Paeds · neurology-neurodisability-and-neuromuscular
Movement disorders, dystonia and chorea
Also known as Paediatric hyperkinetic movement disorders · Dystonia in children · Chorea in children · Status dystonicus · Sydenham chorea · Dyskinetic cerebral palsy
Fellowship guide to paediatric movement disorders. Defines the hyperkinetic movements of chorea, dystonia, myoclonus, tics and tremor against the hypokinetic pole of parkinsonism, applies the Albanese 2013 two-axis dystonia classification and the Sanger 2003 childhood hypertonia framework, traces the basal ganglia direct and indirect pathway mechanism of dyskinesia, and works through Sydenham chorea as a major Jones criterion needing secondary penicillin prophylaxis, dopa-responsive dystonia and its dramatic levodopa response, Wilson disease screening, glutaric aciduria type 1, Lesch-Nyhan, the iatrogenic acute dystonic reversal with an anticholinergic, the status dystonicus escalation ladder from trigger control through oral drugs to sedation and intrathecal baclofen or deep brain stimulation, the dystonic cerebral palsy ladder of trihexyphenidyl, botulinum toxin, intrathecal baclofen and selective dorsal rhizotomy, the PANDAS and PANS controversy, regional rheumatic fever differences, and the family communication and long-term functional outlook.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
Overview & Definition
A child whose hand twists into a fixed posture when they reach for a cup, or who flows with restless, dance-like movements they cannot stop, is showing you a movement disorder. The job in paediatric neurology is to name the movement, work out whether it is a symptom of a treatable disease, and protect the child from the two emergencies that can kill or maim: status dystonicus and the acute dystonic reaction. Movement disorders are conditions in which the motor control system produces either an excess of movement that the child cannot suppress, or a poverty of movement with stiffness and slowness. [3]
The vocabulary is small and worth learning precisely, because the wrong word sends the workup in the wrong direction. Chorea is flowing, unpredictable, semi-purposeful movement. Dystonia is sustained or intermittent involuntary muscle contraction that twists the body into abnormal postures. Athetosis is the slow writhing form, and myoclonus is a brief, sudden, shock-like jerk. Tics are brief, stereotyped, and suppressible movements or sounds. Tremor is a rhythmic oscillation. The opposite pole is parkinsonism, with slowness, rigidity, and a rest tremor. The Albanese 2013 consensus update gives dystonia its modern two-axis classification, and the Sanger 2003 task force gave childhood hypertonia its working framework of spasticity, dystonia, and rigidity that dominates cerebral palsy practice. [1][2]
Three ideas hold this topic together. The movement is named by watching it, not by scanning it, so careful observation and video come before the test list. A single treatable cause, dopa-responsive dystonia, hides among the chronic dystonias and is found only by trialling levodopa in every unexplained case. And the two emergencies, the acute dystonic reaction and status dystonicus, are reversed or controlled by a short, memorable ladder that the exam will reward you for knowing in order. [3][6]
Classification
Movement disorders split cleanly into two families, and the split predicts what you look for next. Hyperkinetic disorders show too much movement, and they are the ones that dominate paediatric practice. Hypokinetic disorders show too little movement, with stiffness and slowness, and parkinsonism is the example that matters. Holding this fork in mind stops you reaching for a dopamine blocker when a child is bradykinetic, and stops you missing a chorea behind what looks like fidgetiness. [3]
Dystonia carries its own deeper classification because the same twist can come from twenty different causes, and the Albanese 2013 consensus update sorts it on two axes. Axis I describes the clinical characteristics: the age at onset, the body distribution from focal through segmental and multifocal to generalised and hemidystonia, the temporal pattern including a diurnal fluctuation that flags dopa-responsive dystonia, and the coexistence of other movement disorders. Axis II describes the aetiology: inherited (a growing list of DYT genes), acquired (perinatal injury, kernicterus, basal ganglia infarct, drug, metabolic), or unknown. The point of the two axes is that the same generalised dystonia in a five-year-old could be dopa-responsive and cured by levodopa, or could be cerebral palsy and managed for life, and only axis II tells them apart. [1]
