Paeds · neurology-neurodisability-and-neuromuscular
Myasthenia gravis and neuromuscular junction disorders
Also known as Juvenile myasthenia gravis · Autoimmune myasthenia gravis · MuSK antibody myasthenia gravis · Congenital myasthenic syndrome · Lambert-Eaton myasthenic syndrome · Myasthenic crisis · Transient neonatal myasthenia
Fellowship guide to myasthenia gravis and the neuromuscular junction disorders of childhood. Covers the fatigable, fluctuating weakness of autoimmune myasthenia gravis with the acetylcholine receptor, muscle-specific kinase, and LRP4 antibody subtypes and their clinical signatures, the complement-mediated destruction of the postsynaptic junctional folds, the myasthenia gravis Foundation of America clinical classification, the bedside fatiguability manoeuvres, the ice pack test, the single-fibre and repetitive nerve stimulation neurophysiology, and the antibody panels. Details pyridostigmine dosing, corticosteroid initiation with its transient worsening, steroid-sparing agents, intravenous immunoglobulin and plasma exchange for crisis, thymectomy after the MGTX trial, and eculizumab for refractory disease. Distinguishes the congenital myasthenic syndromes by gene and by subtype-specific treatment including the subtypes where pyridostigmine worsens the disease, the maternal antibody syndromes of transient neonatal myasthenia and fetal acetylcholine receptor inactivation, the presynaptic Lambert-Eaton myasthenic syndrome and botulism, and the myasthenic crisis with the forced vital capacity thresholds for intensive care.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
Overview & Definition
A child who can hold their arms up for the first minute and watches them droop by the second, whose eyelids lift at rest and fall as they keep talking, and whose swallowing worsens through a meal, is presenting the signature sign of the neuromuscular junction disorders: fatigable weakness that worsens with sustained activity and recovers with rest. Myasthenia gravis is an antibody-mediated attack on the postsynaptic membrane of the neuromuscular junction, and it is the prototypic and the commonest of these disorders. The weakness fluctuates within the day, spares sensation and the pupils, and responds to acetylcholinesterase inhibition in the antibody-mediated forms. [1]
The reason this family of diseases matters above all is that the same process that fatigues the arms can fatigue the respiratory muscles and the bulbar muscles, and the child who is talking in the morning can be in ventilatory failure by evening. The single most important habit at the bedside is to measure the forced vital capacity repeatedly and to escalate to intensive care before the child tires. Symptomatic treatment with pyridostigmine restores strength in the antibody-mediated forms, and disease-modifying immunotherapy with corticosteroids, steroid-sparing agents, intravenous immunoglobulin, and thymectomy controls the underlying attack. [1]
The framing concept is the neuromuscular junction itself, where the motor nerve terminal releases acetylcholine across the synaptic cleft to bind acetylcholine receptors on the folded postsynaptic muscle membrane. Autoimmune myasthenia gravis attacks the postsynaptic side through antibodies against the acetylcholine receptor, the muscle-specific kinase, or LRP4. The congenital myasthenic syndromes are genetic defects of the same structure and are not immune, so they are not antibody-mediated and do not respond to immunotherapy. Lambert-Eaton myasthenic syndrome and botulism attack the presynaptic side, and the maternal antibody syndromes affect the baby. The job is to recognise the fatigable weakness, to treat the autoimmune forms, and to distinguish the genetic and presynaptic disorders that change everything. [1][9]
Classification
The neuromuscular junction disorders are sorted by where the fault lies and by whether the attack is immune or genetic, and both sorts change the treatment. The first sort is anatomical: presynaptic, synaptic, and postsynaptic. The second sort is causal: autoimmune, genetic, and toxin or antibody-mediated. [1]
The autoimmune group dominates and lives on the postsynaptic membrane. Acetylcholine receptor antibody myasthenia gravis is the commonest form, accounts for around eighty percent of generalised disease, and is the form that responds to thymectomy and to complement inhibition. Muscle-specific kinase antibody myasthenia gravis accounts for around five to eight percent and behaves differently, with prominent bulbar, facial, neck, and respiratory weakness, a relative sparing of the limbs, a poorer response to acetylcholinesterase inhibitors, and a higher chance of crisis. LRP4 antibody disease is rarer and often milder. Seronegative disease, with no detectable antibody, rounds out the group. [11]
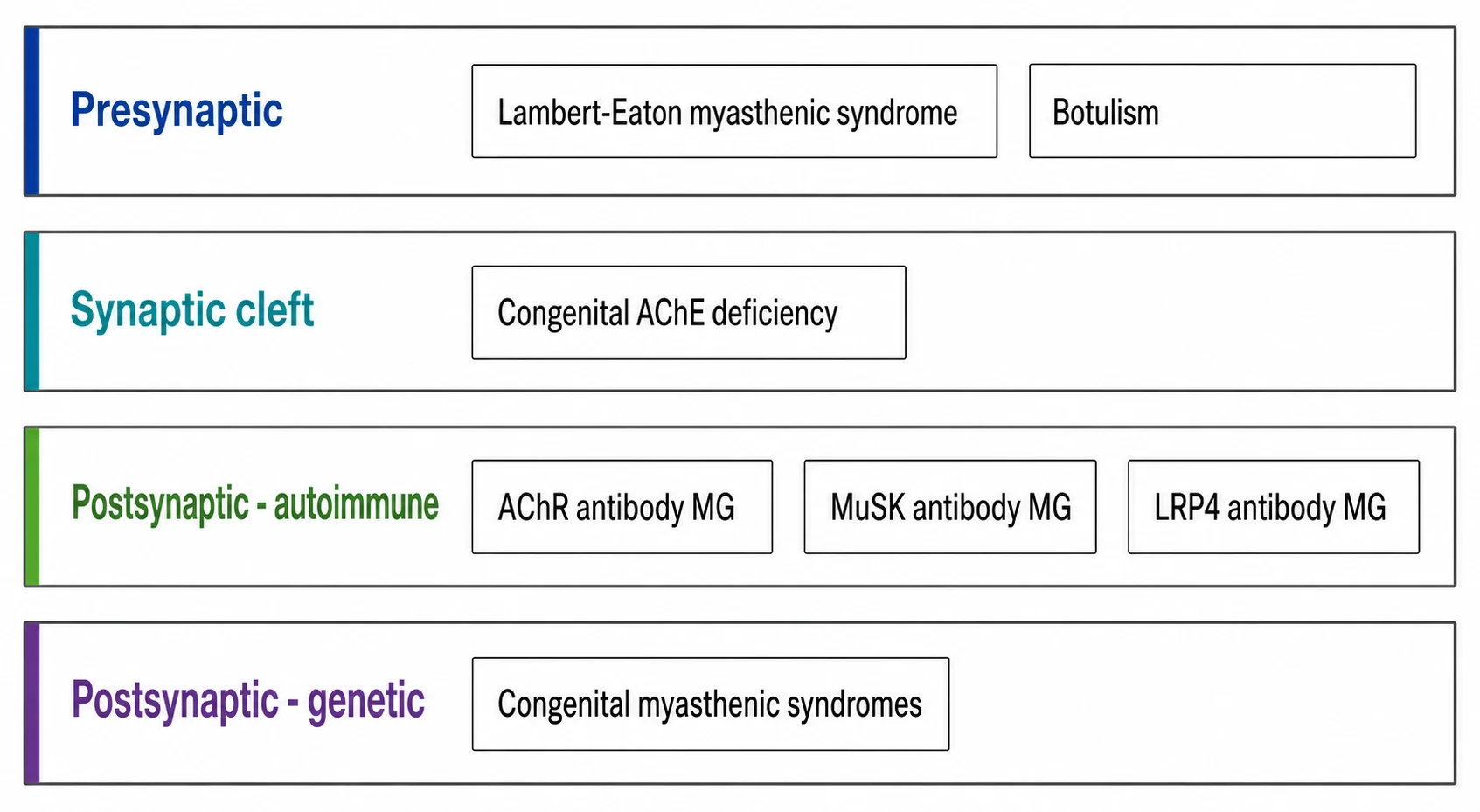
