Paeds · neurology-neurodisability-and-neuromuscular
Neurodegenerative and leukodystrophy disorders
Also known as Leukodystrophies · Inherited white-matter disorders · Genetic leukoencephalopathies · Dysmyelinating disorders · Metabolic neurodegeneration
A fellowship approach to the child with an inherited neurodegenerative white-matter disorder. Read the brain MRI pattern - the symmetric confluent change of metachromatic leukodystrophy, the contrast-enhancing parieto-occipital lesion of cerebral X-linked adrenoleukodystrophy, the tigrid stripes, the diffuse hypomyelination of Pelizaeus-Merzbacher, the vanishing white matter of eIF2B disease, the anterior-temporal cysts of megalencephalic leukoencephalopathy, and the calcification and atrophy of Aicardi-Goutieres - to generate a mechanism-based differential across the lysosomal, peroxisomal, hypomyelinating and astrocytopathic groups, and to drive a tiered workup anchored on MRI, biochemistry and genomic sequencing that identifies the disorders whose only therapy window closes before symptoms begin - the cerebral adrenoleukodystrophy that needs urgent transplant and the presymptomatic metachromatic leukodystrophy and Krabbe disease that newborn screening now lets us treat.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
A three-year-old whose gait has stiffened and whose speech has thinned brings a scan showing symmetric white-matter change, and a six-year-old boy sent for behaviour problems brings a scan with a glowing splenium. In both rooms the question is the same: which leukodystrophy is this, and is there a therapy whose window is already closing? The fellowship task is to convert the magnetic-resonance-imaging pattern into a mechanism-based diagnosis and a time-aware plan that does not forfeit a treatable disorder to a generic degenerative label. [1] [3]
L · E · U · K · O
Overview & Definition
The leukodystrophies are a group of inherited disorders in which a gene defect injures the white matter of the central nervous system - the myelinated axons and their supporting cells - producing progressive neurological dysfunction. They differ from the acquired demyelinating disorders (such as acute disseminated encephalomyelitis and multiple sclerosis) in being genetic, and they differ from the storage disorders that primarily engorge grey-matter neurons in being selective for white matter. The clinical thread that unites them is loss of previously acquired motor and cognitive skills - developmental regression driven by progressive white-matter injury. [1]
Why does white matter deserve its own diagnostic framework? Because the brain's myelinated tracts are built and maintained by specific cell types - oligodendrocytes that make myelin, lysosomes that turn it over, peroxisomes that handle very-long-chain fatty acids, and astrocytes that control the white-matter environment - and each leukodystrophy corresponds to a failure in one of these mechanisms. That correspondence is what lets a single magnetic-resonance-imaging pattern point to a mechanism, a gene, and a therapy. A myelin sheath that is made wrong, broken down wrong, inflamed, never formed, or surrounded by a sick astrocyte each produces a recognisable scan, and the scan is the entry point to the whole field. [1] [3]
What makes this group worth knowing in depth is the same principle that governs the wider field of neurodegeneration: a growing and consequential subset is treatable, and treatment works best - or only - when it is begun before the brain injury becomes irreversible. Haematopoietic stem cell transplant halts the inflammation of early cerebral adrenoleukodystrophy; gene therapy stabilises presymptomatic metachromatic leukodystrophy; and transplant can modify early Krabbe disease. None of these works once the injury is advanced, which is why the speed of recognition and the presymptomatic detection offered by newborn screening are the difference between a halted disease and a fatal one. [4] [9]
Classification
The leukodystrophies are best grouped by the mechanism that injures the white matter, because the mechanism predicts the magnetic-resonance-imaging pattern, the gene, the biochemistry, and whether a disease-modifying therapy exists. The figure below lays out the four mechanism groups - lysosomal and dysmyelinating, peroxisomal and inflammatory, hypomyelinating, and astrocytopathic and interferonopathic - alongside a lower band marking the disorders now detectable by newborn screening, because presymptomatic detection is what makes the treatable subset treatable. [1] [3]
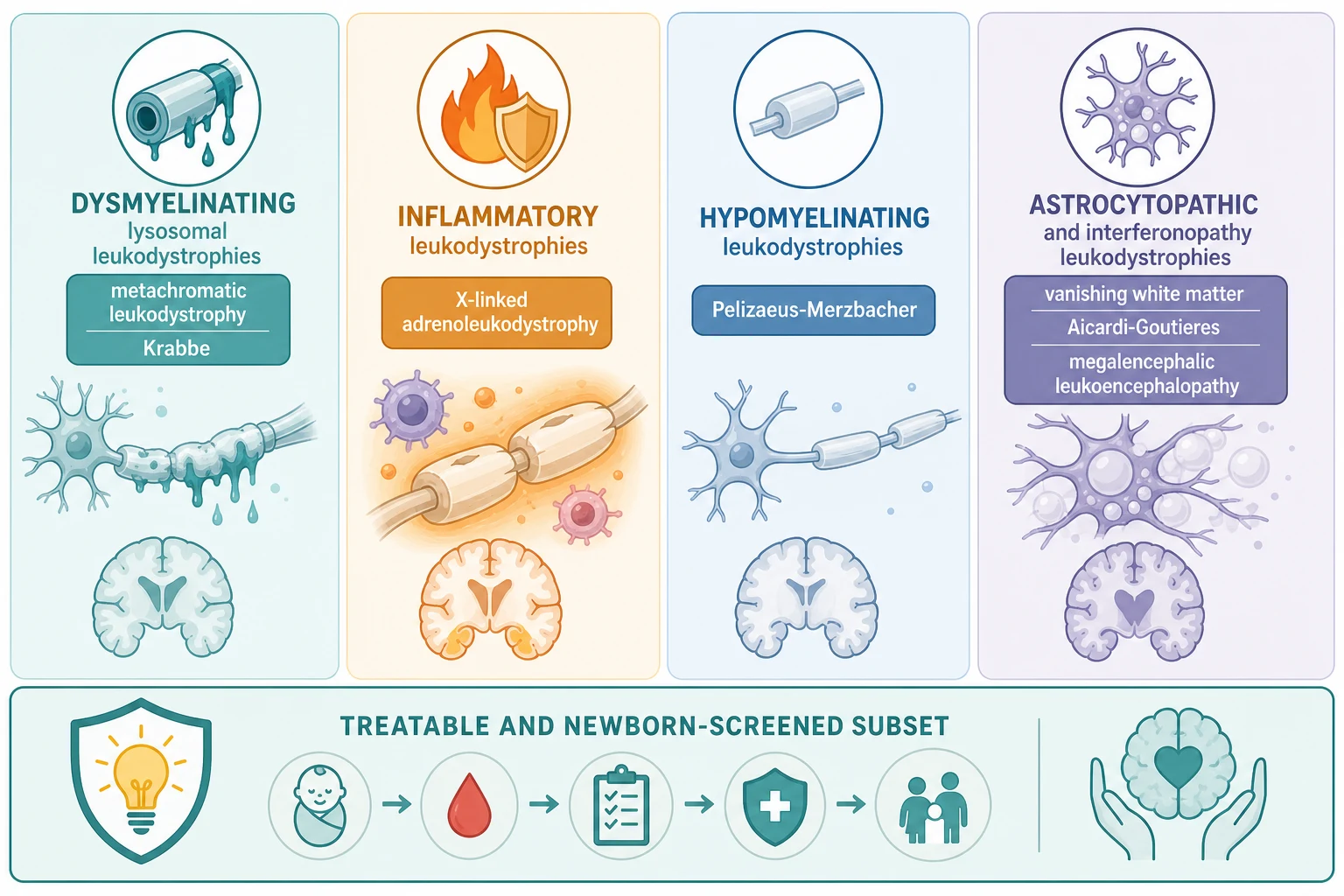
The four groups behave differently at the bedside and on the scan. Lysosomal and dysmyelinating disorders - metachromatic leukodystrophy and Krabbe (globoid cell) disease - are caused by deficiency of a lysosomal enzyme that breaks down myelin lipids, so formed myelin is destroyed and symmetric confluent white-matter change appears. Peroxisomal and inflammatory disease - X-linked adrenoleukodystrophy - is caused by failure to handle very-long-chain fatty acids, which triggers an inflammatory demyelination that lights up with contrast and progresses rapidly. Hypomyelinating disorders - Pelizaeus-Merzbacher and the 4H and HBSL group - fail to form myelin in the first place, so the white matter stays near cerebrospinal-fluid intensity throughout life. Astrocytopathic and interferonopathic disorders - vanishing white matter, Alexander, megalencephalic leukoencephalopathy with subcortical cysts, and Aicardi-Goutieres - injure white matter through a sick astrocyte or a runaway interferon response, producing rarefaction, cavitation, macrocephaly, or calcification. [1] [3]