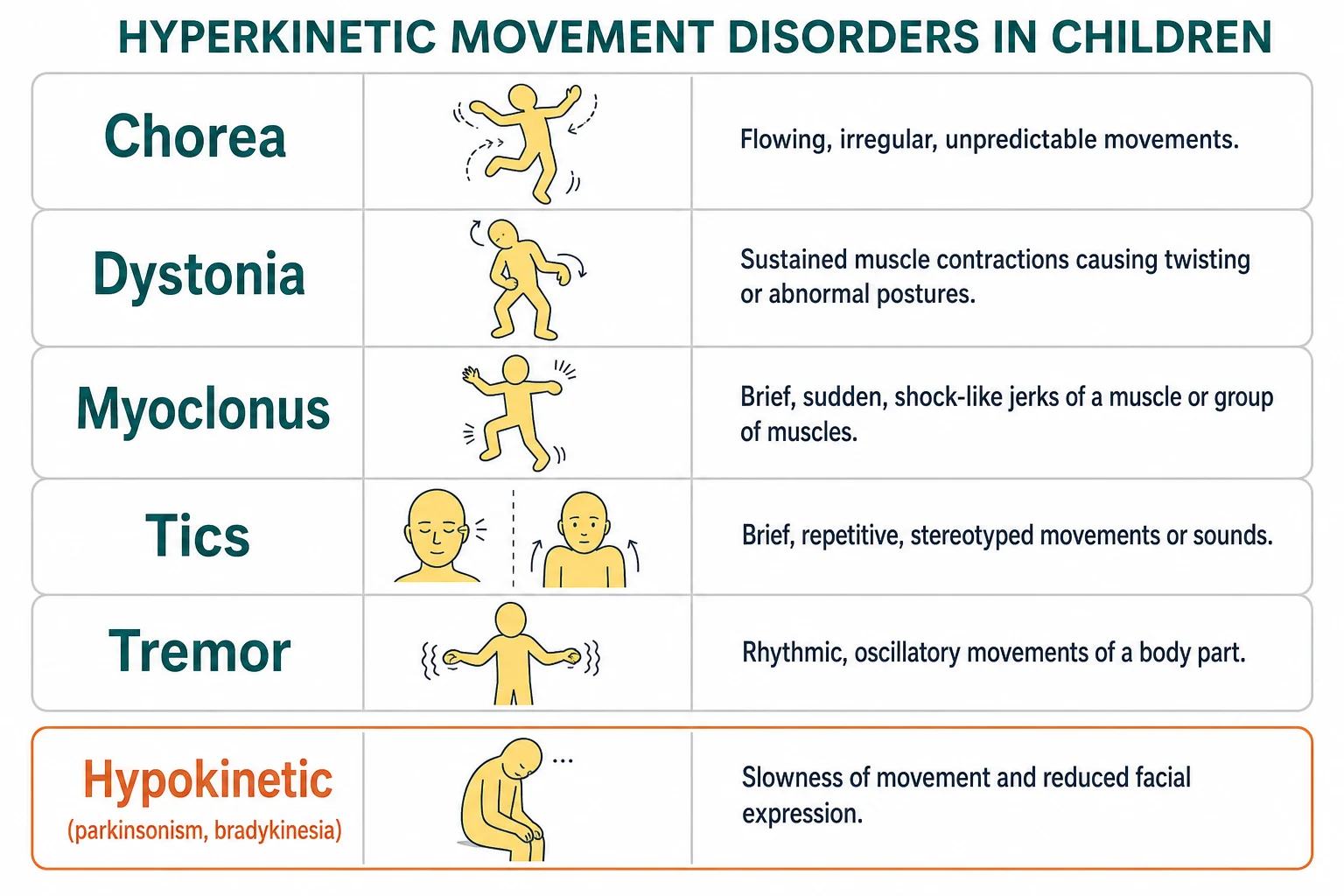
The Sanger 2003 task force gave childhood hypertonia its own working framework, because a child with cerebral palsy rarely has pure spasticity or pure dystonia and the words change what you treat. The framework sorts hypertonia into spasticity (velocity-dependent resistance to stretch), dystonia (involuntary sustained or intermittent contraction that twists), and rigidity (resistance throughout the range that is not velocity-dependent). This is the classification that organises the cerebral palsy examination and the intrathecal baclofen and rhizotomy decisions, and it is the one a fellowship examiner expects you to use at the bedside. [2]
Epidemiology & Risk Factors
Cerebral palsy is the single largest reservoir of childhood dystonia, with a prevalence of around two per thousand live births, and the dyskinetic and mixed types carry the greatest dystonia burden. Sydenham chorea remains the commonest acquired chorea of children worldwide and is the neurologic face of acute rheumatic fever following group A streptococcal infection, with its peak in school-age children. In Australia and Aotearoa New Zealand the burden of acute rheumatic fever falls overwhelmingly on Aboriginal and Torres Strait Islander children and on children in remote communities, where rates are among the highest in the world, so Sydenham chorea is a real and common diagnosis rather than a historical curiosity. [4]
Drug-induced movement disorders are common iatrogenic events, and the leading culprit is the dopamine-blocking agent. Metoclopramide, prochlorperazine, and the antipsychotics precipitate acute dystonic reactions, and the emergency presentation of a locked jaw, oculogyric crisis, or twisted neck hours after a dose is a pattern every paediatric trainee should recognise. The risk factors for a chronic dystonia fan out across perinatal injury and kernicterus, basal ganglia infarct or hypoxic-ischaemic injury, inherited mutations in the DYT genes including GCH1 and TOR1A, and metabolic disease such as glutaric aciduria type 1, Wilson disease, and Lesch-Nyhan syndrome. [3]
Pathophysiology
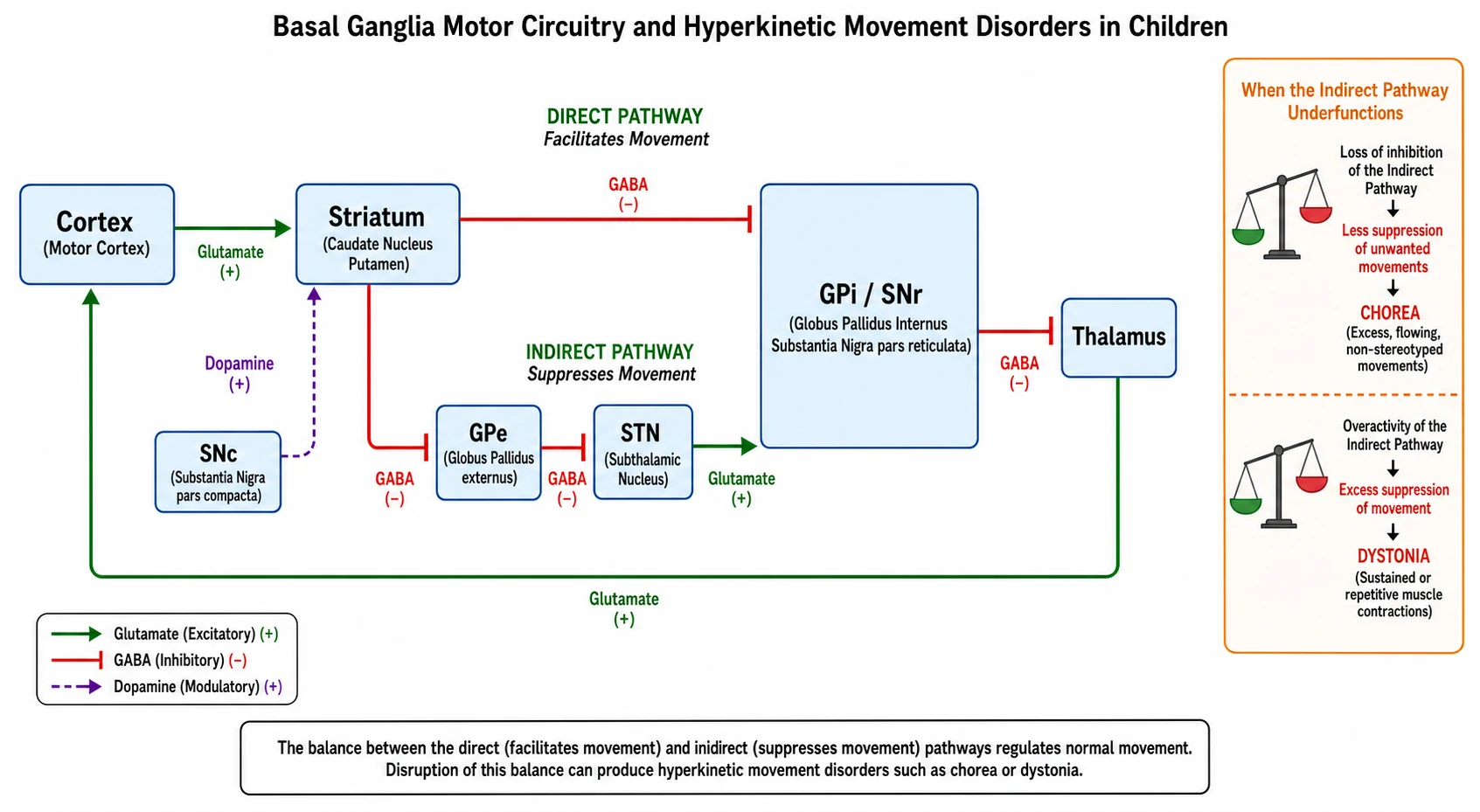
The basal ganglia run a motor loop that gates which movements are allowed to happen, and understanding the loop in one sitting makes the whole topic click. The cortex sends an excitatory signal to the striatum, and the striatum then governs the thalamus through two parallel routes. The direct pathway runs straight from the striatum to the globus pallidus internus and substantia nigra pars reticulata, and it facilitates the movement you want. The indirect pathway detours through the globus pallidus externus and the subthalamic nucleus before reaching the same target, and it suppresses the movements you do not want. Dopamine from the substantia nigra pars compacta tunes the balance, exciting the direct pathway through D1 receptors and braking the indirect pathway through D2 receptors. [3]
When that balance breaks, the hyperkinetic disorders appear, and the direction of the break tells you which one. If the indirect pathway under-functions, its brake on unwanted movement is lost and chorea flows through. If there is excess indirect-pathway suppression or sustained co-contraction and overflow, the body locks into the twisting postures of dystonia. This single insight explains why a dopamine-blocking drug, by over-weighting the indirect pathway, can cause an acute dystonic reaction, and why a dopamine-blocking drug can also bring chorea under partial control. It also explains why dopamine loss produces the poverty and stiffness of parkinsonism. [3]
Sydenham chorea has its own mechanism worth holding. After group A streptococcal infection, antineuronal antibodies cross-react with basal ganglia neurons, and the resulting immune injury disturbs the same loop. This is why chorea follows the infection by weeks to months, why it carries emotional and behavioural features, and why immunomodulation with corticosteroids has a role in severe disease. [4]