The genetic group is the congenital myasthenic syndromes, and it shares the fatigable weakness but is not immune and not antibody-mediated. The subtypes are named for the defective protein: acetylcholine receptor deficiency from CHRNE or RAPSN mutations is commonest, Dok7 and ColQ endplate acetylcholinesterase deficiency and the slow-channel syndrome each carry a treatment that differs from the rest, and each gene points to a specific drug. The presynaptic group is Lambert-Eaton myasthenic syndrome and botulism, which are distinguished by their facilitation and their pupils. [9]
Epidemiology & Risk Factors
Autoimmune myasthenia gravis has a bimodal age distribution that matters in paediatrics. The early-onset form peaks in adolescent girls and overlaps with other autoimmune disease, while the late-onset form affects older adults. Juvenile myasthenia gravis, defined as onset before the sixteenth birthday, is uncommon but not rare, and the UK population study found a childhood prevalence of autoimmune myasthenia of around one to two per one hundred thousand, with a clear female predominance after puberty. The congenital myasthenic syndromes are rarer still but are the dominant cause in infancy and early childhood, where autoimmune disease is uncommon. [8]
The strongest risk marker in juvenile disease is female sex after puberty, and an associated autoimmune condition. Thyroid disease is the commonest accompaniment, and type one diabetes, juvenile idiopathic arthritis, and vitiligo coexist often enough that a thyroid screen is part of the workup. A thymoma is rare in children but must be excluded by chest imaging, because a thymoma changes the management to mandatory surgical resection. A family history of autoimmune disease supports the diagnosis, while a family history of fatigable weakness in infancy or consanguinity points instead to a congenital myasthenic syndrome. [1]
Pathophysiology
The mechanism of autoimmune myasthenia gravis is an antibody-mediated attack on the postsynaptic membrane, and the single sentence to hold is that the antibodies bind the acetylcholine receptor, fix complement, and destroy the junctional folds, so each nerve impulse releases enough acetylcholine to depolarise fewer and fewer receptors as the muscle fatigues. [1]
In acetylcholine receptor antibody disease, IgG1 and IgG3 antibodies bind the receptor and do three things. They block the binding of acetylcholine, they cross-link receptors and speed their removal from the membrane, and they activate complement to form the membrane attack complex on the junctional folds. The folds simplify and flatten, the receptor density falls, and the safety factor for neuromuscular transmission drops. At the start of sustained activity the residual receptors cope, but as acetylcholine release wanes with each impulse the transmission fails, and the weakness appears and deepens until rest allows recovery. This is the physiological basis of fatiguability. [1]
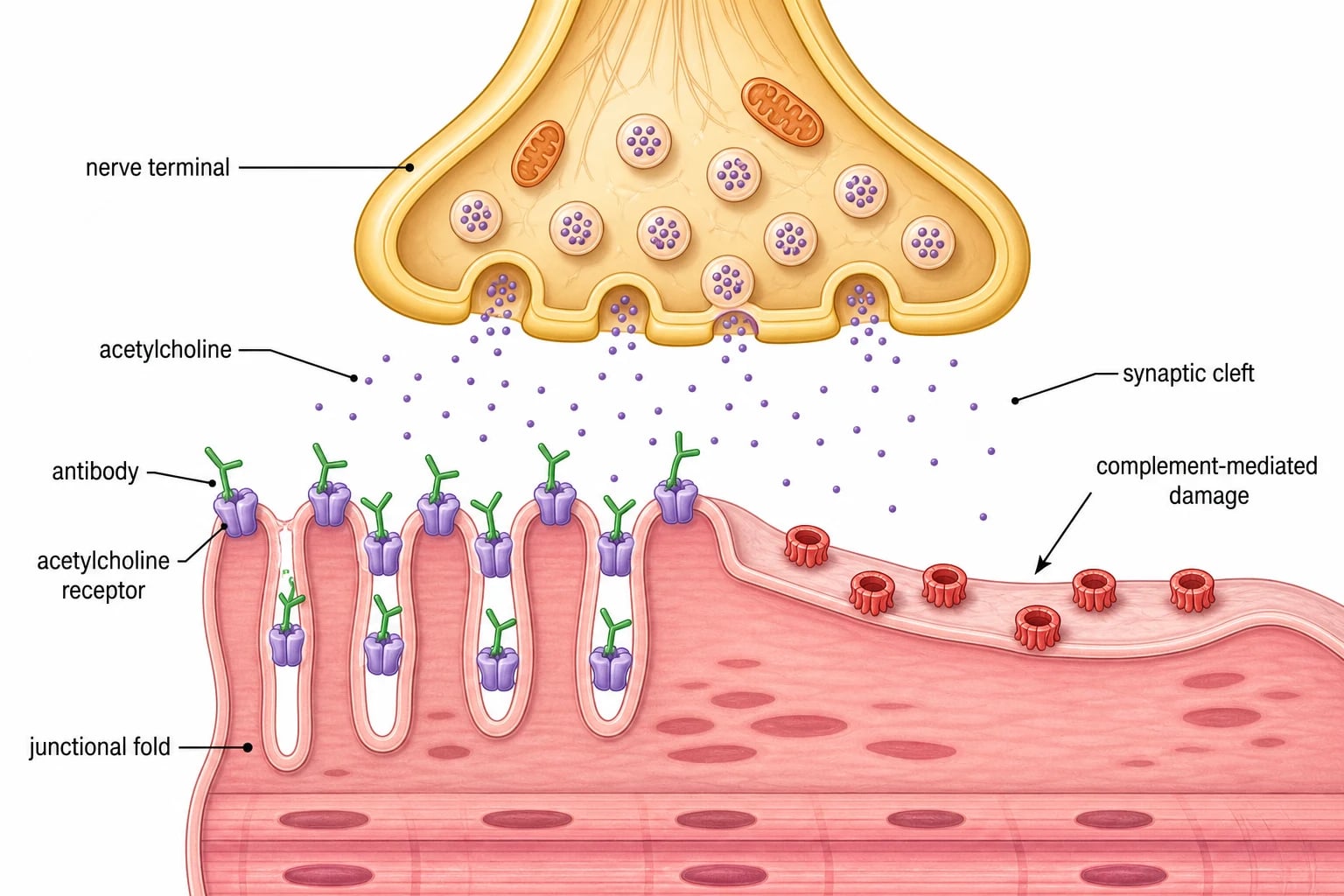
In muscle-specific kinase antibody disease the target is different. The muscle-specific kinase organises the clustering of acetylcholine receptors during development and maintains their density at the junction, and the antibodies are predominantly IgG4 that block this pathway. The receptor density falls without the gross complement-mediated destruction, which explains the different phenotype of severe bulbar and facial weakness and the poor response to acetylcholinesterase inhibitors, because adding more acetylcholine to a junction with too few receptors and a disorganised structure does little. The congenital myasthenic syndromes share the physiology but not the mechanism, because they arise from inherited mutations in the same proteins, so the weakness is present from infancy, often fixidos, and never carries an antibody. [11][9]
Clinical Presentation
The child with autoimmune myasthenia gravis presents with weakness that fluctuates through the day and fatigues with sustained use, and the pattern of the weakness points to the junction. The ocular muscles are involved first in around half of children, with asymmetrical ptosis that varies from hour to hour and diplopia from variable ophthalmoparesis. The bulbar muscles follow, with a nasal voice, dysarthria that worsens with prolonged speech, dysphagia that worsens through a meal, and a weak cough. The proximal limb and neck muscles fatigue, so the child who raises the arms strongly droops within a minute, and neck weakness brings a head droop. Sensation, the deep tendon reflexes, and the pupils are preserved, and this combination is the bedside signature of a junction disorder rather than a neuropathy or a myopathy. [1]
The tempo and the age bend the presentation, and this is where the diagnosis is missed in children. Juvenile disease in the adolescent girl mirrors the adult pattern with ocular onset and a relapsing course, but the younger child may present only as a clumsy or tiring child who falls in the afternoon or struggles to keep up in sport. The prepubertal boy is the classic ocular-only phenotype, which often stays purely ocular for years. MuSK antibody disease, commoner in women and in some populations, presents with dominant bulbar, facial, neck flexor, and respiratory weakness, with little limb involvement, and it is the form most likely to present directly in crisis. [11]