The lower band of the classification is where the therapeutic revolution lives, and it is the band the general paediatrician must understand. Newborn screening for X-linked adrenoleukodystrophy (the ABCD1 gene product) and, in some jurisdictions, for Krabbe disease (galactocerebrosidase, with psychosine as the second-tier marker) lets clinicians identify affected boys and infants before symptoms begin, because haematopoietic stem cell transplant for cerebral adrenoleukodystrophy and gene therapy for presymptomatic metachromatic leukodystrophy work only in that presymptomatic window. Holding this band in mind prevents the most consequential error in the field: labelling a treatable inflammatory leukodystrophy as an untreatable degenerative disorder while the transplant window closes. [4] [5]
Mechanism groups at a glance - pattern, gene, treatment lever
- Lysosomal and dysmyelinating (metachromatic leukodystrophy, Krabbe): enzyme deficiency destroys formed myelin. Key test: arylsulfatase A and urinary sulfatides (MLD); galactocerebrosidase and psychosine (Krabbe). Treatment: gene therapy or transplant for presymptomatic MLD; transplant for early presymptomatic Krabbe.
- Peroxisomal and inflammatory (X-linked adrenoleukodystrophy): very-long-chain fatty acid accumulation drives inflammatory demyelination. Key test: plasma very-long-chain fatty acids, contrasted brain MRI, Loes score. Treatment: allogeneic haematopoietic stem cell transplant for early active cerebral disease.
- Hypomyelinating (Pelizaeus-Merzbacher, 4H, HBSL): myelin never forms. Key test: brain MRI showing persistent near-fluid white-matter signal, PLP1 testing. Treatment: supportive and rehabilitative; no disease-modifying therapy.
- Astrocytopathic and interferonopathic (vanishing white matter, Alexander, megalencephalic leukoencephalopathy, Aicardi-Goutieres): sick astrocyte or interferon response injures white matter. Key test: brain MRI (rarefaction, cysts, calcification), CSF interferon and neopterin, genomic sequencing. Treatment: avoid triggers (VWM); JAK inhibitors for Aicardi-Goutieres; supportive otherwise. [1] [10]
Epidemiology & Risk Factors
Each leukodystrophy is individually rare, but together they are a meaningful cause of progressive neurological disease in childhood, and the probability that a child with unexplained white-matter change and regression has a definable cause is high enough to justify a systematic workup in every case. The combined incidence of the leukodystrophies is in the order of one in several thousand live births, with X-linked adrenoleukodystrophy, metachromatic leukodystrophy, and Krabbe disease among the commonest individually identifiable disorders. Newborn-screening programmes are now refining these estimates by identifying affected children before symptoms, which is itself changing the epidemiology from a fatal-diagnosis registry to a presymptomatic-intervention cohort. [3] [5]
The strongest risk factors for the leukodystrophies are genetic. Most are autosomal recessive, so consanguinity, a previously affected sibling, and a family history of unexplained childhood death or developmental failure raise the probability and should lower the threshold for biochemical and genomic testing. Sex-linked patterns reshape the counselling: X-linked adrenoleukodystrophy affects boys through the ABCD1 gene, and Pelizaeus-Merzbacher disease affects boys through PLP1, while a handful of disorders - LMNB1-related autosomal dominant leukodystrophy and some Aicardi-Goutieres genotypes - are dominantly inherited and produce adult- or later-onset disease that crosses into adult neurology. Founder effects and carrier frequencies matter too: Krabbe and metachromatic leukodystrophy are commoner in specific populations, and Canavan disease has a higher carrier frequency in Ashkenazi Jewish populations, so a careful three-generation pedigree is part of the workup. [3] [11]
The inflammatory leukodystrophies carry an epidemiology that changes the counselling. Cerebral X-linked adrenoleukodystrophy declares itself in roughly a third to forty percent of affected boys, characteristically between the ages of four and ten years, and once the inflammatory phase begins the progression is rapid. Adrenal insufficiency accompanies the disease in a substantial fraction, sometimes as the first manifestation, which is why an adrenal crisis or an Addisonian presentation in a boy can be the entry point to an ABCD1 diagnosis. Adrenomyeloneuropathy, the adult spinal-cord form, affects virtually all affected men over decades, and about half of female carriers develop a milder myelopathy in adulthood. [4]
Pathophysiology
A single upstream gene defect becomes a whole-white-matter disease because the central nervous system depends on specific cell types to build, maintain, and protect myelin, and each leukodystrophy corresponds to the failure of one of them. The cascade runs from a gene variant to a discrete mechanism of white-matter injury - enzyme failure and substrate accumulation, very-long-chain fatty acid overload and inflammation, failed myelin formation, or a sick astrocyte and a runaway interferon response - and that mechanism is what defines the imaging pattern and the clinical tempo. The mechanism, not the gene name, is what the clinician reads on the scan. [1] [3]
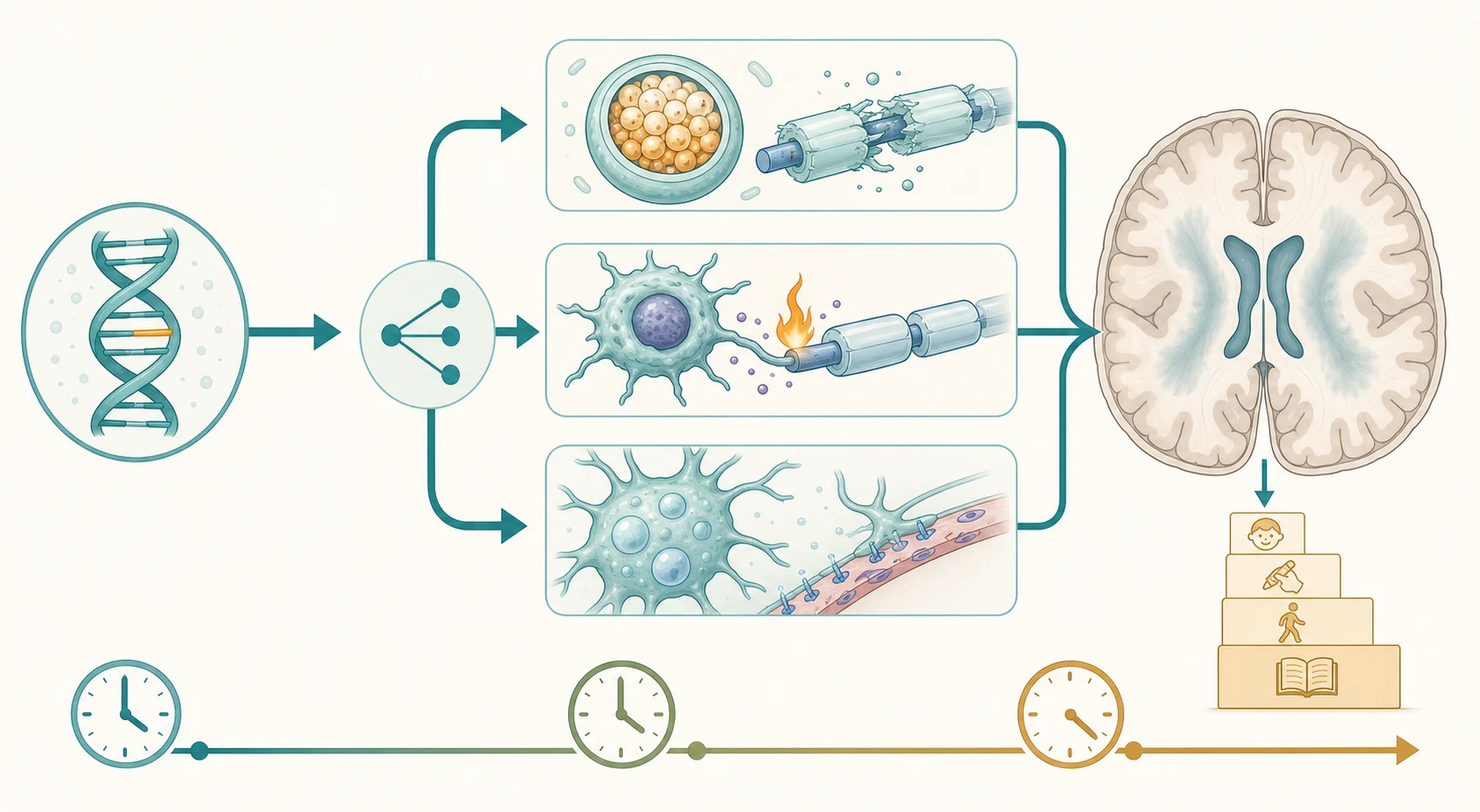
