Paeds · neurology-neurodisability-and-neuromuscular
Infantile spasms and developmental epileptic encephalopathy
Also known as West syndrome · Infantile epileptic spasms syndrome · IESS · Salaam attacks · Developmental and epileptic encephalopathy · DEE · X-linked infantile spasm syndrome
A fellowship approach to infantile spasms (West syndrome) and the developmental and epileptic encephalopathies: recognise the triad of clustered epileptic spasms, hypsarrhythmia and developmental regression as a neurodevelopmental emergency, confirm with a sleep EEG and MRI brain, classify the cause across the ILAE aetiology categories with tuberous sclerosis as the leading identifiable cause, and start vigabatrin first-line when the cause is TSC and hormonal therapy (high-dose ACTH or oral prednisolone) first-line otherwise, because aetiology and the speed of spasm cessation drive developmental outcome.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
The mark-winning candidate holds three things in view at once. The first is the infant and the family in front of you: the clustered salaam attacks, the loss of social smiling and babbling, and the frightened parents who have often filmed the spasms on a phone. The second is the EEG: hypsarrhythmia is a chaotic, high-voltage, disorganised background that tells you this is an epileptic encephalopathy, not reflux or colic. The third is the clock: infantile spasms are not convulsive status epilepticus, but they are a developmental emergency, and the speed of first-line therapy measurably changes cognition. [1] [11]
Overview & Definition
Infantile spasms are first a seizure type and then a syndrome. As a seizure type, an epileptic spasm is a brief, sudden contraction of the neck, trunk and proximal limb muscles that lasts one to three seconds, and the spasms run together in clusters of 20 to 100 over several minutes, most often as the child wakes from sleep. When these clustered spasms sit alongside a chaotic EEG pattern called hypsarrhythmia and a developmental arrest or regression, the triad is West syndrome, the eponymous syndrome William West described in his own son in 1841. [9]
The International League Against Epilepsy now calls this infantile epileptic spasms syndrome, or IESS, defined in the 2022 neonate-and-infant syndromes position statement by epileptic spasms with peak onset in the first year of life, an EEG that often shows hypsarrhythmia or a modified variant, and developmental delay, plateau or regression. West syndrome remains the classic, tightly defined triad form, while IESS is the current broader syndrome term that also covers infants whose full triad is incomplete. [1]
IESS sits inside a larger family, the developmental and epileptic encephalopathies. In a developmental and epileptic encephalopathy the frequent epileptiform activity itself contributes to cognitive and behavioural impairment over and above the underlying cause, so the seizures and the EEG are not merely a symptom of brain disease but an active cause of developmental harm. This is why prompt spasm cessation is neuroprotective, and why IESS shares a conceptual home with Dravet syndrome, Lennox-Gastaut syndrome, CDKL5 deficiency disorder and STXBP1 encephalopathy. [1] [7]
Classification
The useful way to classify IESS is by aetiology, because the cause determines first-line therapy, the developmental outlook, and the surveillance programme. The ILAE aetiology framework has six categories — structural, genetic, metabolic, infectious, immune and unknown — and more than one can apply to a single child. A tuberous sclerosis cortical tuber is simultaneously structural and genetic, for example, which is why the TSC question is the first branch in the management algorithm. [1]
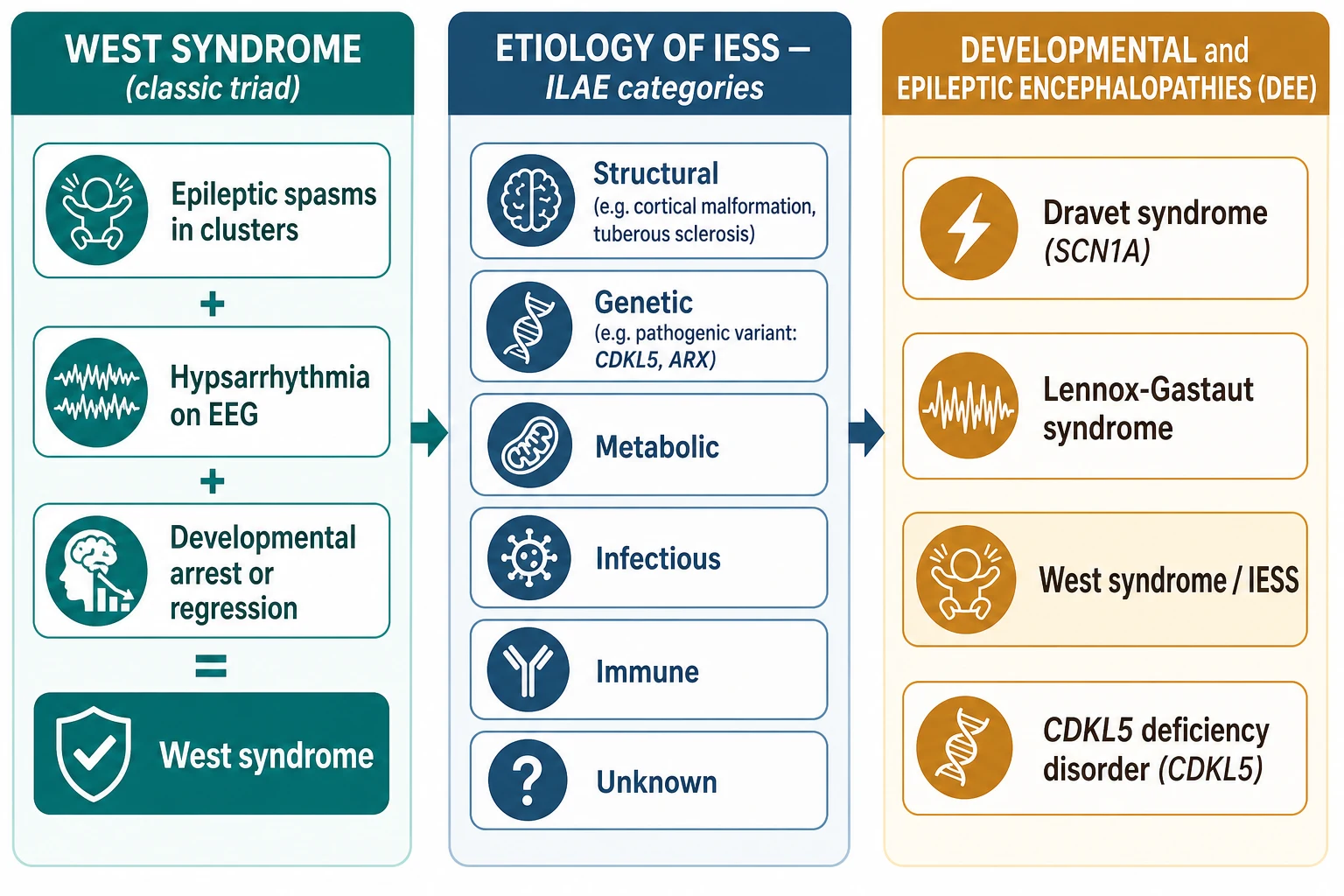
Structural causes include cortical malformations (focal cortical dysplasia, lissencephaly, polymicrogyria), the scar of hypoxic-ischaemic encephalopathy, perinatal arterial stroke, and the tubers of tuberous sclerosis. Genetic causes include pathogenic variants in CDKL5, ARX, STXBP1, KCNQ2, SLC13A5 and the TSC1 and TSC2 genes; metabolic causes include mitochondrial disease, organic acidaemias, and pyridoxine or pyridoxal-phosphate dependency; and a substantial fraction remain unknown even after thorough investigation. The single most clinically useful split is simpler: is the cause tuberous sclerosis complex, or is it not? That one question decides the first-line drug. [5] [10]