The bedside manoeuvres that expose the fatiguability are the core of the clinical assessment and are worth practising. Sustained upgaze for one minute provokes ptosis, a sustained arms-up posture for one minute provokes drooping, repeated squats or counting out loud fatigues the relevant muscle group, and the ice pack test, holding ice over a ptotic eyelid for two minutes, produces a transient improvement by slowing the acetylcholinesterase and is positive when the ptosis lifts. The deep tendon reflexes are preserved, in contrast to the areflexia of Guillain-Barre syndrome, and the pupils are spared, in contrast to the fixed pupils of botulism. [2]
FATIGUE
Differential Diagnosis
The differential of fatigable weakness runs across the junction, the nerve, the muscle, and the brain, and the discriminating features are the fatiguability, the reflex pattern, the pupils, and the fluctuation. The first fork is between a neuromuscular junction disorder and a fixed peripheral process, because the management diverges at once. [1]
Guillain-Barre syndrome produces ascending weakness but it is fixed rather than fatigable, areflexia appears early, and the course is monophasic over days rather than fluctuating through the day. A congenital myopathy or muscular dystrophy produces fixed, slowly progressive proximal weakness with preserved fatiguability pattern, and the reflexes are reduced in proportion to the weakness. Chronic fatigue syndrome and functional weakness are fatigable but lack the objective fatigability on sustained upgaze and the positive ice pack and neurophysiology. Ocular myasthenia must be distinguished from a congenital cranial dysinnervation disorder, a thyroid ophthalmopathy, and a mitochondrial chronic progressive external ophthalmoplegia, all of which produce fixed rather than fatigable ptosis and ophthalmoparesis. [1]
The remaining mimics each carry a specific pitfall. Thyroid eye disease produces fixed proptosis and lid lag rather than fatigable ptosis, and the thyroid function tests settle it. A brainstem stroke or a cranial nerve palsy is acute and fixed and lacks the fluctuation. Tick paralysis is ascending and areflexic, and botulism descends with dilated pupils and constipation. The clinching combination is the fatigable, fluctuating weakness with preserved reflexes and pupils and a positive ice pack or neurophysiology, and the antibody panel confirms the autoimmune form. [1]
Clinical & Bedside Assessment
The bedside assessment runs in parallel with monitoring, because a child with myasthenia gravis can deteriorate quickly into crisis. Secure the airway and assess the bulbar function, the swallow, and the cough, because aspiration and ventilatory failure are the immediate threats. Place the child on cardiac and respiratory monitoring and measure the bedside spirometry in any child with bulbar or proximal weakness, because the respiratory muscles fatigue just as the limbs do. [7]
The respiratory monitoring is the part that saves lives, and it is the single most examined bedside skill in this topic. Measure the forced vital capacity, the negative inspiratory force, and the peak cough flow every two to six hours in any child with progressive or bulbar weakness. Escalate to intensive care for a forced vital capacity under twenty millilitres per kilogram, and intubate when it falls under fifteen millilitres per kilogram or when the negative inspiratory force falls under negative forty centimetres of water. Bulbar weakness with a weak cough, pooled secretions, or aspiration mandates a protected airway. The child who is still talking can have a falling vital capacity, so the numbers, not the bedside appearance, drive the decision to intubate. [2][7]
The focused examination looks for the fatiguability that confirms a junction disorder and for the features that point to a mimic. Perform the sustained-upgaze and arms-up manoeuvres to provoke the weakness, and apply the ice pack test to a ptotic eyelid. Examine the cranial nerves for fatigable ptosis, ophthalmoparesis, facial weakness, and a weak cough, and the limbs for proximal fatigable weakness. Check that the deep tendon reflexes and the pupils are preserved, because their loss argues for Guillain-Barre syndrome or botulism. Look for an associated goitre or vitiligo, take a family history of autoimmune disease and consanguinity, and ask about fatigability from infancy that would point to a congenital myasthenic syndrome. [2]
In ANZ practice, the Australian and New Zealand Child Neurology Network coordinates the care of children with suspected neuromuscular junction disease, and peripheral centres start the workup and the respiratory monitoring on telephone advice while retrieval is arranged. The antibody panels and the specialist neurophysiology are concentrated in tertiary centres, so the threshold to transfer a child with bulbar weakness or a falling forced vital capacity is low, and pyridostigmine is often started empirically once the diagnosis is likely. [2]
