that confirms the syndrome and for the features that argue for a mimic. Confirm the symmetrical ascending weakness and the early areflexia. Examine the cranial nerves for facial diplegia, ophthalmoplegia, and a weak cough. Look hard for a sensory level or for bladder or bowel dysfunction, either of which argues for a cord lesion and triggers an urgent MRI. Palpate the scalp and skin for an embedded tick. Take the history of the antecedent illness, the tempo of progression, and any family history of periodic paralysis or neuromuscular disease. [1]
In ANZ practice, retrieval networks coordinate the transfer of any child with progressive weakness to a tertiary paediatric centre with paediatric neurology, intensive care, and neurophysiology. Peripheral centres start the workup and the respiratory monitoring on telephone advice and plan early retrieval, because the antibody and neurophysiology results may take days. The threshold to transfer a child with a falling forced vital capacity is low. [10]
Investigations
The investigations confirm the syndrome, exclude the mimics, and predict the course, and they run together rather than in sequence. [2]
The cerebrospinal fluid is central and shows the classic albuminocytological dissociation, a raised protein with a normal cell count, usually fewer than ten white cells per microlitre. The dissociation often appears only after the first week, so a normal early cerebrospinal fluid never excludes the diagnosis, and repeating the lumbar puncture after a week is reasonable if the picture is unclear. A high cell count in the cerebrospinal fluid argues for an infectious or infiltrative mimic and against uncomplicated Guillain-Barré syndrome. The cerebrospinal fluid is also sent for viral polymerase chain reaction and culture to identify the antecedent infection. [2]
Neurophysiology distinguishes the subtypes and supports the diagnosis. AIDP shows prolonged distal latencies, conduction block, slowed conduction velocities, and absent or prolonged F-waves, which reflect the proximal nerve root injury. AMAN and AMSAN show reduced compound muscle action potentials with relatively preserved sensory action potentials and conduction velocities, reflecting axonal loss. The neurophysiology may be normal very early, so a normal early study does not exclude the diagnosis. [1]
[10]MRI of the spine is obtained whenever a cord lesion must be excluded, and it may show gadolinium enhancement of the cauda equina and the nerve roots, which supports the diagnosis. Ganglioside antibody panels, including anti-GM1, anti-GD1a, and anti-GQ1b, support the subtype and are especially useful in the Miller Fisher syndrome, where the anti-GQ1b antibody is highly characteristic. The Erasmus Guillain-Barré Syndrome Respiratory Insufficiency Score and the modified Erasmus GBS Outcome Score combine bedside features to predict the need for ventilation and the outcome, and they are worth knowing for the exam. [8][9]
Management — Resuscitation
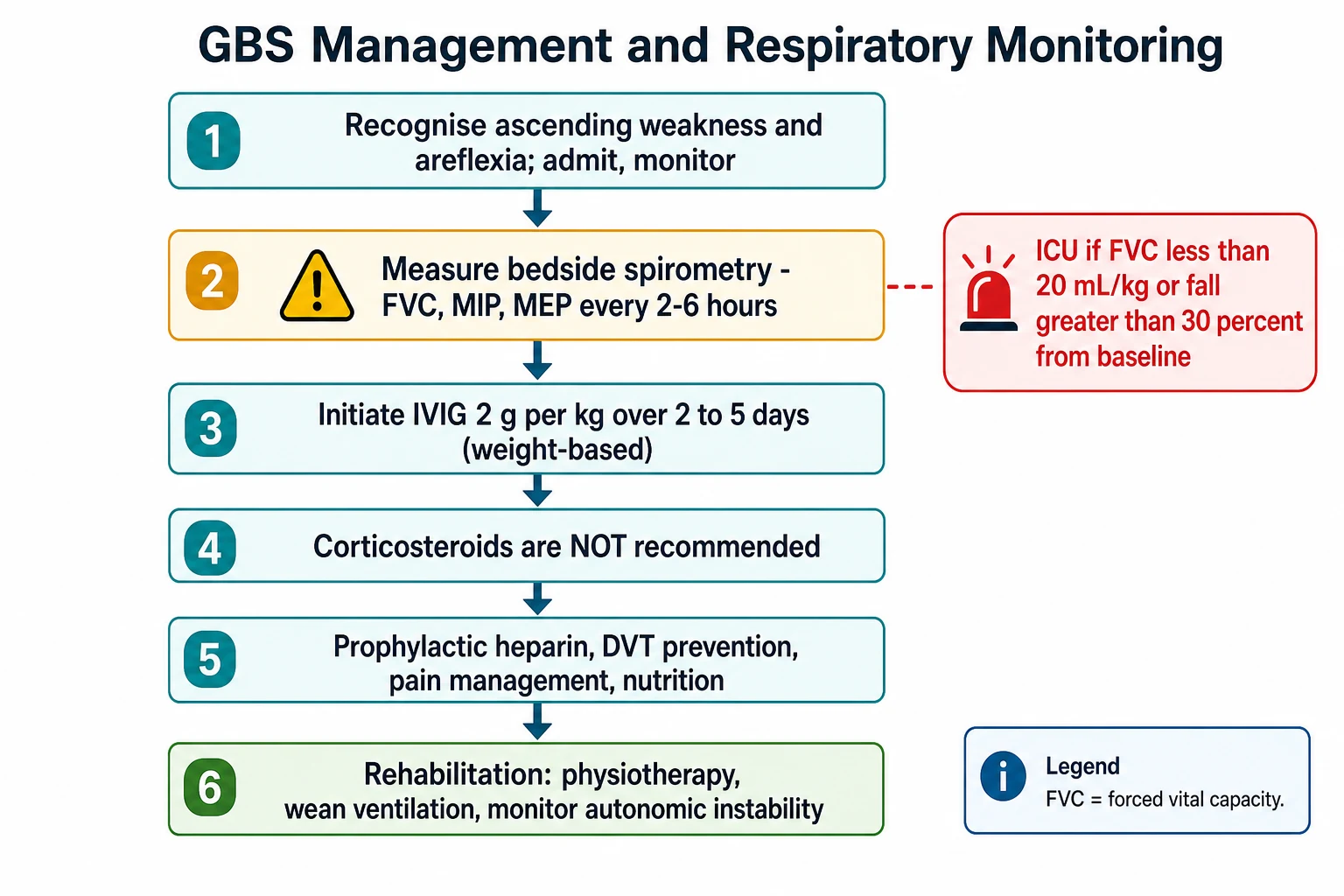
Resuscitation protects the airway and the breathing and manages autonomic instability while the workup runs. Any child with progressive weakness is admitted to a monitored bed with cardiac and haemodynamic monitoring, and the bedside spirometry is set up on a two to six hour cycle. The intensive care team is involved early, and the threshold to intubate is driven by the numbers, not by how the child looks. [12]
Intensive care is indicated for a forced vital capacity under twenty millilitres per kilogram, a fall of greater than thirty percent from baseline, bulbar weakness with aspiration or a weak cough, or autonomic instability with arrhythmia or blood pressure swings. When intubation is needed, the child is preoxygenated and a rapid sequence induction is used, because the weak respiratory muscles and the risk of aspiration make this a high-risk airway. Autonomic instability is managed with careful fluid and vasopressor support and treatment of arrhythmia, and a pacemaker is rarely needed for profound bradycardia. [12]