| ILAE category | Representative causes | First-line implication |
|---|---|---|
| Structural | Tuberous sclerosis, focal cortical dysplasia, hypoxic-ischaemic injury, stroke | Vigabatrin if TSC; consider epilepsy surgery if a resectable focal lesion |
| Genetic | CDKL5, ARX, STXBP1, KCNQ2, chromosomal anomalies | Hormonal first-line; genetic counselling |
| Metabolic | Mitochondrial disease, organic acidaemias, pyridoxine dependency | Treat the metabolic cause; cofactor trial |
| Infectious / Immune | Congenital infection, rare immune encephalitis | Hormonal first-line; treat the infection |
| Unknown | No cause found after MRI and genetic testing | Hormonal first-line; best developmental prognosis of the symptomatic group |
Epidemiology & Risk Factors
Infantile spasms affect roughly two to five of every 10,000 live births, with onset peaking between four and eight months and almost all cases beginning before twelve months. Boys are slightly more often affected than girls. The syndrome accounts for a small fraction of childhood epilepsy by case count but for a disproportionate share of epilepsy-related neurodisability, because it strikes the brain during a critical window of cognitive development. [9] [11]
The risk factors are the causes themselves. Anything that injures or malforms the developing brain during the third trimester or the first postnatal months raises the risk: hypoxic-ischaemic injury, prematurity with intraventricular haemorrhage, congenital brain malformation, perinatal stroke, chromosomal anomalies, and the genetic tuberous sclerosis and single-gene DEEs. A family history of epilepsy, learning difficulty or neurocutaneous stigmata raises the probability of a genetic cause, and an affected sibling raises the question of an inherited or metabolic disorder. [5]
The epidemiological fact that changes practice is the time-to-treatment effect. Infants whose spasms are controlled promptly have better developmental outcomes than those who wait, and the lag from spasm onset to first-line therapy is a modifiable risk factor. Symptomatic (structural or metabolic) aetiology, a longer lag, and failure to achieve spasm cessation at two weeks each predict poorer cognition and a higher risk of evolution to Lennox-Gastaut syndrome. [3] [11]
Pathophysiology
Infantile spasms are a disease of the immature brain, not of the adult brain, and that single fact explains most of their behaviour. During the first year of life the cerebral cortex is in a critical window of synaptogenesis, dendritic refinement and myelination, so a focal lesion or a genetic disturbance does not produce a tidy focal seizure the way it would in an older child. Instead, the abnormal activity recruits a widespread, hypersynchronous cortico-subcortical network that generates the spasm semiology and the chaotic, high-voltage hypsarrhythmia on the EEG. [7]
The cascade begins with a source of abnormal excitability — a cortical malformation, a tuber, an area of gliosis, or an ion-channel or synaptic-protein defect from a pathogenic variant. Because the surrounding cortex is immature and poorly inhibitory, that source spreads rapidly across both hemispheres. The result is a diffusely disorganised cortical network that fires in random, high-amplitude bursts, which is exactly what hypsarrhythmia looks like on paper: chaotic, high-voltage slow waves with multifocal spikes and no recognisable background rhythm. [7] [11]
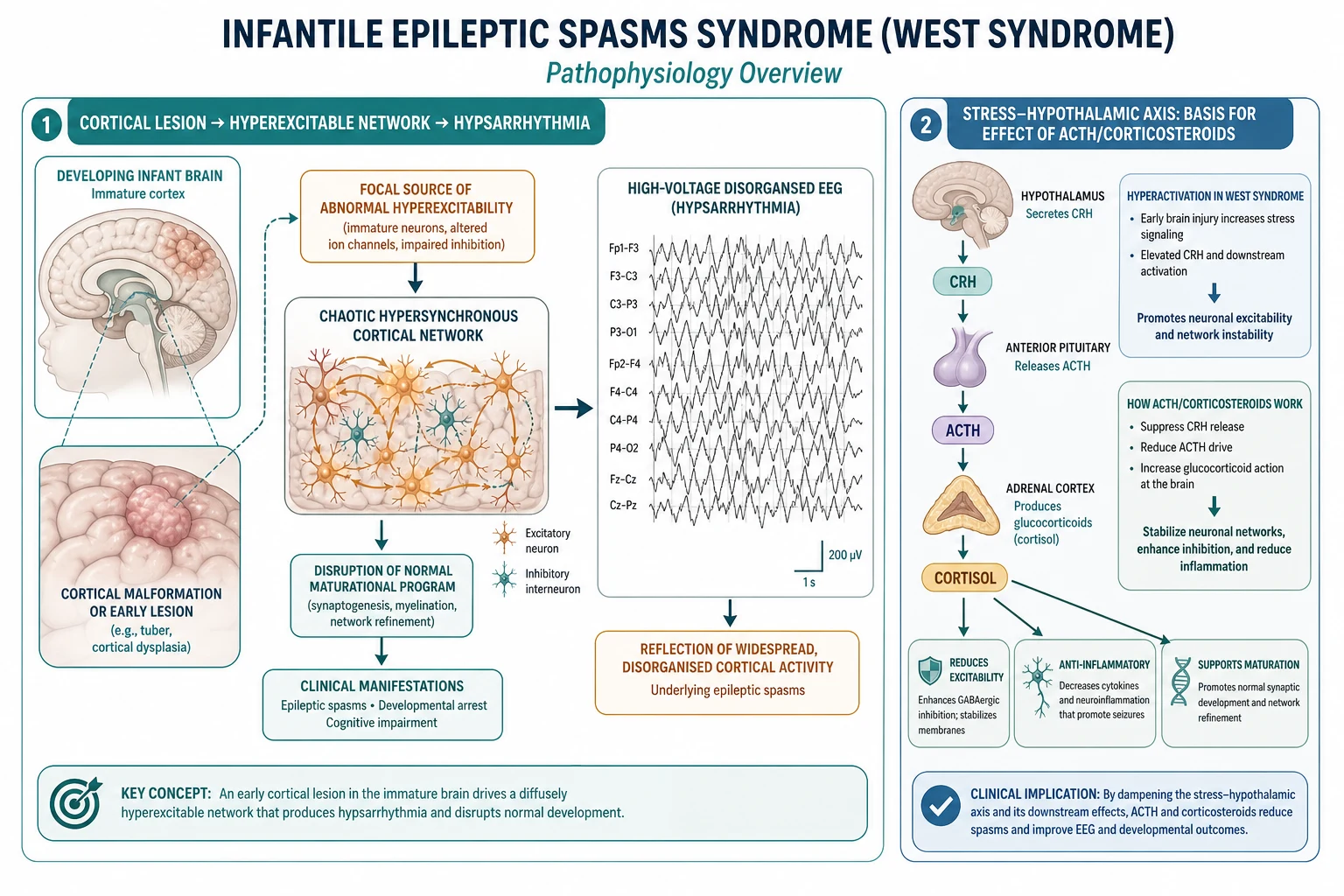
A second mechanism explains why hormonal therapy works. Early brain injury raises signalling in the stress axis, and corticotropin-releasing hormone, or CRH, is proconvulsant specifically in the immature brain, where it increases neuronal excitability during a vulnerable developmental window. ACTH and corticosteroids are thought to act by dampening this CRH drive and by stabilising the network through anti-inflammatory and membrane effects, which is why a drug that looks like a stress hormone can switch off a seizure disorder that standard anti-seizure medicines often fail to control. [7]