Investigations
The investigations confirm a junction disorder, distinguish the autoimmune from the genetic and the postsynaptic from the presynaptic, and exclude the mimics, and they run together rather than in sequence. [1]
The antibody panel is the cornerstone of the autoimmune diagnosis and guides the management. The acetylcholine receptor antibody, measured as binding, blocking, and modulating assays, is positive in around eighty percent of generalised disease and around fifty percent of pure ocular disease, and a positive result is essentially diagnostic. The muscle-specific kinase antibody is tested when the acetylcholine receptor antibody is negative, and it is positive in around five to eight percent of generalised disease, with the bulbar-predominant phenotype. The LRP4 antibody is tested in selected cases. A negative antibody panel with a typical clinical and neurophysiological picture is seronegative myasthenia gravis and is managed as autoimmune disease. [11]
Neurophysiology supports the diagnosis and excludes a myopathy and a neuropathy. Repetitive nerve stimulation at three hertz shows a decremental response of greater than ten percent in the compound muscle action potential in a clinically weak muscle, and is positive in most generalised disease. Single-fibre electromyography, measuring the jitter between two muscle fibres of the same motor unit, is the most sensitive test and is abnormal in nearly all generalised disease, but it is technically demanding and less available in children. A normal early study does not exclude the diagnosis, and a weak muscle should be chosen. [1]
[2]Chest imaging excludes a thymoma and is obtained in every newly diagnosed child. A computed tomography or magnetic resonance imaging of the chest is preferred to a plain radiograph, because a small thymoma is missed on a plain film, and a thymoma mandates surgical resection. The ice pack test and a trial of pyridostigmine support the diagnosis at the bedside when the antibody and neurophysiology results are awaited. The thyroid function tests and a autoimmune screen exclude an associated thyroid disease. Genetic testing, increasingly by a neuromuscular gene panel, confirms a congenital myasthenic syndrome and identifies the subtype that sets the treatment. [1][9]
Management — Resuscitation
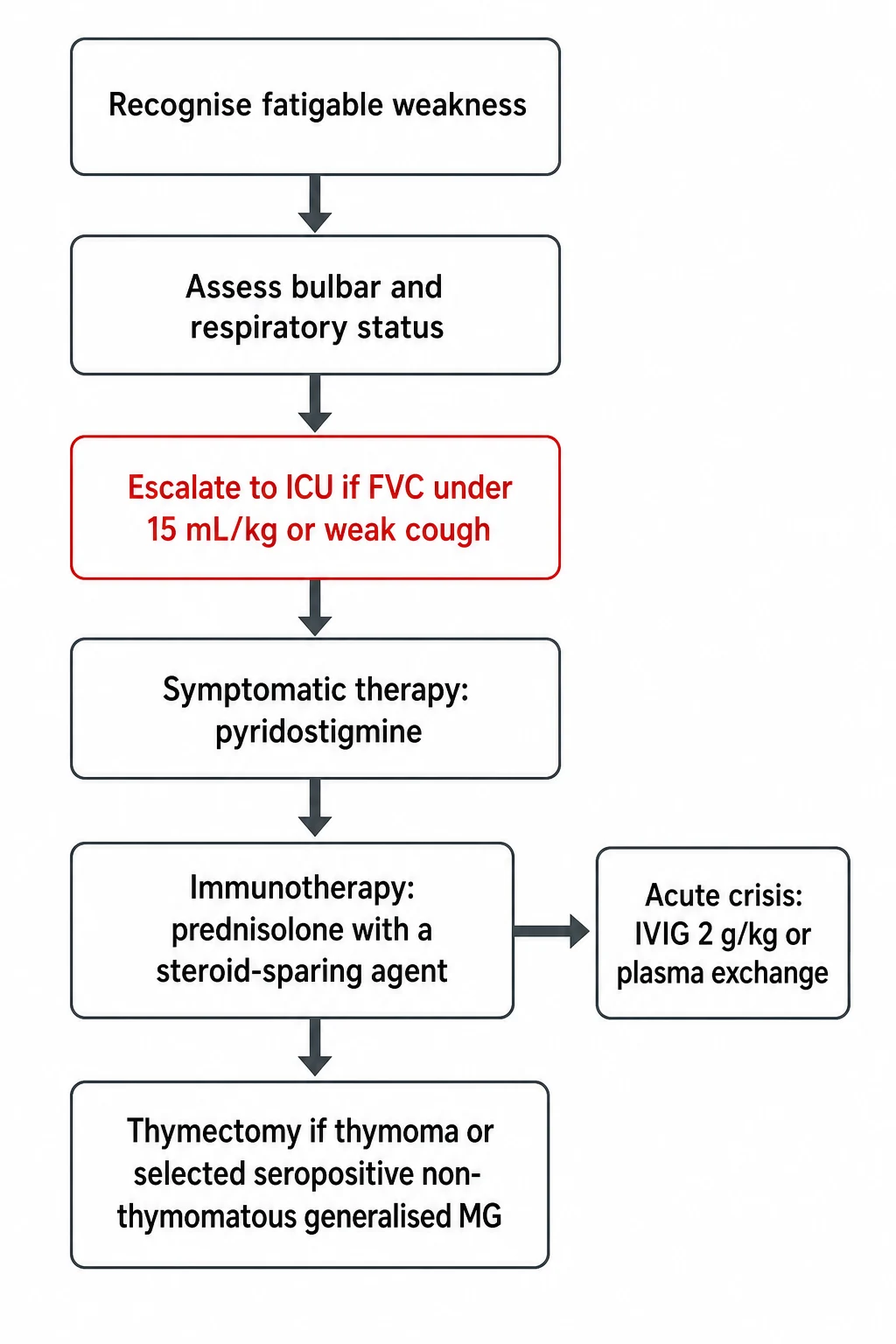
Resuscitation protects the airway and the breathing while the workup runs, and the respiratory monitoring is the central task. Any child with bulbar or progressive weakness is admitted to a monitored bed with bedside spirometry every two to six hours, and the intensive care team is involved early. The decision to intubate is driven by the numbers, not by how the child looks. [7]
Intensive care is indicated for a forced vital capacity under twenty millilitres per kilogram, a negative inspiratory force under negative forty centimetres of water, bulbar weakness with a weak cough or aspiration, or a rapid deterioration. When intubation is needed, a rapid sequence induction is used because the weak respiratory muscles and the risk of aspiration make this a high-risk airway, and the sedative choice avoids prolonged neuromuscular blockade, which can delay weaning in a junction disorder. Non-invasive ventilation has a role in the stable deteriorating child as a bridge, but it does not replace intubation for bulbar failure. [2][7]
The precipitant of a crisis is sought and reversed, because most deteriorations have a trigger. The common precipitants are infection, fever, surgery, emotional stress, and the drugs that worsen the junction, including aminoglycosides, fluoroquinolones, macrolides, beta-blockers, magnesium, and neuromuscular blockers. Pyridostigmine is continued unless secretions are prohibitive, the precipitating drug is stopped, and the fever and the infection are treated. First-line disease-modifying therapy for the crisis is started once the airway is secure, because intravenous immunoglobulin and plasma exchange take days to work. [2]
Management — Definitive & Stepwise
The definitive treatment layers symptomatic therapy with disease-modifying immunotherapy, and the two serve different purposes. Pyridostigmine restores the strength that the surviving receptors can deliver, while the immunotherapy halts the antibody attack that is destroying the receptors. Both are usually needed in generalised disease. [2]