Clinical Presentation
Chorea announces itself as movement that will not settle. The child flows with random, semi-purposeful movements, and a sharp bedside observation is that they often disguise the involuntary movement by folding it into a deliberate act, a trick called parakinesia. Ask the child to hold their arms out and grip your fingers and you feel the rhythmic squeezing and releasing of the milkmaid grip. Ask them to hold their tongue out and it darts back inside. Sydenham chorea develops weeks to months after a streptococcal sore throat or skin infection, is often asymmetric, and arrives with emotional lability, obsessions, anxiety, and a soft, hypotonic motor exam. [4]
Dystonia announces itself as posture. The child twists into sustained or repetitive abnormal postures that are provoked by action at first and appear at rest later, and a distinctive feature is the sensory trick: a light touch to the affected part can briefly abolish the dystonia. A young child who walks on an inturned foot that worsens through the day and is normal on waking has dopa-responsive dystonia until proven otherwise. Dystonia can be focal, as in writer's cramp or torticollis, segmental, hemidystonia, or generalised when it sweeps the trunk and limbs. [1][12]
The two emergencies present dramatically and must not be missed. An acute dystonic reaction appears hours after a dopamine-blocking dose, with an oculogyric crisis in which the eyes roll upward, a twisted neck in torticollis, a clenched jaw in oromandibular spasm, and a frightening but reversible locked posture. Status dystonicus appears as escalating, painful, sustained dystonic spasms that build over hours, threaten the airway and breathing, and precipitate rhabdomyolysis and acute kidney injury from muscle breakdown. Both are reversible if caught early and fatal or maiming if missed. [6][10]
MOVES
Differential Diagnosis
The first fork is between a movement disorder and its mimics, and getting this fork wrong wastes the whole workup. Epilepsy, especially myoclonic and focal motor seizures, can look like a movement disorder, but seizures are stereotyped, episodic, and often accompanied by an electrographic discharge. Stereotypies in autism, self-stimulatory behaviour, shuddering attacks, and Sandifer posture in reflux are patterned but context-bound. The discriminator is that a movement disorder is present during wakefulness, often worsened by action or stress, and is not the discrete, repetitive, electrographic event of a seizure. An EEG settles the question when doubt remains. [3]
Functional, or psychogenic, movement disorder is common in adolescents and is a real and treatable diagnosis, not a dustbin. Its hallmarks are variability, distractibility, and incongruity with organic patterns: the movement changes with attention, eases with distraction, and does not fit a known distribution. The error is to label it as imagined or to dismiss it; the right move is a confident diagnosis and referral to a functional neurology and rehabilitation service. [3]
Within organic dystonia, the differentials are read off the cause. Cerebral palsy sits behind a static encephalopathy with a perinatal history. Dopa-responsive dystonia has its diurnal pattern and dramatic levodopa response. Wilson disease brings hepatic and psychiatric change, and a movement disorder plus any liver or behavioural abnormality demands copper studies and slit-lamp examination. Glutaric aciduria type 1 presents with macrocephaly and an acute encephalopathic crisis that leaves striatal injury. Lesch-Nyhan syndrome adds severe self-injurious behaviour and hyperuricaemia. Within chorea, the differentials are Sydenham chorea, post-streptococcal and autoimmune causes including anti-NMDAR encephalitis, drug-induced and metabolic causes such as hyperthyroidism and hyperglycaemia, and rare genetic causes such as benign hereditary chorea and juvenile Huntington disease. [4][11]
Clinical & Bedside Assessment