The workup is sent early and in parallel. The lumbar puncture, the neurophysiology, and the antibody panels are drawn, the MRI is obtained when a cord lesion must be excluded, and the antecedent infection is sought. First-line disease-modifying therapy is started once the diagnosis is made, without waiting for confirmatory results, because early treatment speeds recovery. [10]
Management — Definitive & Stepwise
The definitive treatment is disease-modifying immunotherapy plus systematic supportive care, and the treatment that changes the course is intravenous immunoglobulin. [4]
First-line therapy is intravenous immunoglobulin 2 g per kg over two to five days. The van der Meche 1992 trial showed that immunoglobulin is as effective as plasma exchange and is easier and safer to give, and the Cochrane review confirmed that it speeds recovery compared with supportive care alone. Plasma exchange, five sessions over one to two weeks, is an equally effective alternative that is used when immunoglobulin is contraindicated or unavailable, but it is less practical in children because of the vascular access required. Combining immunoglobulin with plasma exchange adds no benefit over either alone. [4][5][6]
First-line therapy for Guillain-Barre syndrome (paediatric)
The corticosteroid question is the one examiners ask, and the answer must be crisp. Corticosteroids, whether oral or intravenous, are not recommended for Guillain-Barré syndrome. The Hughes 2007 systematic review showed no benefit, and a randomised trial suggested that a short course of intravenous methylprednisolone combined with immunoglobulin may even slow recovery. The reason steroids fail is that the dominant injury is antibody and complement mediated, and by the time weakness appears the membrane attack complex has already done its work. [7]
The management ladder, day by day
Day 0: Recognise ascending areflexic weakness; admit to a monitored bed; secure ABC and assess bulbar function; start bedside spirometry every 2 to 6 hours
Day 0 to 1: Send lumbar puncture, neurophysiology, antibody panels; obtain spinal MRI if any cord-feature present; involve paediatric neurology and intensive care
Day 0 to 5: Start intravenous immunoglobulin 2 g per kg over 2 to 5 days once the diagnosis is made; do not wait for confirmatory results
Day 0 onwards: Intensive care for FVC under 20 mL/kg, fall over 30 percent, bulbar weakness, or autonomic instability; intubate on the numbers
Throughout: Prophylactic anticoagulation, pressure-area care, pain management with gabapentin or opiates, nasogastric or jejunal nutrition, early physiotherapy
Weeks to months: Wean ventilation, neurorehabilitation, paediatric neurology follow-up, school reintegration, fatigue and pain management
Supportive care is the backbone of the course, and it dominates the hospital stay. Prophylactic anticoagulation and compression devices reduce venous thromboembolism, which is a real risk in an immobile child. Pain, which is often severe and neuropathic, is managed with gabapentin or pregabalin and with opiates where needed, and undertreated pain impedes weaning and rehabilitation. Nutrition is delivered by nasogastric or jejunal tube in any child with bulbar weakness. Early physiotherapy prevents contractures, and speech and language and occupational therapy address the prolonged recovery. [10]
Specific Subtypes & Scenarios
AIDP is the commonest subtype in Western children and is managed on the standard ladder above, with intravenous immunoglobulin and respiratory monitoring. The demyelinating injury usually recovers well, because myelin regrows faster than axons. [1]