The developmental and epileptic encephalopathy concept ties these two ideas together. Because frequent epileptiform activity disrupts ongoing synaptogenesis, the seizures are not merely a marker of brain disease — they are an active cause of the developmental regression. Stopping the spasms and resolving the hypsarrhythmia removes that ongoing injury, which is the neurobiological justification for treating infantile spasms with urgency. [1] [7]
Clinical Presentation
The parents usually tell the story first. A previously developing infant, often around six months old, begins to do sudden jerking movements in runs, typically just after waking from a nap or on falling asleep. Each movement is a brief contraction — a forward bob of the head, a fling-out or pull-in of the arms, and a flexion of the trunk — that lasts only a second or two but repeats every few seconds for several minutes, sometimes dozens of times in a cluster. Parents often film these on a phone, and the video is diagnostically valuable. [9] [11]
The developmental story runs in parallel and is the part most easily missed. Over the same weeks the infant stops smiling responsively, stops babbling, loses head control or reaching, and the developmental trajectory flattens or reverses. This regression is the third arm of the West triad and the one that tells you the spasms are part of an encephalopathy rather than a benign movement disorder. [11]
The atypical and easily-missed presentations are the examiner's traps. Subtle head nods or eye deviations can be the only sign; asymmetric spasms, with one side moving more than the other, point to a focal lesion and demand close MRI scrutiny; late-onset spasms beyond twelve months blur the boundary with Lennox-Gastaut; and the spasms are routinely misattributed to colic, gastro-oesophageal reflux, startle or the Moro reflex by first-contact clinicians. Any infant with developmental regression or repetitive stereotyped movements in clusters in the first year of life deserves an EEG. [9]
West syndrome triad \u2014 the W rule
Differential Diagnosis
The first task is to separate true epileptic spasms from the non-epileptic movements that mimic them, and the EEG does this job. Benign myoclonus of infancy, shuddering attacks, Sandifer syndrome from gastro-oesophageal reflux, hyperekplexia (exaggerated startle), and infantile masturbation or self-gratification all produce repetitive movements in infancy but have a normal interictal EEG and no developmental regression. A normal EEG in a well, developing infant should send you back to the movement diagnosis, not to a spasm diagnosis. [9] [11]
The second task is to distinguish the causes of true infantile spasms, because the cause changes the treatment. Tuberous sclerosis is the single most common identifiable cause and the one that mandates vigabatrin first-line; a focal cortical dysplasia or gliotic lesion may be surgically resectable and changes the trajectory entirely; a metabolic disorder such as pyridoxine dependency responds to a cofactor rather than a standard antiseizure drug; and a genetic DEE such as CDKL5 deficiency disorder carries a guarded prognosis regardless of spasm control. A Wood's lamp examination, an MRI brain, and a genetic panel separate these efficiently. [5] [10]
The third task is to separate IESS from the other early developmental and epileptic encephalopathies that overlap in age. Ohtahara syndrome (early-infantile epileptic encephalopathy) presents earlier, in the neonatal period, with tonic spasms and a burst-suppression EEG; benign myoclonic epilepsy of infancy has briefer myoclonic jerks on a normal background and a good prognosis; Dravet syndrome begins in the first year with febrile hemiclonic seizures and later generalised myoclonia; and epilepsy of infancy with migrating focal seizures presents with focal seizures that shift between regions. Each is defined by its seizure type, its EEG, and its age of onset. [1]