Symptomatic first-line therapy is pyridostigmine, an acetylcholinesterase inhibitor that prolongs the action of acetylcholine at the junction. In children the dose is around one milligram per kilogram per dose given four to five times daily, or the equivalent divided dosing, titrated to the clinical response and limited by the cholinergic side effects of diarrhoea, abdominal cramps, salivation, and bronchial secretions. Pyridostigmine helps acetylcholine receptor and LRP4 antibody disease and the symptomatic congenital syndromes, but it does little for muscle-specific kinase antibody disease, where excess secretions can worsen the bulbar weakness, and it is contraindicated in several congenital subtypes. The neostigmine and atropine adjuncts have a limited place. [2]
Myasthenia gravis pharmacotherapy (paediatric)
Corticosteroids are the mainstay of disease-modifying immunotherapy, but the way they are started matters. Prednisolone started at a high dose can transiently worsen the myasthenia in up to a half of patients within the first two weeks and can precipitate crisis, so it is begun at a low dose of around half a milligram per kilogram per day or on alternate days and titrated upward over weeks to around one to one and a half milligrams per kilogram, then tapered to the lowest effective dose. A steroid-sparing agent, such as azathioprine, mycophenolate, or methotrexate, is introduced early to permit the steroid taper and to control the chronic disease, with the caveat that azathioprine takes six to twelve months to work and that the thiopurine methyltransferase activity is checked. Rituximab has an increasing role in refractory disease, especially in muscle-specific kinase antibody disease. [2]
The crisis-specific therapies work faster than the oral immunotherapy and are reserved for a rapid deterioration, a bulbar or respiratory crisis, and the preparation for surgery. Intravenous immunoglobulin 2 g per kg over two to five days and plasma exchange, three to five sessions over one to two weeks, are equally effective, and the Cochrane review found the intravenous immunoglobulin speeds recovery in an exacerbation. Plasma exchange works faster and is often preferred for a severe crisis, while the intravenous immunoglobulin is easier and safer in children with poor venous access. The two are not combined. [5]
The management ladder, week by week
Day 0: Recognise fatigable weakness; admit if bulbar or respiratory; start bedside spirometry every 2 to 6 hours; secure the airway on the numbers
Day 0 to 1: Send acetylcholine receptor and muscle-specific kinase antibodies, repetitive nerve stimulation, chest imaging, and thyroid function tests; start pyridostigmine ~1 mg/kg per dose four to five times daily
Week 1 to 4: Start prednisolone low and titrate upward; add a steroid-sparing agent early; involve paediatric neurology and plan long-term immunotherapy
Crisis at any time: Intravenous immunoglobulin 2 g per kg over 2 to 5 days or plasma exchange 3 to 5 sessions; continue in the intensive care unit
Months 1 to 6: Consider thymectomy for thymoma and for selected seropositive non-thymomatous generalised disease; consider rituximab or eculizumab for refractory disease
Long-term: Taper the steroid to the lowest effective dose, monitor the bone density and growth, vaccinate on immunosuppression guidance, and plan the transition to adult care
Thymectomy is considered in selected children, and the decision rests on the MGTX trial and the thymoma status. A thymoma mandates surgical resection in every age group. In non-thymomatous generalised acetylcholine receptor antibody disease, the MGTX trial in adults showed that extended transsternal thymectomy plus prednisone improved the clinical status and reduced the immunosuppression over three years, and the benefit is extrapolated to the adolescent with generalised disease. Thymectomy is not helpful in muscle-specific kinase antibody disease and in the congenital myasthenic syndromes. Eculizumab, a terminal complement inhibitor, is reserved for refractory anti-acetylcholine receptor generalised disease after the REGAIN trial, and it requires meningococcal vaccination and lifelong vigilance for meningococcal infection. [4][6]
Supportive care runs throughout and protects the child from the complications of the disease and the treatment. Ocular symptoms are helped by eyelid crutches and prisms, the bulbar weakness by thickened fluids and nasogastric nutrition, and the fatigue by pacing the activity. The corticosteroid side effects of growth suppression, glucose intolerance, osteopenia, and weight gain are monitored, and the bone density and the growth are charted. The immunosuppression requires live-vaccine avoidance and pneumococcal and influenza cover. The psychological burden of a chronic, fluctuating, visible disease is real, and the school and the family are supported with a written management plan. [2]
Specific Subtypes & Scenarios
Juvenile myasthenia gravis is autoimmune disease with onset before the sixteenth birthday, and it follows the adult pattern in the adolescent but has its own features in the younger child. The prepubertal boy often has pure ocular disease that may remit, while the adolescent girl has generalised disease with a relapsing course and a strong association with thyroid disease. The treatment is pyridostigmine and immunotherapy, and thymectomy is considered in the adolescent with generalised acetylcholine receptor antibody disease, with evidence of good outcomes in juvenile series. [8]