AMAN and AMSAN are the axonal subtypes, commoner after Campylobacter jejuni and in East Asian children, and linked to anti-GM1 and anti-GD1a antibodies. AMAN is a pure motor axonal neuropathy that may recover rapidly, through reversible conduction failure at the nodes, or may leave residual deficit if axonal degeneration is severe. AMSAN adds sensory axonal injury. The recovery depends on axonal regrowth and is measured in months rather than weeks, and these children more often need prolonged intensive care. [1]
The Miller Fisher syndrome presents with the triad of ophthalmoplegia, ataxia, and areflexia and carries the anti-GQ1b antibody. It overlaps with Bickerstaff brainstem encephalitis, which adds altered consciousness, and both are treated with intravenous immunoglobulin. The pharyngeal-cervical-brachial variant presents with bulbar and upper-limb weakness and sparing of the legs, and it is easily missed because the classic ascending pattern is absent. [1]
The acute flaccid paralysis presentation in a child under fifteen years is a notifiable public-health event. It triggers stool testing for poliovirus, the exclusion of acute flaccid myelitis by MRI, and the consideration of transverse myelitis. The paediatrician who sees acute flaccid paralysis is the front line of polio surveillance, and the notification and the stool sample are part of the management, not an afterthought. [1]
Complications & Pitfalls
The life-threatening complications are the direct features of the disease and the prolonged intensive care course. Ventilatory failure from respiratory muscle weakness needs mechanical ventilation in around ten to fifteen percent of children, often for weeks. Aspiration from bulbar weakness and a weak cough, and autonomic instability with arrhythmia and blood pressure swings, are the immediate threats. The intensive care complications, including ventilator-associated pneumonia, thrombosis, and critical-illness myopathy, add to the burden of a long admission. [12]
The classic pitfalls are the ones examiners reward a candidate for naming. The first is failing to monitor bedside spirometry and missing the falling forced vital capacity. The second is performing a single early lumbar puncture that misses the albuminocytological dissociation and then dismissing the diagnosis. The third is attributing the weakness to a viral illness or myalgia before the reflexes are checked. The fourth is missing a spinal cord compression, a transverse myelitis, or an acute flaccid myelitis because the MRI was not obtained. The fifth is giving corticosteroids in the belief that they help. The sixth is under-treating the pain and the prolonged rehabilitation. [1]
The long-term complications dominate the lived burden of the disease. Residual weakness, especially of the proximal muscles, fatigue that limits school and play, chronic neuropathic pain, and the psychological burden of a prolonged and frightening illness can outlast the acute admission by months. A child who looks recovered may still fatigue quickly, so a structured return to school and activity is part of treatment, not an afterthought. [11]
Prognosis & Disposition
Children with Guillain-Barré syndrome recover faster and more completely than adults, and that fact is the one to give the family. Most children reach the clinical nadir within two to four weeks and begin to recover within weeks. A substantial majority walk independently by six months, and near-complete recovery by one year is the rule in children, in contrast to the slower and less complete recovery in adults. [11]