| Condition | Movement / seizure | EEG | Key discriminator |
|---|---|---|---|
| West syndrome (IESS) | Clustered epileptic spasms | Hypsarrhythmia or modified variant | Developmental regression in first year |
| Benign myoclonus of infancy | Brief myoclonic jerks | Normal | Normal development; resolves |
| Sandifer syndrome (reflux) | Tonic posturing, often after feeds | Normal | Responds to reflux treatment |
| Hyperekplexia | Excessive startle to stimuli | Normal | Genetic; responds to clonazepam |
| Ohtahara syndrome | Tonic spasms in neonate | Burst-suppression | Onset in first weeks of life |
| Dravet syndrome | Febrile hemiclonic, later myoclonia | Normal early; later spike-wave | SCN1A; fever-sensitive onset |
Clinical & Bedside Assessment
The history is built around three questions: describe the movements, describe the development, and describe the perinatal and family background. Ask the parents to describe a single spasm and then a cluster — the timing on waking, the duration of a cluster, and whether the spasms are symmetric — and ask them to bring any phone video. Document the developmental trajectory in detail, including exactly which skills were present and have now been lost, and record the perinatal history, birthweight, neonatal complications and a three-generation family history of epilepsy, learning difficulty, skin lesions and consanguinity. [5] [11]
The examination is general before it is neurological, because the skin and the dysmorphic features often hold the aetiology. Examine the whole skin under a Wood's lamp for the hypomelanotic macules of tuberous sclerosis, and look for facial angiofibroma, shagreen patch, ash-leaf patches and any neurocutaneous stigmata. Measure and plot the head circumference, look for asymmetry of tone or movement that points to a focal lesion, and assess the developmental milestones against age using a standardised tool, because the developmental profile is part of the diagnosis and the baseline against which treatment response is judged. [10]
Synthesise the findings into a problem representation before you leave the bedside: an infant with new-onset clustered epileptic spasms and developmental regression has an epileptic encephalopathy that needs an urgent EEG to confirm hypsarrhythmia, an MRI brain to find a structural cause, and a skin examination and genetic workup to identify tuberous sclerosis or a single-gene DEE. That single sentence frames every investigation and the first-line treatment decision. [11]
Investigations
The single most important investigation is the EEG, and it should be requested as a sleep-deprived or sleep study the same day the diagnosis is suspected. Classic hypsarrhythmia is a chaotic, high-voltage pattern with random, asynchronous slow waves of more than 200 microvolts, intermixed with multifocal spikes, and a complete loss of normal background organisation. Modified variants are common and do not exclude the diagnosis: an asymmetric pattern suggests a focal lesion, a pattern with focal epileptiform discharges points to a resectable region, and a pattern with periods of voltage attenuation can appear after partial treatment. [1] [11]
The MRI brain is the core structural investigation. Look for a cortical malformation such as focal cortical dysplasia, the tubers and subependymal nodules of tuberous sclerosis, signal change from prior hypoxic-ischaemic injury, or a focal gliotic lesion. A resectable focal lesion is the finding that most changes management, because early epilepsy surgery can be curative and improves developmental outcome, so any focal or asymmetric MRI abnormality warrants urgent referral to a paediatric epilepsy surgery centre. [5]
A genetic workup is now part of the standard evaluation. Start with chromosomal microarray and move to an epilepsy gene panel or whole-exome sequencing, because identifying a pathogenic variant — in CDKL5, ARX, STXBP1, KCNQ2, SLC13A5 or the TSC1 and TSC2 genes — confirms the aetiology, refines the prognosis and enables genetic counselling and cascade testing. Add targeted metabolic testing (plasma lactate, urine organic acids, plasma amino acids) when the presentation or family history suggests an inborn error, and consider a therapeutic trial of pyridoxine or pyridoxal phosphate where dependency is suspected. [1] [7]