The examination rests on watching the child before you touch them. Ask the child to undress, walk across the room, reach for a toy, and sit and stand, and catch the movement at its natural peak. Video the examination, because serial comparison at follow-up is how you judge whether a treatment is working and whether a chronic dystonia is worsening toward status dystonicus. Define the movement by its speed, rhythm, and suppressibility, its distribution from focal to generalised, and its triggers, including action, stress, fatigue, and a diurnal pattern that flags dopa-responsive dystonia. [2]
Test for the sensory trick by lightly touching the affected part and watching the dystonia ease, and test for motor impersistence with a sustained grip and tongue protrusion. Examine the eyes for an oculogyric crisis in the acute presentation and for the Kayser-Fleischer rings of Wilson disease at the slit lamp. Examine the abdomen for hepatosplenomegaly, the skin and joints for rheumatic and neurocutaneous stigmata, and the developmental and cognitive profile. Always assess swallowing, secretions, and respiratory reserve, because the threat in severe dystonia is airway compromise and aspiration, and a child whose chest wall is involved in status dystonicus can lose the ability to breathe. [6]
The history is where the cause usually declares itself. Take a three-generation family history for dystonia, tremor, and parkinsonism, a perinatal history that includes prematurity, neonatal jaundice and kernicterus, and hypoxic-ischaemic injury, a careful drug exposure history that names metoclopramide, prochlorperazine, antipsychotics, and any recent change or withdrawal of an anti-dystonia drug, and a recent illness history that covers streptococcal sore throat, viral infection, and vaccination. In an adolescent with new chorea or dystonia, ask about pregnancy, substance use, and the insidious onset of Wilson disease. [4]
Investigations
Investigation is guided by the clinical pattern and tempo, and the cardinal principle is that an empiric levodopa trial is both diagnostic and therapeutic and should be done early. Dopa-responsive dystonia responds dramatically and sustainedly to a low dose of levodopa combined with a decarboxylase inhibitor, and the trial costs days and changes a life, so every dystonia of unknown cause gets one before the workup closes. [12]
For a first presentation of dystonia, the metabolic and genetic workup runs in parallel with the levodopa trial. Send serum copper and caeruloplasmin, a 24-hour urinary copper, and arrange slit-lamp examination for Kayser-Fleischer rings to screen for Wilson disease, because missed Wilson disease causes irreversible neurologic injury. Send lactate, ammonia, plasma amino acids, urine organic acids to catch glutaric aciduria type 1, creatine kinase, thyroid function, and a blood film for acanthocytes. MRI brain looks for the characteristic basal ganglia signal change of kernicterus, glutaric aciduria type 1 striatal injury, Wilson disease, or hypoxic-ischaemic injury, and for structural lesions. CSF glucose is sent when GLUT1 deficiency is suspected, where a low CSF glucose against a normal plasma glucose is diagnostic. [3]
For chorea, the workup targets the likely cause. Send anti-streptolysin O and anti-DNase B titres and a throat swab for evidence of recent group A streptococcal infection, and arrange echocardiography and electrocardiography because Sydenham chorea is a major Jones criterion and rheumatic carditis may be silent. Add an autoimmune and infection panel including anti-NMDAR antibodies when the picture is subacute and multidomain, thyroid function, and a pregnancy test in an adolescent girl. An EEG separates a movement disorder from ongoing seizure, and is essential when the movement is episodic and stereotyped. [4][11]
Management — Resuscitation
The two time-critical emergencies are the acute dystonic reaction and status dystonicus, and each has a short, memorable response that the exam rewards. The acute dystonic reaction is reversed immediately with an anticholinergic. Benztropine, procyclidine, or diphenhydramine is given intramuscularly or intravenously, the causative dopamine-blocking drug is withheld, and the frightening locked posture and oculogyric crisis resolve within minutes. The child is monitored because the effect can wear off, and the family is warned to avoid the offending drug in future. [10]
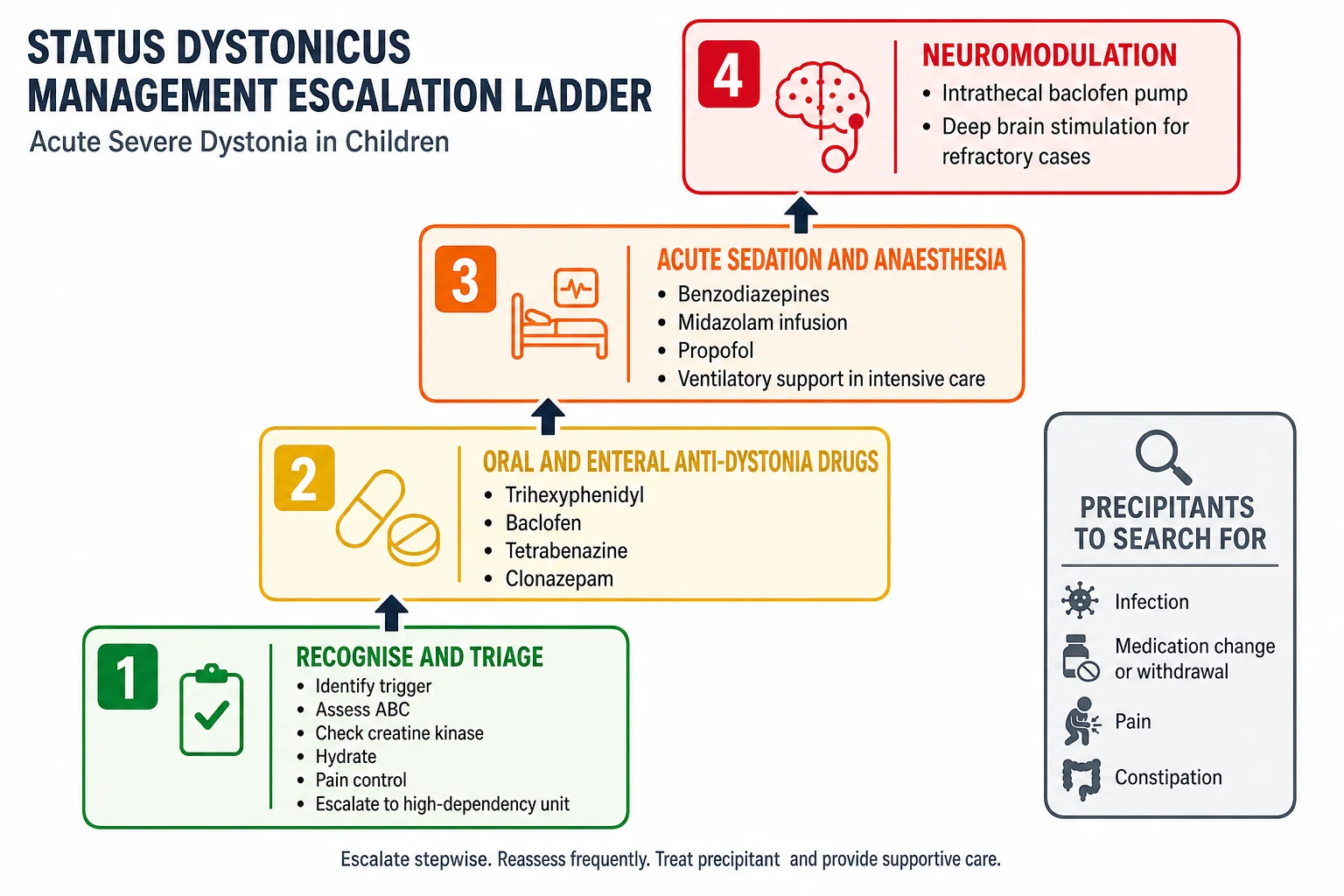