MuSK antibody myasthenia gravis is the form that demands recognition, because it behaves differently and is more dangerous. The weakness is dominated by the bulbar, facial, neck flexor, and respiratory muscles, with relative limb sparing, and the response to acetylcholinesterase inhibitors is poor and can worsen the secretions. The disease is more refractory to first-line immunotherapy, and rituximab has a particular role. The crisis risk is higher, and the diagnosis is made on the muscle-specific kinase antibody when the acetylcholine receptor antibody is negative. Thymectomy is not helpful in this subtype. [11]
The congenital myasthenic syndromes are genetic disorders of the junctional proteins that share the fatigable weakness but are not immune and not antibody-mediated. Acetylcholine receptor deficiency from CHRNE mutations is the commonest and responds to pyridostigmine. RAPSN syndrome presents in infancy with multiple joint contractures and a striking response to pyridostigmine. Dok7 syndrome causes a limb-girdle pattern and is worsened by pyridostigmine, which is treated instead with ephedrine or salbutamol. ColQ endplate acetylcholinesterase deficiency is worsened by pyridostigmine, because the enzyme it inhibits is already absent, and is treated with ephedrine or salbutamol. The slow-channel syndrome is treated with quinidine or fluoxetine to block the overactive channel. The message is to confirm the gene before treating, because the wrong drug worsens the child. [9]
The maternal antibody syndromes affect the baby and are distinguished by whether the antibodies are against the adult or the fetal acetylcholine receptor. Transient neonatal myasthenia occurs in around ten to fifteen percent of babies of myasthenia gravis mothers, is caused by transplacental acetylcholine receptor antibodies, presents with hypotonia, weak suck, and respiratory difficulty in the first days, and resolves over weeks as the antibody clears, with pyridostigmine and supportive care. Fetal acetylcholine receptor inactivation syndrome is caused by maternal antibodies against the fetal gamma-subunit acetylcholine receptor, which the mother lacks, and it produces arthrogryposis and a fixed permanent myopathy that does not resolve, so a baby of a myasthenia gravis mother with arthrogryposis and fixed weakness has this syndrome and not transient neonatal myasthenia. [10]
Complications & Pitfalls
The life-threatening complications are the myasthenic crisis and the prolonged intensive care course. A myasthenic crisis is an exacerbation severe enough to need intubation, and it is driven by ventilatory failure from respiratory muscle weakness and by aspiration from bulbar weakness. Around fifteen to twenty percent of children with myasthenia gravis will have a crisis at some point, and it is commonest in the first year after diagnosis, in muscle-specific kinase antibody disease, and after a precipitant. Autonomic instability and the intensive care complications of ventilator-associated pneumonia and thrombosis add to the burden. [7]
The classic pitfalls are the ones examiners reward a candidate for naming. The first is failing to monitor bedside spirometry and missing the falling forced vital capacity. The second is starting corticosteroids at a high dose and precipitating a crisis through the transient steroid-induced worsening. The third is treating a congenital myasthenic syndrome, especially Dok7, ColQ, or the slow-channel syndrome, with pyridostigmine and worsening the child. The fourth is mistaking a seronegative myasthenia for a functional disorder because the antibody is negative. The fifth is missing a thymoma on chest imaging. The sixth is forgetting the associated thyroid disease and the live-vaccine contraindication on immunosuppression. [2][9]
The long-term complications dominate the lived burden of the disease and the treatment. The chronic relapsing course, the visible ptosis and the fatigability, and the psychological burden of a fluctuating and unpredictable illness can limit school, sport, and social life. The corticosteroid side effects of growth suppression, weight gain, osteopenia, and glucose intolerance demand monitoring and steroid-sparing. The immunosuppression carries the infection and the live-vaccine risks. A child who looks well between relapses may still fatigue quickly, so a structured activity plan and school support are part of treatment, not an afterthought. [2]
Prognosis & Disposition
Children with autoimmune myasthenia gravis have a better prognosis than adults when the disease is well managed, and many achieve remission. With modern symptomatic therapy and immunotherapy, most children gain good functional control, ocular-only disease often remits, and a substantial proportion achieve pharmacological remission on low-dose immunotherapy. The muscle-specific kinase antibody disease and the seronegative disease are more refractory and demand closer follow-up and more aggressive immunotherapy, including rituximab. The congenital myasthenic syndromes run a lifelong course determined by the subtype, with a good outlook for the responsive forms and a guarded outlook for the refractory forms. [8][9]