[1]Management — Resuscitation
Infantile spasms are a neurodevelopmental emergency, not a convulsive status epilepticus, and holding that distinction prevents two opposite errors. The child with routine spasm clusters is alert and interactive between clusters and does not need rescue benzodiazepines for the clusters themselves; instead, the urgency is to confirm the diagnosis and start definitive first-line therapy within days. The exception is a prolonged cluster that evolves to a convulsive seizure or status epilepticus, which is managed with standard paediatric rescue therapy. [4] [11]
The resuscitation step is therefore diagnostic and aetiological. Secure the diagnosis with an EEG and MRI, examine the skin for tuberous sclerosis, and look for any reversible cause — a surgically resectable focal lesion, a metabolic disorder, or an infection — because each changes the management plan. Establish intravenous access only if a prolonged seizure requires it, and do not delay first-line therapy while awaiting every test result: the EEG confirms the syndrome, and the first-line drug can be started once hypsarrhythmia is documented and the TSC question is addressed. [5]
The decision that must be made before the first dose is whether the cause is tuberous sclerosis complex. That single decision sets the first-line agent, because vigabatrin is preferred first-line for TSC-associated spasms and hormonal therapy is preferred for every other cause. If the Wood's lamp examination is positive for hypomelanotic macules, or if an MRI shows tubers, treat as TSC and start vigabatrin while confirming the diagnosis with TSC1 and TSC2 testing. [10] [6]
Management — Definitive & Stepwise
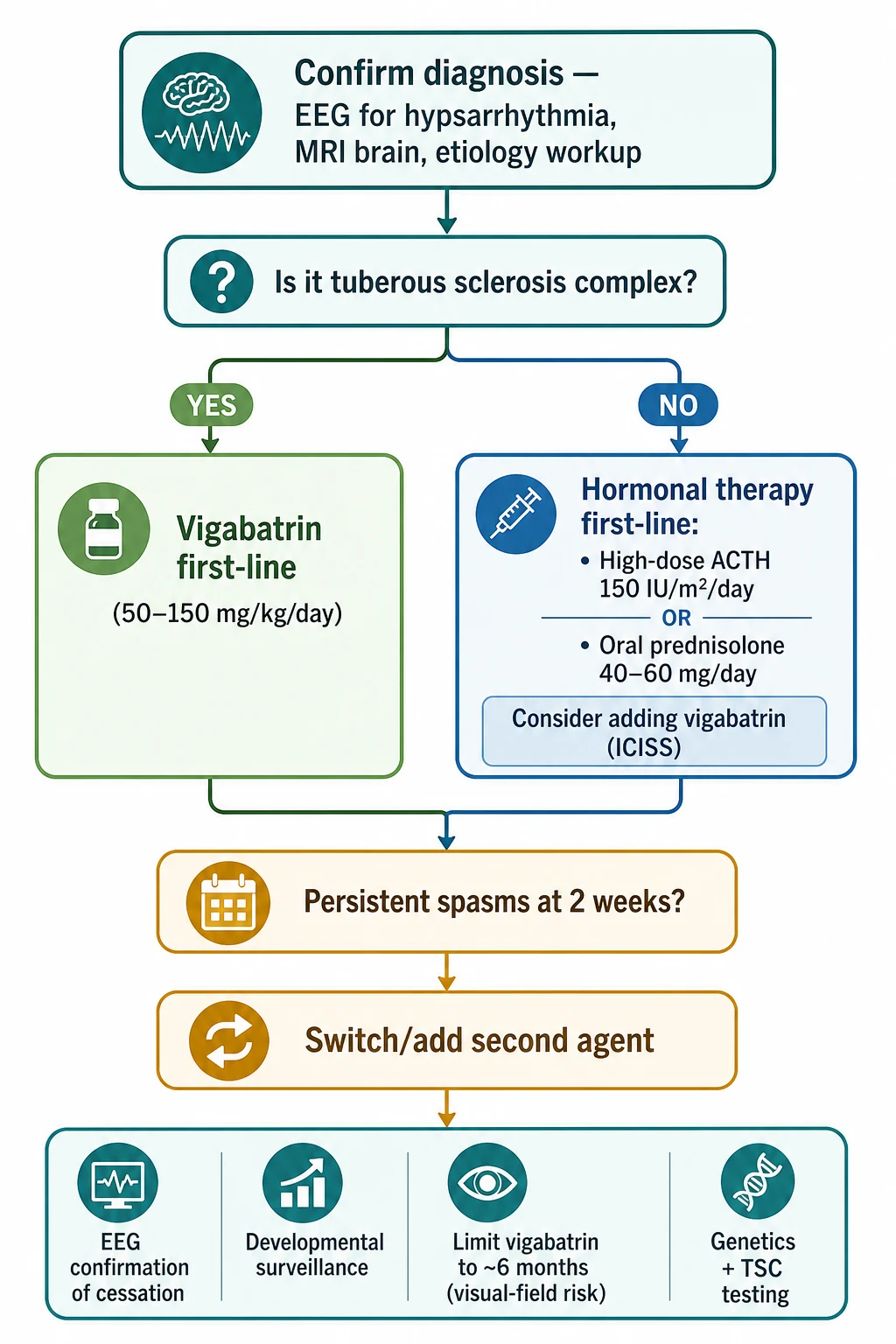
The first-line rule is simple to state and exact in its doses. For tuberous-sclerosis-associated infantile spasms, vigabatrin is the first-line drug, started at 50 mg per kg per day and titrated up by 25 to 50 mg per kg per day every three days to a target of 100 to 150 mg per kg per day. For every other cause, hormonal therapy is first-line: high-dose ACTH at 150 IU per square metre per day by intramuscular injection, or oral prednisolone at 40 to 60 mg per day (approximately 2 mg per kg per day). The evidence behind these doses is the now-superseded 2012 American Academy of Neurology guideline, the ILAE paediatric task-force summary and the US consensus report. [3] [4] [5]
The ICISS trial refined this picture by showing that adding vigabatrin to hormonal therapy improved short-term spasm cessation over hormonal therapy alone. In ICISS, combination hormonal plus vigabatrin therapy stopped the spasms in a higher proportion of infants at 13 to 14 days than hormonal therapy alone, so for a non-TSC infant many specialists now add vigabatrin to the hormonal course. The 18-month developmental outcome was not significantly different between the two arms, which tempers but does not negate the case for combination therapy. [2]
If spasms persist at two weeks, switch or add the second agent: a child started on hormonal therapy who still has spasms is given vigabatrin, and vice versa. Confirm cessation with a repeat EEG, because clinical cessation without electrographic resolution is incomplete, and an unresolved hypsarrhythmia predicts relapse and poorer outcome. Build the developmental surveillance programme alongside the drug treatment — early intervention services, physiotherapy, occupational and speech therapy, and psychology — and complete the genetic and TSC workup so the family has an aetiology and a prognosis. [4] [11]
The vigabatrin duration matters because of retinal toxicity. Vigabatrin can cause an irreversible bilateral concentric constriction of the visual field, with an attributable risk that rises with cumulative dose and duration, so the total course is usually limited to around six months once spasm control is achieved. Arrange ophthalmological surveillance where the child can cooperate with perimetry, and balance the visual-field risk against the seizure-control benefit, which in TSC-associated spasms is substantial enough to justify use. [6] [8]