Status dystonicus is triaged to a high-dependency or intensive care setting without delay, because it is life-threatening and the threat is airway compromise, respiratory failure, and rhabdomyolysis with acute kidney injury. Secure the airway and breathing, establish intravenous access, and check creatine kinase, renal function, and urine myoglobin, because the muscle breakdown of sustained dystonic spasm can precipitate acute kidney injury and demands aggressive intravenous hydration. Control pain, because dystonia hurts, and uncontrolled pain feeds the spasm. Identify and treat the precipitant, which is usually an infection, a medication change or withdrawal, pain, or constipation, because removing the trigger is part of the cure. [6][7]
Deploy temporising measures while preparing for escalation. A benzodiazepine such as diazepam or midazolam settles the spasm and buys time, and enteral anti-dystonia drugs such as trihexyphenidyl, baclofen, and tetrabenazine are started or up-titrated. If the spasms will not settle, the child is sedated, paralysed, and ventilated in intensive care while the dystonia-directed therapy takes effect, and early consideration is given to intrathecal baclofen or deep brain stimulation for refractory disease. [6][7]
Management — Definitive & Stepwise
Chronic dystonia is built up on an oral ladder, and the order is the order of evidence and tolerability. Trihexyphenidyl is the first oral agent in many children with dystonic cerebral palsy, started at a low dose and titrated to effect, and the Cochrane review found low-certainty evidence of benefit from a single crossover trial of sixteen children, so the expectation is modest and the watch is for anticholinergic side effects of sedation, dry mouth, blurred vision, and urinary retention. Baclofen is added for its effect on spasticity and dystonia, tetrabenazine is reserved for severe chorea and dystonia where the dopamine-depleting effect is wanted, and clonazepam is useful for myoclonus and a dystonia with an anxiety component. [8][9]
[6] [7]Focal dystonia is treated with botulinum toxin injected into the overactive muscle, repeated every three to four months, and this is the mainstay for a painful and function-limiting focal dystonia such as torticollis or a tight adductor group. Dyskinetic cerebral palsy with severe generalised dystonia may respond to intrathecal baclofen delivered by an implanted pump, which reduces both spasticity and dystonia and improves comfort, care, and ease of handling. Deep brain stimulation of the globus pallidus internus is offered to selected children, most effectively those with primary generalised dystonia, and is considered in some secondary dystonias. Selective dorsal rhizotomy has a role in the spastic-dominant child with cerebral palsy but is not a primary dystonia treatment. [9]
Dopa-responsive dystonia is the one cause that is simply cured. Levodopa combined with a peripheral decarboxylase inhibitor is given at a low dose, the response is dramatic and sustained, and the child who walked on an inturned foot walks normally. This is why the empiric levodopa trial is not optional in a dystonia of unknown cause. Sydenham chorea is managed supportively with rest and a calm environment, with symptomatic drugs such as valproate, carbamazepine, or a neuroleptic for moderate or severe chorea, and short-course corticosteroids for severe or refractory disease. Secondary penicillin prophylaxis follows the rheumatic fever regimen, with benzathine penicillin G 600,000 units intramuscularly for a child weighing 27 kilograms or less and 1,200,000 units for a heavier child every four weeks, continued into early adulthood. [4][5][12]
Specific Subtypes & Scenarios
Dyskinetic cerebral palsy is the dominant chronic dystonia of childhood, and it is managed across a ladder that runs from oral trihexyphenidyl through botulinum toxin for focal problem areas to intrathecal baclofen for generalised dystonia, with parallel attention to pain, posture, hip surveillance, and communication aids. The Bohn 2021 systematic review collated the pharmacological and neurosurgical evidence and confirmed that the certainty is modest and the choice is individualised, which is exactly the counselling the family needs to hear. [8][9]