The mechanism of injury differs by group and explains both the scan and the bedside picture. In metachromatic leukodystrophy, deficiency of arylsulfatase A (or its activator saposin B) blocks the breakdown of sulfatides, which accumulate in oligodendrocytes and trigger demyelination; the late-infantile form is aggressive because the residual enzyme activity is near zero. In Krabbe disease, deficiency of galactocerebrosidase allows the toxic lipid psychosine to build up in oligodendrocytes, killing them and producing a rapidly progressive demyelination - and it is psychosine, measurable as a second-tier newborn-screen marker, that predicts which screen-positive infants will develop the disease. In X-linked adrenoleukodystrophy, defective ABCD1 prevents very-long-chain fatty acid transport into peroxisomes; accumulation triggers a CD8 T-cell-mediated inflammatory demyelination whose leading edge enhances vividly with gadolinium, which is why the contrasted scan is the transplant decision. [4] [12]
The hypomyelinating and astrocytopathic mechanisms explain the scans that look different from the confluent demyelination of the lysosomal group. In Pelizaeus-Merzbacher disease, a PLP1 mutation (most often a duplication) produces abnormal proteolipid protein that disrupts oligodendrocyte function, so myelin is never properly laid down and the white matter retains a near-cerebrospinal-fluid signal for life. In vanishing white matter disease, mutations in the eukaryotic initiation factor 2B genes make astrocytes fragile; stress - fever, infection, minor head trauma, even fright - tips them into a failed stress response, the white matter rarefies and cavitates, and the child deteriorates in steps triggered by illness. In Aicardi-Goutieres syndrome, a defect in nucleic-acid handling (TREX1, RNASEH2, SAMHD1, ADAR1, IFIH1 among others) drives a chronic type I interferon response that mimics congenital infection with basal-ganglia calcification, white-matter change, and cerebral atrophy. [1] [10]
Tempo - the dimension that triages transplant urgency
The tempo is itself diagnostic and triages urgency. Acute or subacute deterioration in a boy with white-matter change demands a contrasted magnetic resonance imaging scan the same day, because the cerebral adrenoleukodystrophy inflammatory phase is the one leukodystrophy in which delay is measured in lost brain. Stepwise deterioration provoked by fever or minor head trauma points to vanishing white matter, where the trigger is avoidable even though the disease is not yet curable. Insidious, steady progression over months to years points to the lysosomal and hypomyelinating disorders, where the workup is deliberate but the same governing principle applies: name the disorder before a therapy window closes. [4] [9]
Clinical Presentation
The presenting complaint is usually progressive loss of motor and cognitive skills, but the accompanying features and the age at onset are what point to the group. The first pattern is the infant or young child with a lysosomal leukodystrophy. Late-infantile metachromatic leukodystrophy presents between six months and four years with loss of motor milestones, a progressive spastic or ataxic gait, hypotonia giving way to hypertonia, and cognitive and speech decline, often with optic atrophy; the juvenile and adult forms present later with declining school performance, behaviour change, and a slower spastic or psychiatric course. Infantile Krabbe disease presents in the first six months with irritability, feeding difficulty, unexplained low-grade fever, hypertonia, seizures, and optic atrophy, and the course is rapid. [1] [3]
The second pattern is the school-age boy with cerebral X-linked adrenoleukodystrophy, and this is the presentation that must never be missed. A boy aged four to ten develops behavioural change, declining school performance, inattention, then visual and auditory processing difficulty, gait disturbance, and new seizures, with adrenal insufficiency in a substantial fraction. The bedside picture of a previously well boy with new behavioural or visual decline and skin pigmentation or a known adrenal diagnosis is the entry point to a contrasted brain scan, and the scan - a symmetric, contrast-enhancing splenium-based parieto-occipital white-matter lesion - is the transplant decision. The tempo from first symptom to severe disability is often only months once the inflammatory phase is established. [4] [6]
The third pattern is the hypomyelinating and astrocytopathic group, declared by the scan as much as by the bedside. Pelizaeus-Merzbacher presents in male infants with nystagmus, hypotonia, and developmental delay, evolving into a severe spastic and ataxic disorder with a diffusely unmyelinated brain. Megalencephalic leukoencephalopathy with subcortical cysts presents with macrocephaly in the first year, seizures, and a slow motor and cognitive decline, with anterior-temporal subcortical cysts on imaging. Alexander disease presents with megalencephaly, seizures, and bulbar dysfunction, with frontal-predominant white-matter change and a periventricular rim. Vanishing white matter presents with ataxia and spasticity that deteriorate in steps after fever or minor head trauma. Aicardi-Goutieres presents in early infancy with irritability, feeding difficulty, a severe encephalopathy, chilblain-like skin lesions, and intracranial calcification that mimics congenital infection. [1] [10]
Differential Diagnosis