Specific Subtypes & Scenarios
The infant with tuberous-sclerosis-associated spasms is the scenario where the treatment most clearly changes outcome. Vigabatrin is first-line and produces a higher response rate in TSC than in non-TSC spasms, so start it promptly once the diagnosis is confirmed, arrange serial EEGs, and begin the lifelong TSC surveillance programme — brain MRI for a subependymal giant cell astrocytoma, renal imaging, developmental review, and consideration of everolimus for a growing lesion or refractory epilepsy downstream. The TSC diagnosis also opens cascade testing of relatives and reproductive counselling. [6] [10]
The infant with a surgically resectable focal lesion is the scenario where surgery can be curative. A focal cortical dysplasia or a gliotic lesion that generates asymmetric spasms, a focal EEG abnormality, and a matching MRI change is the substrate for paediatric epilepsy surgery, and early resection can stop the spasms and improve developmental outcome. Refer urgently to a paediatric epilepsy surgery centre for presurgical evaluation, because the window for developmental benefit closes with ongoing seizures. [5]
The infant with a genetic developmental and epileptic encephalopathy is the scenario where the prognosis is often guarded regardless of spasm control. A pathogenic variant in CDKL5, ARX, STXBP1, KCNQ2 or SLC13A5 typically produces early-onset, refractory spasms with severe developmental impairment, and while spasm cessation remains the goal, the family needs honest counselling that the underlying genetic disorder drives much of the outcome. Genetic confirmation enables accurate counselling, recurrence-risk discussion and reproductive options. [1] [7]
The older infant or toddler with refractory spasms or evolution to Lennox-Gastaut syndrome is the scenario of the chronic DEE. Roughly a third to a half of children with infantile spasms evolve to Lennox-Gastaut syndrome, defined by multiple seizure types (tonic, atonic, atypical absence), a slow spike-wave EEG, and cognitive impairment, so ongoing surveillance for this evolution is part of long-term care. Management then shifts to the Lennox-Gastaut toolkit, including broad-spectrum anti-seizure medicines, the ketogenic diet, and consideration of corpus callosotomy or vagus-nerve stimulation for drop attacks. [12]
Complications & Pitfalls
The complications of the disease are developmental and epileptic. Refractory epilepsy, intellectual disability and autism spectrum disorder are the dominant long-term comorbidities and the ones that most shape quality of life, and evolution to Lennox-Gastaut syndrome is the commonest epileptic evolution. The developmental outcome is driven by the aetiology and by the speed and completeness of spasm cessation, which is why delay is the single most preventable cause of harm. [11] [12]
The complications of therapy are drug-specific and worth naming precisely. Vigabatrin causes irreversible bilateral concentric visual-field constriction in a meaningful fraction of treated patients, which is why the course is limited to around six months and ophthalmological surveillance is arranged; the risk is dose- and duration-dependent and lower in infants than in adults but never negligible. Hormonal therapy causes irritability, hypertension, hyperglycaemia, immunosuppression with infection risk, and gastrointestinal irritation or bleeding, so a course of ACTH or high-dose prednisolone needs blood-pressure and glucose monitoring, gastric protection, and vigilance for intercurrent infection. [3] [8]
The cognitive traps are where candidates lose marks and clinicians lose time. The first trap is attributing subtle head nods or clustered movements to colic or reflux and never ordering an EEG; the second is delaying first-line therapy while completing non-essential tests; the third is giving vigabatrin first-line to a non-TSC infant when hormonal therapy is the preferred first-line agent; the fourth is forgetting to examine the skin under a Wood's lamp and missing a treatable TSC diagnosis; and the fifth is assuming that clinical cessation of spasms means the EEG has resolved, when an unresolved hypsarrhythmia predicts relapse. [5] [11]