Dopa-responsive dystonia, or Segawa disease, is the textbook curable dystonia and deserves its own paragraph because missing it is the cardinal error. It is caused in the commonest form by a mutation in the GCH1 gene that reduces dopamine synthesis, presents in early childhood with lower-limb dystonia that worsens through the day and is relieved by sleep, and responds dramatically to low-dose levodopa. The message for the exam and for the child is identical: trial levodopa in every dystonia of unknown cause, because the cost of the trial is a few days and the cost of missing it is a lifetime of unnecessary disability. [12]
Sydenham chorea presents weeks to months after group A streptococcal infection, is often asymmetric, and carries emotional lability, obsessions, and behavioural change alongside the movement. It is managed supportively, with symptomatic drugs for moderate or severe chorea, corticosteroids for severe or refractory disease, and secondary penicillin prophylaxis to prevent recurrence and protect the heart. The Dean and Singer 2017 evidence review confirmed that the evidence base is thin, with a single placebo-controlled steroid study and small series for intravenous immunoglobulin and plasma exchange, so treatment is individualised and immunotherapy is reserved for severe or refractory disease. [4][5]
Wilson disease presents with hepatic, psychiatric, and movement features, and the movement disorder is a mix of dystonia, tremor, and parkinsonism. Screen every dystonia and chorea of unknown cause with serum copper and caeruloplasmin, a 24-hour urinary copper, and slit-lamp examination for Kayser-Fleischer rings, and treat with chelation and zinc because delayed treatment causes irreversible neurologic injury. Glutaric aciduria type 1 presents with macrocephaly and an acute encephalopathic crisis, usually during an intercurrent illness, that leaves striatal injury and severe dystonia, and the crisis is preventable by aggressive emergency management of catabolic stress. The acute dystonic reaction is the common iatrogenic emergency, precipitated by metoclopramide or an antipsychotic, and reversed by an anticholinergic. [10]
Complications & Pitfalls
The life-threatening complications of status dystonicus are rhabdomyolysis with acute kidney injury, respiratory compromise when the chest wall is involved, aspiration, deep vein thrombosis, and prolonged intensive care dependency. The long-term complications of a chronic dystonia are pain, contracture and joint deformity, hip subluxation and dislocation, scoliosis, swallowing difficulty and malnutrition, impaired communication, and reduced participation with a direct effect on school, social life, and mental health. A child with severe generalised dystonia is a child whose comfort, care, and dignity depend on the vigilance of the treating team. [6]
The classic pitfalls are the ones an examiner probes deliberately. Mistaking a movement disorder for a seizure, or for a primary psychiatric disorder, sends the workup the wrong way. Failing to trial levodopa in an unexplained dystonia misses the one curable cause. Attributing Sydenham chorea to a behavioural disorder misses the cardiac assessment and the prophylaxis that prevents recurrence and rheumatic heart disease. Prescribing a dopamine-blocking antiemetic to a child precipitates the very acute dystonic reaction that fills the emergency department. Treating status dystonicus with inadequate escalation or delayed intensive care transfer costs the child their kidneys, their airway, or their life. Withholding botulinum toxin from a child whose focal dystonia is painful and function-limiting is a common under-treatment error that leaves the child in pain for months. [6][10]
Prognosis & Disposition
Prognosis is set by the cause, and the range is wide. Dopa-responsive dystonia has an excellent prognosis with a sustained levodopa response, and is the diagnosis that turns a wheelchair prediction into a child who walks. Sydenham chorea usually resolves over weeks to months but recurs in a minority and demands ongoing rheumatic prophylaxis and cardiac surveillance, because the long-term threat is rheumatic heart disease rather than the chorea itself. Dystonic cerebral palsy is a lifelong condition whose outlook is set by the functional burden, the control of pain, and the success of posture, hip, and communication management, and intrathecal baclofen and deep brain stimulation improve comfort and care in carefully selected children. [9][12]