The differential of progressive white-matter disease is broad, and several treatable non-leukodystrophic causes must be actively excluded because each changes the trajectory if missed. The acquired demyelinating disorders - acute disseminated encephalomyelitis, multiple sclerosis, neuromyelitis optica spectrum disorder, and MOG-antibody disease - produce white-matter change and regression but are immune-mediated and steroid-responsive, and a contrasted scan with lesion distribution and a CSF oligoclonal band panel separates them. Nutritional deficiency (such as vitamin B12 in an exclusively breastfed infant of a deficient mother), chronic illness, and treatable endocrine disease can all mimic regression and are excluded by a targeted screen. [1]
Why does a normal initial screen not exclude a leukodystrophy? Because several disorders are intermittent, tissue-specific, or not on the standard panel. A single very-long-chain fatty acid measurement is usually diagnostic for X-linked adrenoleukodystrophy but female carriers can have normal values, so a carrier with a compatible phenotype needs ABCD1 sequencing. Enzyme assays can be affected by sample handling and pseudodeficiency alleles, which is why a low arylsulfatase A is confirmed by urinary sulfatides and molecular testing rather than accepted alone. When the clinical phenotype and the imaging are highly suggestive but the targeted biochemistry is unrevealing, the answer is a leukodystrophy and neurodegeneration gene panel or trio exome sequencing, not reassurance. [1] [3]
Clinical & Bedside Assessment
The single most informative bedside act is to combine the developmental trajectory with the magnetic-resonance-imaging pattern, because together they name the group before any blood test returns. Ask what the child could do at defined earlier ages - sit, walk, speak in phrases, follow a two-step instruction - and whether the loss is steady, stepwise with triggers, or acute. A trajectory that rises then falls with a progressive spastic ataxia and symmetric confluent white-matter change points to a lysosomal leukodystrophy; stepwise deterioration after fever with cavitating white matter points to vanishing white matter; and acute behavioural and visual decline in a school-age boy with a contrast-enhancing parieto-occipital lesion points to cerebral adrenoleukodystrophy. [1] [4]
Three questions frame every consultation. First, which white-matter pattern is on the scan, and is there contrast enhancement - because enhancement is the transplant decision. Second, what is the age and tempo, and is there a trigger - because a boy aged four to ten with acute decline, or an infant with stepwise post-fever deterioration, is a different urgency from a child with insidious progression. Third, is there consanguinity, a previously affected sibling, a family history of unexplained childhood death, adrenal disease in a male relative, or a known carrier status - because these reshape the pre-test probability and the counselling. [1] [3]
The examination is then directed and recorded. Measure growth and head circumference and plot them against earlier values - macrocephaly suggests megalencephalic leukoencephalopathy, Alexander, or Canavan disease. Examine the eyes for nystagmus (Pelizaeus-Merzbacher), optic atrophy (metachromatic leukodystrophy, Krabbe), a cherry-red spot (the storage disorders), and chilblain-like lesions on the extremities (Aicardi-Goutieres). Assess tone and movement for the spasticity, ataxia, dystonia, and extrapyramidal signs that localise the white-matter injury, and look for skin pigmentation or signs of adrenal insufficiency in any boy with white-matter change. The discriminating neuro-ophthalmic and adrenal findings localise the disorder before any test is sent, which is why a focused examination is part of the diagnostic act. [1]
Investigations
The investigation strategy is magnetic-resonance-imaging-led, and each tier either names the disorder or justifies the next. The first tier is a high-quality brain magnetic resonance imaging scan with contrast and, where available, magnetic resonance spectroscopy, because the pattern of white-matter change is the entry point to the whole field. Symmetric confluent change with a tigrid pattern raises a lysosomal leukodystrophy; a contrast-enhancing parieto-occipital splenium-based lesion raises cerebral adrenoleukodystrophy; diffuse persistent hypomyelination raises a hypomyelinating disorder; rarefied or cavitating white matter raises vanishing white matter; anterior-temporal subcortical cysts with macrocephaly raise megalencephalic leukoencephalopathy; and basal-ganglia calcification with atrophy raises Aicardi-Goutieres. [1] [4]
The second tier is the targeted biochemistry matched to the imaging pattern. Plasma very-long-chain fatty acids diagnose X-linked adrenoleukodystrophy and are the anchor for newborn screening; arylsulfatase A activity with urinary sulfatides diagnose metachromatic leukodystrophy; leucocyte galactocerebrosidase activity with plasma psychosine diagnose Krabbe disease; and a lysosomal enzyme panel covers the wider storage group. Magnetic resonance spectroscopy adds a metabolic dimension - a towering N-acetylaspartate peak is the signature of Canavan disease, and a lactate peak raises a mitochondrial overlap. A cerebrospinal-fluid analysis is reserved for the phenotype that demands it: a chronic lymphocytosis with raised neopterin and interferon activity points to Aicardi-Goutieres, and a low glucose with a low cerebrospinal-fluid-to-plasma ratio raises GLUT1 overlap. [1] [10]
The magnetic-resonance-imaging-led tiered workup
- Read the contrasted brain MRI - the pattern names the mechanism group; gadolinium enhancement in a parieto-occipital lesion is the transplant decision in cerebral adrenoleukodystrophy, scored with the Loes score.