[4]Prognosis & Disposition
The developmental outcome is driven mainly by the aetiology and by the speed and completeness of spasm cessation. Infants with an unknown (cryptogenic) cause who are controlled promptly have the best prognosis, with a meaningful proportion achieving normal or near-normal cognition; infants with a structural or metabolic cause, a long lag to treatment, or refractory spasms have the worst, with high rates of intellectual disability, autism and chronic epilepsy. The aetiology and the treatment response together set the trajectory, which is why both the workup and the therapy are time-critical. [3] [11]
A substantial minority evolve to other epilepsies, most often Lennox-Gastaut syndrome, and intellectual disability and autism are common long-term comorbidities even when the spasms are controlled. Counsel the family early and honestly: even with prompt and effective treatment the developmental outcome is often guarded, and a hopeful but realistic conversation supports adherence, early-intervention engagement and long-term planning. Avoid both false reassurance and therapeutic nihilism. [12]
Disposition is to a paediatric neurology service with a multidisciplinary developmental team. Early intervention services, physiotherapy, occupational and speech therapy, psychology and educational support are the backbone of long-term care, and structured surveillance of seizure control, development and behaviour continues through childhood. A confirmed aetiology enables genetic counselling, and for the surgically amenable focal lesion, early referral to an epilepsy surgery centre can be life-changing. [5]
Special Populations
Access to diagnosis and treatment is unequal, and that inequality changes outcome. In remote, Indigenous, migrant and refugee families, delayed recognition of the spasms, reduced access to urgent EEG, paediatric neurology and genetic testing, and longer travel times to specialist care all lengthen the lag from onset to treatment and worsen the developmental prognosis. Telehealth-supported diagnostic pathways, visiting neurology outreach, and culturally appropriate genetic counselling with an interpreter or Indigenous health worker narrow this gap and should be offered proactively. [4]
Children with disability and neurodiversity form the majority of this population, and their care must be strengths-based as well as deficit-focused. A formal developmental and behavioural assessment, early referral to early-intervention services, educational planning, and screening for autism spectrum disorder are part of the core management, because neurocognitive comorbidity dominates quality of life. Engage the family as partners in goal-setting rather than passive recipients of a neurological workup. [5]
The infant with a complex chronic or technology-dependent presentation needs coordinated care across neurology, metabolic medicine, epilepsy surgery and community services, with a single identified care coordinator and an emergency-care plan. Genetic counselling follows a confirmed pathogenic variant: the family receives a recurrence-risk discussion, reproductive options including prenatal and preimplantation genetic testing once the familial variant is known, and cascade testing of at-risk relatives. [1] [7]
In Australia and Aotearoa New Zealand, specialist paediatric neurology and epilepsy services are concentrated in major paediatric centres, and infants in rural and remote areas rely on retrieval, visiting neurology outreach or telehealth for the urgent EEG and first-line therapy that the time-to-treatment effect demands. Engaging an Aboriginal and Torres Strait Islander health worker or Maori kaiawhina, and offering culturally appropriate genetic counselling, improves diagnostic uptake and long-term engagement. Families should be connected to the relevant epilepsy foundations and to TSC support organisations, and access to ACTH, genetic testing and epilepsy surgery is coordinated through the specialist neurology service.