Status dystonicus carries significant mortality and morbidity and demands early escalation, because the threats are airway, breathing, and rhabdomyolysis. Disposition runs from intensive care for status dystonicus or airway compromise, to the ward for titration of oral or enteral therapy, to the outpatient multidisciplinary clinic for the lifelong management of chronic dystonia, and to a tertiary neurosciences centre for intrathecal baclofen, deep brain stimulation, and a full genetic and metabolic workup. A written dystonia action plan and a family safety-net for escalation are essential parts of discharge, because the family is the first responder when a chronic dystonia worsens toward status dystonicus. [6][7]
Special Populations
The child with dyskinetic cerebral palsy and complex disability is the largest single group and needs a coordinated plan that reads dystonia as one thread in a fabric of posture, pain, hip health, nutrition, and communication. The non-verbal child needs a movement assessment that treats pain and discomfort as drivers of worsening dystonia, and the team needs a low threshold for treatable precipitants such as constipation, gastro-oesophageal reflux, urinary infection, dental disease, or an evolving hip subluxation. A worsening dystonia in a child with cerebral palsy is a hip or a pain problem until proven otherwise. [9]
The Aboriginal and Torres Strait Islander child and the child from a remote setting carries the highest burden of acute rheumatic fever and Sydenham chorea in Australia and Aotearoa New Zealand, so the stakes of recognising chorea are high. Secondary prophylaxis adherence, household contact tracing, and access to echocardiography are central to preventing rheumatic heart disease, and a culturally safe model of care with Aboriginal health workers and remote retrieval pathways is part of the treatment. The adolescent with new chorea or dystonia needs a pregnancy test, a drug and alcohol history, and screening for Wilson disease, because the adolescent is the age band where Wilson disease declares itself. [4]
The child from a refugee or migrant family needs early interpreter use, an expanded infection screen, and attention to the possibility of an inherited metabolic disease with a high carrier rate in some populations. The child in out-of-home care and the child with a trauma history needs a trauma-informed approach to the assessment of a possible functional movement disorder, with a confident diagnosis and a clear path to functional neurology and rehabilitation rather than a dismissive label. [11]
Evidence, Guidelines & Regional Differences
The dystonia classification rests on the Albanese 2013 consensus update, which set the two-axis framework that still organises practice, and the childhood hypertonia framework rests on the Sanger 2003 task-force statement that split spasticity, dystonia, and rigidity for the cerebral palsy examination. Status dystonicus management is guided by the Allen 2014 practice guide and the 2024 international consensus recommendations for the initial and refractory paediatric disease from the Boston, Toronto, and international group, which set out a stepwise triage and escalation pathway and integrate genomics and neuromodulation. [1][2][7]
The treatment evidence is humbler than the classification, and the candour matters at viva. The Cochrane review of trihexyphenidyl for dystonia in cerebral palsy found only low-certainty evidence from a single crossover trial of sixteen children, and the Bohn 2021 systematic review update collated the pharmacological and neurosurgical evidence across oral drugs, botulinum toxin, intrathecal baclofen, and deep brain stimulation. Sydenham chorea treatment rests on the Dean and Singer 2017 evidence review and the Gewitz 2015 Jones criteria revision, which confirmed Sydenham chorea as a major criterion and codified the secondary prophylaxis regimen. [4][5][8]