- Targeted biochemistry by pattern - very-long-chain fatty acids (X-linked adrenoleukodystrophy), arylsulfatase A and urinary sulfatides (metachromatic leukodystrophy), galactocerebrosidase and psychosine (Krabbe), with magnetic resonance spectroscopy for the metabolic signature.
- Genomic confirmation - a leukodystrophy gene panel or trio exome or genome sequencing confirms the gene, distinguishes true deficiency from pseudodeficiency, and enables family counselling and prenatal testing.
- Newborn-screening linkage - for X-linked adrenoleukodystrophy and Krabbe, confirm the newborn-screen result and arrange urgent surveillance imaging, because therapy works only when given presymptomatically. [1] [6]
The third tier is genomic, and it has transformed the diagnosis of the leukodystrophies. A targeted leukodystrophy and neurodegeneration gene panel confirms the imaging-based hypothesis and distinguishes a true deficiency from a pseudodeficiency allele, which is a common source of biochemical ambiguity. When the panel and the biochemistry are unrevealing, trio exome or genome sequencing identifies a diagnostic variant in a substantial fraction of cases and often surfaces an unexpected gene, including a dominantly acting disorder such as LMNB1-related leukodystrophy that changes the counselling. An accurate molecular diagnosis brings prognosis, reproductive counselling, carrier testing, and the possibility of trial or therapy eligibility, which is why the workup is completed even when the immediate treatment options are limited. [3] [11]
Management — Resuscitation
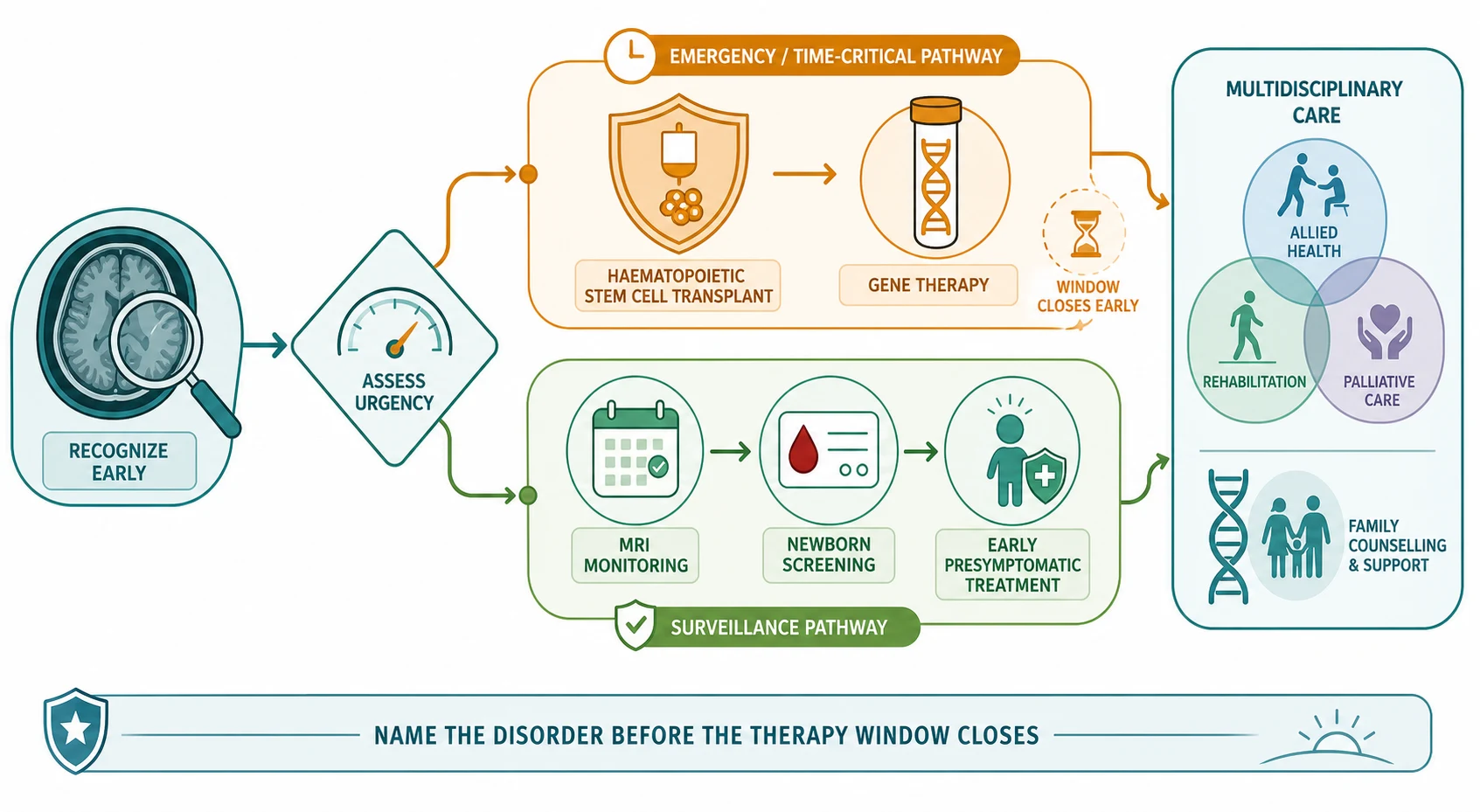
When a leukodystrophy presents as acute or subacute encephalopathy - altered consciousness, seizures, a movement disorder, or an adrenal crisis - the response is resuscitation first and the diagnostic workup in parallel. Secure the airway, breathing, and circulation; treat seizures early; correct any adrenal insufficiency with parenteral hydrocortisone, because adrenal crisis can be the first presentation of X-linked adrenoleukodystrophy and untreated Addisonian crisis is fatal; and move immediately to obtain the contrasted brain scan that decides the transplant question. [4]
The two resuscitation-pace decisions are the cerebral adrenoleukodystrophy inflammatory phase and the acute decompensation of vanishing white matter, because they demand opposite immediate acts. For suspected cerebral adrenoleukodystrophy, the urgency is to stage the disease - Loes score, gadolinium enhancement, neurological examination - and to refer urgently for haematopoietic stem cell transplant, because transplant halts the inflammation only when the burden is still low and the neurological examination still preserved. For an acute vanishing-white-matter decompensation, the urgency is to remove the trigger - treat the fever, manage the infection, avoid unnecessary stress and procedures - because the deterioration is driven by the stress response and there is, as yet, no curative therapy. [4] [10]
Status epilepticus and acute raised intracranial pressure are managed in parallel with a first-line benzodiazepine followed by a second-line antiseizure agent, and with neurocritical-care support as needed. Any child under investigation for a possible leukodystrophy or metabolic disorder needs a peri-procedure safety plan, because prolonged fasting before an imaging study or an anaesthetic can trigger a metabolic decompensation that becomes the first presentation of the disorder. These resuscitation decisions are owned by the general paediatrician in the first hours, with the specialist metabolic and neurology service taking over as the diagnosis is confirmed. [2] [9]
Management — Definitive & Stepwise
Definitive management means naming the disorder and matching it to the disease-modifying or syndrome-specific therapy that fits its mechanism. For early active cerebral adrenoleukodystrophy, the lever is allogeneic haematopoietic stem cell transplant, which halts the inflammatory demyelination by replacing the microglial lineage with donor cells; it works when the Loes score is still low and the neurological examination preserved, and it carries a real mortality and morbidity that the family must weigh. Lentiviral gene therapy is a developing option for cerebral adrenoleukodystrophy, offering an autologous alternative that avoids donor availability and graft-versus-host disease but carries a recognised risk of haematological malignancy. [4] [6]
For metachromatic leukodystrophy, the lever is lentiviral haematopoietic stem cell gene therapy, which delivers a functional ARSA gene to the child's own haematopoietic stem cells. The seminal trials showed that gene therapy given presymptomatically - or very early in the late-infantile and early-juvenile disease - can halt or slow the demyelination and preserve cognition and function, whereas the same therapy given after symptoms are established has limited benefit. This is the biological basis for the urgency of presymptomatic detection: a child identified by newborn screening or by an affected sibling can receive gene therapy before the demyelination begins, and that window is measured in months. Allogeneic transplant retains a role for juvenile and adult metachromatic leukodystrophy identified early. [7] [8]
For Krabbe disease, the lever is haematopoietic stem cell transplant for the presymptomatic infant identified by newborn screening, where it can modify the course when performed in the neonatal period before symptoms; once the infantile disease is symptomatic the outcome is poor. For the astrocytopathic and interferonopathic group, disease-modifying therapy is limited and the emphasis is on trigger avoidance for vanishing white matter, emerging JAK-inhibitor treatment for Aicardi-Goutieres aimed at suppressing the interferon response, and supportive and rehabilitative care across the group. Symptom management is itself a major treatment stream - antiseizure medication, spasticity and dystonia management, feeding and nutrition support, and management of adrenal insufficiency with regular hydrocortisone replacement in X-linked adrenoleukodystrophy - and the consensus care frameworks structure it into a planned, auditable programme. [2] [9]