Evidence, Guidelines & Regional Differences
The current syndromic definition is the 2022 ILAE neonate-and-infant syndromes position statement (Zuberi and colleagues), which formalises infantile epileptic spasms syndrome with its age of onset, EEG and developmental features. The management consensus draws on the 2015 ILAE paediatric task-force summary (Wilmshurst) and the 2010 US consensus report (Pellock), and on the 2012 American Academy of Neurology evidence-based guideline (Go), which has since been retired but remains the historical reference for the dose evidence supporting ACTH and vigabatrin. [1] [3] [4] [5]
The key randomised evidence is the ICISS trial (O'Callaghan and colleagues), which showed that adding vigabatrin to hormonal therapy improved short-term spasm cessation over hormonal therapy alone, with 18-month developmental outcomes not significantly different between arms. The earlier Elterman trial established vigabatrin for infantile spasms, and the Biswas meta-analysis quantified the visual-field risk that limits the drug's duration. The Stafstrom review synthesises the current understanding of mechanism, including the CRH stress axis that explains why ACTH and corticosteroids work. [2] [6] [7] [8]
The live controversies are practical ones. Vigabatrin versus hormonal therapy as first-line for non-TSC spasms, high-dose versus low-dose ACTH, the optimal duration of vigabatrin given the visual-field risk, and the timing of epilepsy surgery for a resectable lesion each remain matters of judgement. Regional pathways converge on the TSC decision fork — vigabatrin first-line for TSC and hormonal therapy first-line otherwise — but access to ACTH preparations, oral prednisolone, genetic testing and epilepsy surgery varies by health system, with ANZ, UK and North American pathways broadly aligned through their specialist epilepsy services. [4] [11]
Exam Pearls
The mnemonic that pays off most often is the West syndrome W triad: Waves on the EEG (hypsarrhythmia), salaam Attacks (clustered epileptic spasms), and psychomotor Regression, all in the first year of life. Any infant losing skills with repetitive clustered jerks gets an EEG the same day. The triad defines the syndrome and triggers the time-critical first-line therapy that protects the developing brain. [9]
The dose facts are worth memorising exactly, because they appear in written and viva questions. Vigabatrin is 50 mg per kg per day, titrated up by 25 to 50 mg per kg per day every three days to a target of 100 to 150 mg per kg per day, with a total course of around six months. High-dose ACTH is 150 IU per square metre per day by intramuscular injection, and oral prednisolone is 40 to 60 mg per day (approximately 2 mg per kg per day). Vigabatrin is first-line for TSC; hormonal therapy is first-line for every other cause. [3] [4]
The diagnostic rule that catches the most marks: an infant with new clustered spasms gets an urgent EEG, and an infant with spasms and hypomelanotic macules on Wood's lamp examination has tuberous sclerosis complex until proven otherwise and gets vigabatrin first-line. The prognostic rule: aetiology and speed of spasm cessation drive developmental outcome, so an unknown cause controlled promptly does best, and a symptomatic cause with refractory spasms does worst. [5] [10]
Infantile epileptic spasms syndrome (West syndrome) is the triad of clustered epileptic spasms, hypsarrhythmia and developmental regression in infancy, and a developmental and epileptic encephalopathy in which ongoing epileptiform activity harms the developing brain. Confirm with a sleep EEG and MRI, examine the skin for TSC, and complete a genetic workup. First-line therapy turns on one question — is the cause TSC? Vigabatrin (50–150 mg/kg/day, limit ~6 months) is first-line for TSC; hormonal therapy (ACTH 150 IU/m²/day or prednisolone 40–60 mg/day) is first-line otherwise, with ICISS supporting the addition of vigabatrin to hormonal. Outcome is driven by aetiology and the speed and completeness of spasm cessation.
References
- [1]Zuberi SM, Wirrell E, Yozawitz E, Wilmshurst JM, Oka E, Kahn S, et al. ILAE classification and definition of epilepsy syndromes with onset in neonates and infants: Position statement by the ILAE Task Force on Nosology and Definitions. Epilepsia, 2022.PMID 35503712
- [2]O'Callaghan FJK, Edwards SW, Alber FD, Cortina Borja M, Hancock E, Johnson AL, et al. Vigabatrin with hormonal treatment versus hormonal treatment alone (ICISS) for infantile spasms: 18-month outcomes of an open-label, randomised controlled trial. Lancet Child Adolesc Health, 2018.PMID 30236380
- [3]Go CY, Mackay MT, Weiss SK, Stephens D, Adams-Webber T, Ashwal S, et al. Evidence-based guideline update: medical treatment of infantile spasms. Report of the Guideline Development Subcommittee of the American Academy of Neurology and the Practice Committee of the Child Neurology Society. Neurology, 2012.PMID 22689735
- [4]Wilmshurst JM, Gaillard WD, Vinayan KP, Tsuchida TN, Plouin P, Van Bogaert P, et al. Summary of recommendations for the management of infantile seizures: Task Force Report for the ILAE Commission of Pediatrics. Epilepsia, 2015.PMID 26122601
- [5]Pellock JM, Hrachovy R, Shinnar S, Baram TZ, Bettis D, Dlugos DJ, et al. Infantile spasms: a U.S. consensus report. Epilepsia, 2010.PMID 20608959
- [6]Elterman RD, Shields WD, Mansfield KA, Nakagawa J, the US Infantile Spasms Vigabatrin Study Group. Randomized trial of vigabatrin in patients with infantile spasms. Neurology, 2001.PMID 11673582
- [7]Stafstrom CE. Infantile epileptic spasms syndrome: mechanisms and therapeutic approaches. Neurotherapeutics, 2026.PMID 41419420
- [8]Biswas A, Yossofzai O, Vincent A, Go C. Vigabatrin-related adverse events for the treatment of epileptic spasms: systematic review and meta-analysis. Expert Rev Neurother, 2020.PMID 33078964
- [9]Wheless JW, Gibson PA, Rosbeck KL, Hardin M, O'Dell C, Whittemore V, et al. Infantile spasms (West syndrome): update and resources for pediatricians and providers to share with parents. BMC Pediatr, 2012.PMID 22830456
- [10]Northrup H, Krueger DA; International Tuberous Sclerosis Complex Consensus Group. Tuberous sclerosis complex diagnostic criteria update: recommendations of the 2012 International Tuberous Sclerosis Complex Consensus Conference. Pediatr Neurol, 2013.PMID 24053982
- [11]D'Alonzo R, Rigante D, Mencaroni E, Esposito S. West Syndrome: a review and guide for paediatricians. Clin Drug Investig, 2018.PMID 29086890
- [12]Arzimanoglou A, French J, Blume WT, Cross JH, Ernst JP, Feucht M, et al. Lennox-Gastaut syndrome: a consensus approach on diagnosis, assessment, management, and trial methodology. Lancet Neurol, 2009.PMID 19081517