The predictors of outcome are the levers worth knowing, because they set the prognosis and the intensive care course. Around ten to fifteen percent of children need mechanical ventilation. The predictors of a poorer outcome are a rapid onset to nadir, the axonal AMAN and AMSAN subtypes, severe weakness at the nadir, the need for mechanical ventilation, and a older age within childhood. The Erasmus Guillain-Barré Syndrome Respiratory Insufficiency Score predicts the need for ventilation from the admission features, and the modified Erasmus GBS Outcome Score predicts the outcome at six months. [8][9]
recovery trajectory
Sustained recovery toward independent walking in a substantial majority of children
Disposition follows the tempo and the respiratory status. Any child with a falling forced vital capacity, bulbar weakness, or autonomic instability goes to intensive care. A treated child with stable physiology and rising spirometry is managed on the ward with paediatric neurology input and continued spirometric monitoring. As the acute phase settles, the child moves to neurorehabilitation, which may be inpatient or community-based, and to paediatric neurology follow-up for residual deficit, fatigue, and a structured school return. [10]
Special Populations
The young child often cannot report weakness and instead presents with pain, irritability, or refusal to walk, so a low threshold for examining the reflexes and for spirometry is essential. The child with an existing neurodisability is at particular risk of being mislabelled as a behavioural or functional change, and any deviation from the baseline needs the same workup as a new presentation. [11]
The Aboriginal and Torres Strait Islander child and the child from a remote setting may present late after prolonged weakness, because of distance and access, so retrieval pathways and early specialist input are central, and the threshold to treat and transfer is low. The child from a refugee or migrant family needs an early interpreter and a broad workup that includes the infections and nutritional deficiencies common in the country of origin. The child who presents with acute flaccid paralysis under fifteen years is a public-health notifiable event that triggers stool virology for poliovirus and the exclusion of acute flaccid myelitis. [1]
Evidence, Guidelines & Regional Differences
The diagnostic framework rests on the Asbury and Cornblath 1990 criteria, which codified the clinical picture, and on the validated Brighton criteria of Fokke 2014, which tiered diagnostic certainty into levels one, two, and three and are now the standard in epidemiological studies. The comprehensive Willison 2016 Lancet review set the modern synthesis of the disease. [3][2][1]
The treatment evidence is dominated by the van der Meche 1992 trial, which showed that intravenous immunoglobulin is as effective as plasma exchange and is easier and safer to give. The Cochrane reviews of intravenous immunoglobulin and of plasma exchange confirmed that both speed recovery compared with supportive care alone, and the Hughes 2007 systematic review showed that corticosteroids confer no benefit and may slow recovery, which is why they are not recommended. The Erasmus Respiratory Insufficiency Score and the modified Erasmus Outcome Score were developed by van Doorn and colleagues to predict ventilation and outcome from bedside features. [4][5][6][7][8][9]
van der Meche 1992 — IVIG vs plasma exchange
Multicentre randomised trial of 150 adults with severe Guillain-Barre syndrome comparing intravenous immune globulin 0.4 g per kg per day for five days with plasma exchange
Key finding
Intravenous immunoglobulin was at least as effective as plasma exchange for speeding recovery and had a better safety and convenience profile.
Practice change
Intravenous immunoglobulin 2 g per kg over two to five days became the first-line therapy worldwide and remains the standard for children.