The care plan is multidisciplinary and lifelong, built around the specialist neurology, metabolic, and genetics service. Developmental and educational support, allied health (physiotherapy, occupational therapy, speech and language), orthopaedics and respiratory for complications, and palliative care involved early in the most severe disorders all coexist with disease-modifying treatment rather than replacing it. Once a molecular diagnosis is made, genetic counselling, carrier testing, prenatal diagnosis, and preimplantation genetic testing reshape the reproductive risk for the whole family, and this is part of management, not an afterthought. [2] [11]
Specific Subtypes & Scenarios
Metachromatic leukodystrophy is the prototypic lysosomal leukodystrophy and every candidate must know its course. The late-infantile form, the commonest and most aggressive, presents between six months and four years with loss of motor milestones, a progressive spastic or ataxic gait, optic atrophy, and cognitive and speech decline, with symmetric confluent periventricular white-matter change and a tigrid pattern on imaging. The juvenile and adult forms present later with declining school performance, behaviour change, and a slower course. Arylsulfatase A deficiency (or saposin B) with urinary sulfatide accumulation confirms the diagnosis, and the management question is the therapy window: gene therapy for presymptomatic or very early disease, transplant for early juvenile and adult disease, and supportive care once the disease is established. [9] [12]
X-linked adrenoleukodystrophy is the leukodystrophy whose timing defines the transplant decision. An ABCD1 defect in a boy leads to very-long-chain fatty acid accumulation, and roughly a third to forty percent develop the cerebral inflammatory form between four and ten years of age, presenting with behavioural change, visual and auditory processing difficulty, gait disturbance, and seizures, with adrenal insufficiency in a substantial fraction. The contrasted magnetic resonance imaging shows a symmetric, gadolinium-enhancing splenium-based parieto-occipital lesion, and the Loes score - a standardised severity scale - guides the transplant decision: allogeneic haematopoietic stem cell transplant for active early disease halts the inflammation, while advanced disease with a high Loes score and neurological deficit carries a poor outcome and a high transplant mortality. Newborn screening now identifies affected boys before symptoms, and consensus surveillance imaging detects the cerebral form at its earliest, most treatable stage. [4] [5]
Krabbe (globoid cell) disease is the lysosomal leukodystrophy defined by psychosine. Galactocerebrosidase deficiency allows the toxic lipid psychosine to accumulate and kill oligodendrocytes, and the infantile form presents in the first six months with irritability, feeding difficulty, unexplained fever, hypertonia, seizures, and optic atrophy, with a rapid course. The treatment lever is haematopoietic stem cell transplant for the presymptomatic neonate identified by newborn screening - where it can modify the course - because symptomatic infantile disease responds poorly. Psychosine as a second-tier newborn-screen marker helps distinguish the infants who will develop disease from those with a milder later-onset course, which is central to the transplant counselling. [6]
The astrocytopathic group is named by the scan. Vanishing white matter disease, caused by eukaryotic initiation factor 2B mutations, presents with ataxia and spasticity that deteriorate in steps after fever, infection, minor head trauma, or fright, with white matter that rarefies and cavitates on serial imaging; management is trigger avoidance and supportive care, with no curative therapy. Megalencephalic leukoencephalopathy with subcortical cysts presents with macrocephaly, seizures, and slow decline, with anterior-temporal subcortical cysts. Aicardi-Goutieres syndrome, a type I interferonopathy caused by defects in nucleic-acid handling, presents in early infancy with a severe encephalopathy, intracranial calcification, cerebral atrophy, and a chronic cerebrospinal-fluid lymphocytosis that mimics congenital infection; JAK inhibitors aimed at the interferon response are an emerging therapy. [1] [10]
Complications & Pitfalls
The most consequential complications are the consequences of delay: a forfeited transplant or gene-therapy window, irreversible white-matter injury, and the lost opportunity for accurate genetic counselling. Every week matters for the disorders whose therapy works only early - cerebral adrenoleukodystrophy, presymptomatic metachromatic leukodystrophy, neonatal Krabbe - and the single biggest contributor to delay is the failure to obtain a contrasted scan or to recognise the enhancing lesion. A boy whose cerebral adrenoleukodystrophy is diagnosed after the neurological examination has deteriorated has crossed the threshold beyond which transplant carries more harm than benefit, and that threshold is measured in weeks of inflammation. [4] [6]
Over-reliance on an unenhanced scan is the cardinal imaging pitfall. The leading edge of cerebral adrenoleukodystrophy enhances with gadolinium, and an unenhanced scan can look deceptively bland in the early inflammatory phase, so a child with behavioural or visual decline and white-matter change on an unenhanced scan must have the contrasted study before a degenerative label is accepted. A second pitfall is accepting a low enzyme activity as diagnostic without excluding a pseudodeficiency allele, which is common for arylsulfatase A and can mislabel a healthy child; the safeguard is urinary sulfatides and molecular confirmation. A third pitfall is underestimating the anaesthetic and fasting risk in a child with an undiagnosed leukodystrophy, where prolonged fasting or certain agents can trigger a metabolic decompensation that becomes the first presentation. [1] [2]
Prognosis & Disposition