The predictors of a poorer outcome are the muscle-specific kinase antibody subtype, the early generalised onset, a crisis in the first year, and a poor early response to immunotherapy. Around fifteen to twenty percent of children will have a crisis, and the intensive care course is dominated by weaning ventilation and managing the precipitant. The MGFA classification, derived from the Jaretzki research standards, tiers the severity and the post-intervention status and is used to follow the response to treatment. [3][7]
disease trajectory
Maintenance: low-dose immunotherapy, monitor bone and growth, school support
Disposition follows the severity and the respiratory status. Any child with a falling forced vital capacity, bulbar weakness, or a crisis goes to intensive care. A treated child with stable physiology and a clear precipitant is managed on the ward with paediatric neurology input and continued spirometric monitoring. As the disease stabilises, the child moves to the outpatient setting with paediatric neurology follow-up, a written school and crisis plan, and a structured transition to adult care in adolescence. The congenital myasthenic syndromes demand lifelong genetic and neurodisability follow-up. [2]
Special Populations
The neonate of a myasthenia gravis mother needs assessment at birth, because around ten to fifteen percent develop transient neonatal myasthenia from the transplacental antibodies. The presentation is hypotonia, a weak suck, a weak cry, and respiratory difficulty in the first two to three days, and the management is supportive with pyridostigmine and tube feeding until the antibody clears over weeks. A baby with arthrogryposis or fixed weakness, in contrast, has fetal acetylcholine receptor inactivation syndrome from maternal antibodies against the fetal gamma-subunit, which is permanent, and the mother should be counselled and treated in a future pregnancy to prevent recurrence. [10]
The adolescent girl with juvenile myasthenia gravis faces the dual burden of a chronic disease and the developmental tasks of adolescence. Adherence to a complex immunotherapy regimen, the cosmetic effects of the corticosteroids, and the reproductive implications of the immunosuppression all bear on the management, and a structured transition to adult care is essential. The Aboriginal and Torres Strait Islander child and the child from a remote setting may present late after prolonged fatigable weakness, because of distance and access, so retrieval pathways and early specialist input are central. The child from a refugee or migrant family needs an early interpreter and a broad workup that considers the congenital syndromes and consanguinity. [8]
Evidence, Guidelines & Regional Differences
The diagnostic framework rests on the myasthenia gravis Foundation of America clinical classification and research standards of Jaretzki 2000, which codified the fatigable weakness and tiered the severity and the post-intervention status, and which remain the common language of the field. The comprehensive Gilhus 2016 New England Journal of Medicine review set the modern synthesis of the autoimmune disease, and the Engel 2015 Lancet Neurology review did the same for the congenital myasthenic syndromes. [3][1][9]
The treatment evidence is dominated by the MGTX trial of Wolfe 2016, the Cochrane review of intravenous immunoglobulin of Gajdos 2012, and the REGAIN trial of eculizumab of Howard 2017. The MGTX trial showed that extended transsternal thymectomy plus prednisone improved the clinical status of adults with acetylcholine receptor antibody non-thymomatous generalised disease over three years, and the benefit is extrapolated to the adolescent. The Cochrane review confirmed that intravenous immunoglobulin is as effective as plasma exchange for an exacerbation and speeds recovery. The REGAIN trial showed that eculizumab benefits refractory anti-acetylcholine receptor generalised disease, with the caveat of the meningococcal risk. [4][5][6]
MGTX — Wolfe 2016, thymectomy plus prednisone versus prednisone alone
Multicentre randomised trial of 126 adults with acetylcholine receptor antibody non-thymomatous generalised myasthenia gravis comparing extended transsternal thymectomy plus prednisone with prednisone alone over three years.
Key finding
Thymectomy plus prednisone improved the quantitative myasthenia gravis score and reduced the immunosuppression requirement, with more time in remission.
Practice change
Thymectomy is a rational option for the adolescent with generalised acetylcholine receptor antibody disease, while the muscle-specific kinase and congenital forms do not benefit.