The European paediatric guideline of Korinthenberg 2020 translates the adult evidence into child-specific practice and endorses intravenous immunoglobulin as first-line, respiratory monitoring with bedside spirometry, and the exclusion of the acute flaccid paralysis mimics. The prospective Korinthenberg 2007 multicentre study established the faster and more complete recovery of children and the common paediatric features of pain, cranial nerve involvement, and the young age at presentation. [10][11]
Controversies remain. The role of a second course of intravenous immunoglobulin for a poor early response, the place of complement inhibition with eculizumab in severe disease, the threshold for plasma exchange in children, and the management of the prolonged intensive care course are all active areas. The evidence for eculizumab is limited and it is not yet standard care. [10]
Regional differences are small in principle because the framework is international, but access matters in practice. Rapid neurophysiology, intravenous immunoglobulin, and paediatric intensive care are available promptly in tertiary centres in ANZ, the UK, and North America, but retrieval from remote settings can stretch to days, reinforcing the rule to monitor the forced vital capacity and to treat on suspicion. The threshold to treat and the choice of first-line agent are broadly consistent across the RACP, RCPCH, ABP, and RCPSC contexts. [10]
Exam Pearls
Guillain-Barré syndrome is the commonest cause of acute flaccid paralysis in children in the post-polio era. The weakness is symmetrical, ascending, and areflexic, reaching its nadir within four weeks. The deep tendon reflexes disappear early, often before the weakness is severe, and their loss in a weak limb is a defining sign. [1]
Albuminocytological dissociation is the cerebrospinal fluid hallmark, but it may be absent in the first week, so a normal early lumbar puncture never excludes the diagnosis. The Brighton criteria tier the diagnostic certainty into levels one, two, and three, and a Brighton level three child is treated on the clinical picture. Neurophysiology distinguishes the demyelinating AIDP from the axonal AMAN and AMSAN. [2]
First-line therapy is intravenous immunoglobulin 2 g per kg over two to five days. Plasma exchange is an equally effective alternative. Corticosteroids are not recommended, because the systematic review showed no benefit and possible harm. Intensive care is indicated for a forced vital capacity under twenty millilitres per kilogram or a fall of greater than thirty percent from baseline, and the decision to intubate is driven by the numbers, not by the bedside appearance. [4][7][12]
Campylobacter jejuni precedes the axonal AMAN and AMSAN subtypes. The Miller Fisher syndrome is the triad of ophthalmoplegia, ataxia, and areflexia with the anti-GQ1b antibody. The key mimic to exclude is acute flaccid myelitis, which shows anterior horn cell T2 change on spinal MRI and is polio-like, and any sensory level, bladder or bowel dysfunction, or asymmetric weakness triggers an urgent spinal MRI. The two rules that save children are to monitor the forced vital capacity and to treat early with intravenous immunoglobulin. [1][9]
References
- [1]Willison HJ, Jacobs BC, van Doorn PA Guillain-Barré syndrome. Lancet, 2016.PMID 26948435
- [2]Fokke C, van den Berg B, Drenthen J, et al Diagnosis of Guillain-Barré syndrome and validation of Brighton criteria. Brain, 2014.PMID 24163275
- [3]Asbury AK, Cornblath DR Assessment of current diagnostic criteria for Guillain-Barré syndrome. Ann Neurol, 1990.PMID 2194422
- [4]van der Meché FGA, Schmitz PIM, Dutch Guillain-Barré Study Group A randomized trial comparing intravenous immune globulin and plasma exchange in Guillain-Barré syndrome. N Engl J Med, 1992.PMID 1552913
- [5]Hughes RAC, Swan AV, van Doorn PA Intravenous immunoglobulin for Guillain-Barré syndrome. Cochrane Database Syst Rev, 2014.PMID 25238327
- [6]Raphaël JC, Chevret S, Hughes RAC, Annane D Plasma exchange for Guillain-Barré syndrome. Cochrane Database Syst Rev, 2012.PMID 22786475
- [7]Hughes RAC, Swan AV, Raphaël JC, et al Immunotherapy for Guillain-Barré syndrome: a systematic review. Brain, 2007.PMID 17337484
- [8]van Koningsveld R, Steyerberg EW, Hughes RAC, et al A clinical prognostic scoring system for Guillain-Barré syndrome. Lancet Neurol, 2007.PMID 17537676
- [9]van Doorn PA, Jacobs BC Predicting the course of Guillain-Barré syndrome. Lancet Neurol, 2006.PMID 17110272
- [10]Korinthenberg R, Trollmann R, Felderhoff-Müser U, et al Diagnosis and treatment of Guillain-Barré Syndrome in childhood and adolescence: An evidence- and consensus-based guideline. Eur J Paediatr Neurol, 2020.PMID 31941581
- [11]Korinthenberg R, Schessl J, Kirschner J, Mönting JS Clinical presentation and course of childhood Guillain-Barré syndrome: a prospective multicentre study. Neuropediatrics, 2007.PMID 17607598
- [12]Hu MH, Chen CM, Lin KL, et al Risk factors of respiratory failure in children with Guillain-Barré syndrome. Pediatr Neonatol, 2012.PMID 23084721