Prognosis is determined by the specific disorder, the age and tempo of onset, the genotype, and - critically - the timing of treatment. The early-treated treatable leukodystrophies have a transformed prognosis: a boy with cerebral adrenoleukodystrophy transplanted early, with a low Loes score and a preserved neurological examination, can halt the disease and retain function; a child with metachromatic leukodystrophy given gene therapy presymptomatically can be stabilised; and a presymptomatic neonate with Krabbe disease transplanted early can be modified. The untreated or late-treated disorders - symptomatic infantile Krabbe, advanced cerebral adrenoleukodystrophy, the severe hypomyelinating disorders, and progressive vanishing white matter - have a guarded outlook measured in months to a few years, and the task shifts to symptom control, developmental support, and palliative care. [4] [7]
Disposition is structured around the specialist neurology, metabolic, and genetics service, with lifelong surveillance and a named coordinator. The general paediatrician holds the whole child, coordinates the multidisciplinary team, and owns the emergency sick-day plan, the peri-procedure metabolic safety plan, and the adrenal replacement for the boy with adrenoleukodystrophy. For boys identified by newborn screening, a structured magnetic-resonance-imaging surveillance programme - the consensus schedules recommend regular monitoring through the highest-risk childhood years - is the safeguard that detects the cerebral form at its earliest, most treatable stage. [5] [6]
The conversation about goals of care is iterative, revisited as the disease declares its trajectory, and it is held with honesty and without abandoning the search for a treatable subset. Palliative care is involved early in the most severe disorders, coexisting with disease-modifying treatment rather than replacing it: symptom control for seizures, spasticity, feeding difficulty, and respiratory compromise improves the child's quality of life and the family's capacity to cope. Even when a disorder is not yet treatable, an accurate molecular diagnosis brings prognosis, family planning, and the possibility of future trial eligibility, which is why the workup is completed even when the immediate options are limited. [2] [11]
Special Populations
The approach differs across the age span and the family structure. Neonates identified by newborn screening for X-linked adrenoleukodystrophy or Krabbe disease are the population in whom the therapeutic window is widest, and their management is presymptomatic surveillance and early therapy rather than treatment of established disease. Infants and toddlers present with the aggressive lysosomal and astrocytopathic disorders, where the tempo is fast and the imaging decision is urgent. School-age children and adolescents present with cerebral adrenoleukodystrophy, the juvenile leukodystrophies, and the adult-onset forms that cross into adult neurology, including adrenomyeloneuropathy and LMNB1-related autosomal dominant leukodystrophy. Recognising the age-typical presentation sharpens the differential and the first test. [3] [11]
Consanguineous and founder populations carry a higher burden of the autosomal recessive leukodystrophies, and the consultation reframes reproductive risk for the whole family: carrier testing, prenatal diagnosis, and preimplantation genetic testing become central to management once a molecular diagnosis is made. For Indigenous, migrant, refugee, and remote populations, equitable and culturally safe access to specialist neurology, metabolic, and genomic services is a real challenge, concentrated as those services are in tertiary centres, and newborn-screening coverage varies by jurisdiction. The principles are constant - read the contrasted scan, exclude cerebral adrenoleukodystrophy, deploy the tiered workup - but the pathway must be adapted to geography, language, and cultural safety, often with telehealth and a local coordinator. [1]
The technology-dependent and complex-chronic child, and the female carrier of an X-linked adrenoleukodystrophy allele, need a plan that adapts across the lifespan. Female carriers can develop a milder adult-onset myelopathy and need surveillance, and the family of an affected boy faces cascade testing of at-risk male relatives and carrier testing of female relatives. A structured transition to adult metabolic and neurology services, begun in early adolescence, is the safeguard for the young person who has lived with a leukodystrophy diagnosis since childhood and now faces new questions of adherence, transfer of care, and reproductive decisions. [3] [11]
Evidence, Guidelines & Regional Differences
The conceptual backbone of the field is a set of consensus frameworks that let the clinician act on the imaging before the confirmatory gene returns. The clinical approach to the leukodystrophies and genetic leukoencephalopathies of Parikh and colleagues standardised the imaging-led, mechanism-based diagnostic pathway that anchors this whole topic. The consensus care statement of Adang and colleagues structures the preventive and symptomatic care that is the daily reality for affected families, and the international adrenoleukodystrophy consensus of Engelen and colleagues, the metachromatic leukodystrophy monitoring consensus, the vanishing white matter recommendations, and the LMNB1 clinical practice guidelines together provide disorder-specific management pathways that converge across regions on the same imaging-led and presymptomatic approach. [1] [2]
The disease-modifying evidence is now mature enough to justify the urgency of diagnosis and presymptomatic detection. The seminal lentiviral gene-therapy trials in metachromatic leukodystrophy showed that gene therapy given before symptoms preserves central nervous system function, establishing the principle that presymptomatic treatment is the standard of care for affected siblings and screen-positive infants. The consensus that haematopoietic stem cell transplant halts the inflammation of early cerebral adrenoleukodystrophy, and that newborn screening with magnetic-resonance-imaging surveillance detects the cerebral form at its most treatable stage, is the evidence base for the screening programmes now expanding across jurisdictions. [4] [7]