The international consensus guidance of Narayanaswami 2021, the 2020 update of the Sanders 2016 executive summary, translates the evidence into practice and endorses pyridostigmine as symptomatic therapy, a cautious corticosteroid start with early steroid-sparing, intravenous immunoglobulin or plasma exchange for crisis, thymectomy for thymoma and selected non-thymomatous disease, and eculizumab and rituximab for refractory disease. The Alshekhlee 2009 study set the modern incidence and mortality of the myasthenic crisis in the United States. [2][7]
Regional differences are small in principle because the framework is international, but access matters in practice. Rapid antibody testing, specialist neurophysiology, intravenous immunoglobulin, and paediatric intensive care are available promptly in tertiary centres in ANZ, the UK, and North America, but retrieval from remote settings can stretch to days, reinforcing the rule to monitor the forced vital capacity and to treat on suspicion. The threshold to treat and the choice of the first-line agent are broadly consistent across the RACP, RCPCH, ABP, and RCPSC contexts, with the rituximab and eculizumab access varying by funding and the national immunisation guidance. [2]
Exam Pearls
Myasthenia gravis is an antibody-mediated attack on the postsynaptic membrane of the neuromuscular junction, and its signature is fatigable, fluctuating weakness that worsens with sustained activity and recovers with rest. The ocular, bulbar, and proximal muscles are involved, and the sensation, the deep tendon reflexes, and the pupils are preserved. The acetylcholine receptor antibody is positive in around eighty percent of generalised disease, and the muscle-specific kinase antibody is the second-line test in the seronegative case. [1]
First-line symptomatic therapy is pyridostigmine around one milligram per kilogram per dose four to five times daily. Corticosteroids are started low and titrated upward, because a high starting dose can transiently worsen the disease and precipitate crisis, and a steroid-sparing agent is added early. Intravenous immunoglobulin 2 g per kg over two to five days or plasma exchange treats a crisis, and the two are equally effective. Intensive care is indicated for a forced vital capacity under twenty millilitres per kilogram, and intubation is for a value under fifteen millilitres per kilogram or a negative inspiratory force under negative forty centimetres of water. [2][5][7]
The congenital myasthenic syndromes are genetic and not antibody-mediated, and their treatment is subtype-specific. Pyridostigmine helps acetylcholine receptor deficiency and RAPSN syndrome, but it worsens Dok7, ColQ endplate acetylcholinesterase deficiency, and the slow-channel syndrome, which are treated with ephedrine, salbutamol, quinidine, or fluoxetine. The message is to confirm the gene before treating. The maternal antibody syndromes distinguish transient neonatal myasthenia, which resolves over weeks, from fetal acetylcholine receptor inactivation syndrome, which causes arthrogryposis and a permanent myopathy. [9][10]
Thymectomy is mandatory for a thymoma and is an option for the adolescent with generalised acetylcholine receptor antibody disease after the MGTX trial, but it is not helpful in muscle-specific kinase disease or in the congenital syndromes. Eculizumab, a terminal complement inhibitor, is reserved for refractory anti-acetylcholine receptor generalised disease after the REGAIN trial, and it requires meningococcal vaccination. Lambert-Eaton myasthenic syndrome is a presynaptic disorder with facilitation and a paraneoplastic association that is treated with amifampridine. The two rules that save children are to monitor the forced vital capacity and to confirm the subtype before treating with pyridostigmine. [4][6][12]
References
- [1]Gilhus NE Myasthenia Gravis. N Engl J Med, 2016.PMID 28029925
- [2]Narayanaswami P, Sanders DB, Wolfe G, et al International Consensus Guidance for Management of Myasthenia Gravis: 2020 Update. Neurology, 2021.PMID 33144515
- [3]Jaretzki A 3rd, Barohn RJ, Ernstoff RM, et al Myasthenia gravis: recommendations for clinical research standards. Task Force of the Medical Scientific Advisory Board of the Myasthenia Gravis Foundation of America. Neurology, 2000.PMID 10891897
- [4]Wolfe GI, Kaminski HJ, Aban IB, et al Randomized Trial of Thymectomy in Myasthenia Gravis. N Engl J Med, 2016.PMID 27509100
- [5]Gajdos P, Chevret S, Toyka KV Intravenous immunoglobulin for myasthenia gravis. Cochrane Database Syst Rev, 2012.PMID 23235588
- [6]Howard JF Jr, Utsugisawa K, Benatar M, et al Safety and efficacy of eculizumab in anti-acetylcholine receptor antibody-positive refractory generalised myasthenia gravis (REGAIN): a phase 3, randomised, double-blind, placebo-controlled, multicentre study. Lancet Neurol, 2017.PMID 29066163
- [7]Alshekhlee A, Miles JD, Katirji B, et al Incidence and mortality rates of myasthenia gravis and myasthenic crisis in US hospitals. Neurology, 2009.PMID 19414721
- [8]Parr JR, Andrew MJ, Finnis M, et al How common is childhood myasthenia? The UK incidence and prevalence of autoimmune and congenital myasthenia. Arch Dis Child, 2014.PMID 24500997
- [9]Engel AG, Shen XM, Selcen D, Sine SM Congenital myasthenic syndromes: pathogenesis, diagnosis, and treatment. Lancet Neurol, 2015.PMID 25792100
- [10]Hacohen Y, Jacobson LW, Byrne S, et al Fetal acetylcholine receptor inactivation syndrome: A myopathy due to maternal antibodies. Neurol Neuroimmunol Neuroinflamm, 2015.PMID 25566546
- [11]Guptill JT, Sanders DB Update on muscle-specific tyrosine kinase antibody positive myasthenia gravis. Curr Opin Neurol, 2010.PMID 20613516
- [12]Titulaer MJ, Lang B, Verschuuren JJ Lambert-Eaton myasthenic syndrome: from clinical characteristics to therapeutic strategies. Lancet Neurol, 2011.PMID 22094130