The chief controversy is PANDAS and PANS, the putative post-streptococcal autoinflammatory neuropsychiatric syndrome. The evidence base is contested, the diagnostic criteria are broad, and the role of antibiotic and immunomodulatory therapy is not settled, so most paediatric neurology services restrict the diagnosis to tightly defined presentations and treat the obsessive-compulsive and behavioural features with standard cognitive-behavioural and pharmacological therapy. Regional differences matter most in rheumatic fever: in Australia and Aotearoa New Zealand the burden falls on Aboriginal and Torres Strait Islander and remote communities, with secondary prophylaxis extended per the local rheumatic fever guideline, whereas in regions of low incidence Sydenham chorea is rare and the diagnosis is easily missed. [4][11]
Exam Pearls
Chorea is flowing, unpredictable, semi-purposeful movement with motor impersistence, shown by the milkmaid grip and the darting tongue. Dystonia is sustained or intermittent involuntary contraction causing twisting and abnormal postures, often eased by a sensory trick. Myoclonus is a sudden shock-like jerk. Tics are brief, stereotyped, and suppressible, with a premonitory urge. The Albanese 2013 classification runs axis I for clinical characteristics and axis II for aetiology. The Sanger 2003 framework splits childhood hypertonia into spasticity, dystonia, and rigidity. [1][2]
Every unexplained childhood dystonia gets an empiric low-dose levodopa trial, because dopa-responsive dystonia, or Segawa disease, with its GCH1 mutation, diurnal worsening, and lower-limb onset, responds dramatically. Sydenham chorea is a major Jones criterion, follows group A streptococcal infection, and needs secondary penicillin prophylaxis with benzathine penicillin G at 600,000 units for a child weighing 27 kilograms or less and 1,200,000 units for a heavier child every four weeks. The acute dystonic reaction from metoclopramide or an antipsychotic is reversed with an anticholinergic such as benztropine, procyclidine, or diphenhydramine. [4][12]
Status dystonicus escalation runs trigger control, oral anti-dystonia drugs, benzodiazepine and sedation, then intrathecal baclofen or deep brain stimulation, with creatine kinase and rhabdomyolysis surveillance throughout. Screen every dystonia and chorea of unknown cause for Wilson disease with serum copper, caeruloplasmin, 24-hour urinary copper, and slit-lamp examination. The Cochrane evidence for trihexyphenidyl in cerebral palsy dystonia is of low certainty from a single small crossover trial. The basal ganglia direct pathway facilitates wanted movement and the indirect pathway suppresses unwanted movement, with dopamine tuning the balance, which is why dopamine-blocking drugs cause dystonia and dopamine loss causes parkinsonism. [6][8]
References
- [1]Albanese A, Bhatia K, Bressman SB, et al Phenomenology and classification of dystonia: a consensus update. Mov Disord, 2013.PMID 23649720
- [2]Sanger TD, Delgado MR, Gaebler-Spira D, Hallett M, Mink JW Classification and definition of disorders causing hypertonia in childhood. Pediatrics, 2003.PMID 12509602
- [3]Sanger TD Pediatric movement disorders. Curr Opin Neurol, 2003.PMID 12869814
- [4]Gewitz MH, Baltimore RS, Tani LY, et al Revision of the Jones Criteria for the diagnosis of acute rheumatic fever in the era of Doppler echocardiography: a scientific statement from the American Heart Association. Circulation, 2015.PMID 25908771
- [5]Dean SL, Singer HS Treatment of Sydenham's Chorea: A Review of the Current Evidence. Tremor Other Hyperkinet Mov (N Y), 2017.PMID 28589057
- [6]Allen NM, Lin JP, Lynch T, King MD Status dystonicus: a practice guide. Dev Med Child Neurol, 2014.PMID 24304390
- [7]Vogt LM, Yang K, Tse G, et al Recommendations for the Management of Initial and Refractory Pediatric Status Dystonicus. Mov Disord, 2024.PMID 38619077
- [8]Harvey AR, Baker LB, Reddihough DS, Scheinberg A, Williams K Trihexyphenidyl for dystonia in cerebral palsy. Cochrane Database Syst Rev, 2018.PMID 29763510
- [9]Bohn E, Goren K, Switzer L, Falck-Ytter Y, Fehlings D Pharmacological and neurosurgical interventions for individuals with cerebral palsy and dystonia: a systematic review update and meta-analysis. Dev Med Child Neurol, 2021.PMID 33772789
- [10]Fong AT, Jacob JG, Carroll B, Fernando SL Acute dystonic reaction mimicking angioedema secondary to metoclopramide ingestion. Emerg Med Australas, 2021.PMID 33873238
- [11]Wilbur C, Bitnun A, Kronenberg S, Laxer RM, Levy DM, Logan WJ, et al PANDAS/PANS in childhood: Controversies and evidence. Paediatr Child Health, 2019.PMID 30996598
- [12]Segawa M Dopa-responsive dystonia. Handb Clin Neurol, 2011.PMID 21496606