In Australia and Aotearoa New Zealand, newborn-bloodspot screening for X-linked adrenoleukodystrophy is expanding across jurisdictions, and funded access to gene therapy, enzyme replacement, and transplant services is governed by national and state programmes that evolve over time. Specialist metabolic and neurology services are concentrated in tertiary paediatric centres, with outreach to regional and remote areas via telehealth and retrieval networks, and the contrasted magnetic resonance imaging and the Loes score are the shared decision tools between the referring general paediatrician and the transplant service. State the local screening panel and the local funded pathway rather than assuming a universal list, and involve the regional metabolic and neurology service at the earliest suspicion of an enhancing lesion or a presymptomatic screen-positive infant. [5] [9]
The strength of evidence varies across the field. The evidence for early transplant in cerebral adrenoleukodystrophy and for presymptomatic gene therapy in metachromatic leukodystrophy is mature enough to justify the urgency of diagnosis. The evidence for JAK inhibitors in Aicardi-Goutieres syndrome and for gene therapy in cerebral adrenoleukodystrophy is still maturing, and the long-term outcomes of gene therapy and the natural history of the ultra-rare leukodystrophies remain areas of active study. Uncertainty changes counselling: a family should be told what is known, what is likely, and what is genuinely unknown, so that decisions rest on real information rather than false reassurance or undue pessimism. [7] [10]
Exam Pearls
Remember the golden rule that frames every consultation: a leukodystrophy is named by its magnetic-resonance-imaging pattern, and the treatable leukodystrophies are treated before symptoms begin. The contrasted scan excludes the transplant emergency of cerebral adrenoleukodystrophy, and newborn screening plus presymptomatic gene therapy or transplant is what converts a fatal diagnosis into a halted one for metachromatic leukodystrophy and Krabbe disease. The diagnostic rate-limiting step is reading the scan early, and the Loes score is the tool that decides the transplant. [1] [4]
Self-test: a seven-year-old boy with school decline and a white-matter lesion
A previously well seven-year-old boy is referred for declining school performance, inattention, and difficulty following the ball at sport. An unenhanced brain magnetic resonance imaging shows symmetric parieto-occipital white-matter change. He has subtle increased skin pigmentation. What is the most likely diagnosis, what single addition to the scan settles the transplant question, and what is the first step? [4] [6]
Answer: This is cerebral X-linked adrenoleukodystrophy until proved otherwise. The single addition that settles the transplant question is gadolinium contrast - the enhancing leading edge of the inflammatory phase confirms active cerebral disease, and the Loes score plus the neurological examination guide the transplant decision. The first step is an urgent contrasted brain magnetic resonance imaging and paediatric neurology and metabolic referral, plasma very-long-chain fatty acids to confirm the ABCD1 defect, a synacthen test to exclude adrenal insufficiency with hydrocortisone replacement if deficient, and urgent discussion with a haematopoietic stem cell transplant service - because the window halts the disease only when the burden is still low and the examination still preserved. [4] [5]
References
- [1]Parikh S, Bernard G, Leventer RJ, van der Knaap MS, van Hove J, Pizzino A, et al. A clinical approach to the diagnosis of patients with leukodystrophies and genetic leukoencephelopathies. Mol Genet Metab, 2015.PMID 25655951
- [2]Adang LA, Sherbini O, Ball L, Brunel-Guitton C, Cooper JD, Kecskemethy N, et al. Revised consensus statement on the preventive and symptomatic care of patients with leukodystrophies. Mol Genet Metab, 2017.PMID 28863857
- [3]Kohler W, Curiel J, Vanderver A. Adulthood leukodystrophies. Nat Rev Neurol, 2018.PMID 29302065
- [4]Engelen M, van Ballegoij WJC, Mallack EJ, van Haelst MM, van Engelen CE, Vaz FM, et al. International Recommendations for the Diagnosis and Management of Patients With Adrenoleukodystrophy: A Consensus-Based Approach. Neurology, 2022.PMID 36175155
- [5]Moser AB, Seeger E, Raymond GV, Engelen M, Kuester M, Kohler W, et al. Newborn Screening for X-Linked Adrenoleukodystrophy: Past, Present, and Future. Int J Neonatal Screen, 2022.PMID 35225938
- [6]Mallack EJ, Turk BR, Yan H, Millington DS, Jordan R, Ozen A, et al. MRI surveillance of boys with X-linked adrenoleukodystrophy identified by newborn screening: Meta-analysis and consensus guidelines. J Inherit Metab Dis, 2021.PMID 33373467
- [7]Sessa M, Lorioli L, Fumagalli F, Benedicenti F, De Amici M, Galimberti S, et al. Lentiviral haemopoietic stem-cell gene therapy in early-onset metachromatic leukodystrophy: an ad-hoc analysis of a non-randomised, open-label, phase 1/2 trial and an extension study. Lancet, 2016.PMID 27289174
- [8]Fumagalli F, Calbi V, Natali Sora MG, Consiglieri G, Canale S, Sessa M, et al. Lentiviral haematopoietic stem-cell gene therapy for early-onset metachromatic leukodystrophy: long-term results from a non-randomise, open-label, phase 1/2 trial and expanded access. Lancet, 2022.PMID 35065785
- [9]Adang LA, Bonkowsky JL, Boelens JJ, Bley AE, Braverman N, Bhatt A, et al. Consensus guidelines for the monitoring and management of metachromatic leukodystrophy in the United States. Cytotherapy, 2024.PMID 38613540
- [10]van Voorst RJ, Schoenmakers DH, Bonkowsky JL, Dietrich A, Fattal-Valevski A, Kaczmarek E, et al. Consensus-Based Expert Recommendations for Diagnosis and Clinical Management of Vanishing White Matter. Neurology, 2025.PMID 41232062
- [11]Dhamija R, Tobin WO, Cortelli P, Filla A, Klebe S, Koelliger C, et al. Clinical Practice Guidelines for the Diagnosis, Management, and Surveillance of LMNB1-Related Autosomal Dominant Leukodystrophy. Neurol Genet, 2025.PMID 40933505
- [12]Biffi A, Montini E, Lorioli L, Cesani M, Fumagalli F, Plati T, et al. Lentiviral hematopoietic stem cell gene therapy benefits metachromatic leukodystrophy. Science, 2013.PMID 23845948