Paeds · neurology-neurodisability-and-neuromuscular
Neurogenetic conditions and precision diagnosis
Also known as Genomic diagnosis of paediatric neurological disease · Precision neurogenomics · Tiered genomic testing in paediatric neurology · Ending the diagnostic odyssey · Phenotype-driven and genome-first diagnosis
Fellowship guide to precision diagnosis in paediatric neurogenetic disease. Covers the diagnostic odyssey and why tiered genomic testing ends it, the test ladder from chromosomal microarray through trio exome and genome sequencing with their diagnostic yields around fifteen to forty per cent, rapid whole-genome sequencing in the acutely ill infant that returns an answer in days, phenotype-driven deep phenotyping against genome-first reverse phenotyping, the American College of Medical Genetics five-tier variant classification framework and the management of the variant of uncertain significance, the inheritance patterns with de novo dominant change dominant in sporadic severe neurodevelopmental disease, and the treatable conditions unlocked by a molecular diagnosis including glucose transporter one deficiency and the ketogenic diet, creatine transporter deficiency and creatine, pyridoxine-dependent epilepsy, dopa-responsive dystonia, biotinidase deficiency, Wilson disease, and the gene-modifying therapies for spinal muscular atrophy and Duchenne muscular dystrophy, with genetic counselling, periodic reanalysis, and regional practice.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
A four-year-old with global developmental delay, a movement disorder, and three years of normal blood tests, normal brain imaging, and a string of single-gene tests finally receives a chromosomal microarray and a trio exome. The exome finds a pathogenic variant in SLC2A1, the diagnosis is glucose transporter one deficiency, and within weeks the child is on a ketogenic diet and the seizures and movement disorder improve. The single most important shift in paediatric neurology over the last decade is the move from serial single-gene testing to tiered genomic testing that ends this diagnostic odyssey early. A confirmed molecular diagnosis changes everything: it stops the testing, it unlocks therapy, it counsels the family on recurrence, and it links them to a community and to trials. [3][9][11]
Overview & Definition
Precision diagnosis in paediatric neurogenetic disease is the pairing of detailed clinical phenotyping with tiered genomic testing to convert a long diagnostic odyssey into a molecular answer that changes management. The target is the child with an unexplained neurodevelopmental disorder, regression, early-onset or drug-resistant epilepsy, hypotonia, a movement disorder, or a combination of these with congenital anomalies, in whom a single underlying gene is suspected but unknown. The toolset is chromosomal microarray for copy-number variants, exome sequencing for single-nucleotide and small insertion-deletion variants in the coding genome, and genome sequencing for the whole sequence including non-coding and structural change. [3][4]
The framing shift is that exome and genome sequencing are now first-tier tests for neurodevelopmental disorders rather than a last resort after years of negative investigations. A meta-analysis of more than thirty thousand individuals found a pooled diagnostic yield of around thirty-six per cent for exome in neurodevelopmental disorders, and the yield rises when the test is performed as a trio and when the result is periodically reanalysed as new gene-disease associations are discovered. The diagnosis is valuable in its own right, but its real worth is that it directs precision therapy for the treatable conditions and ends further invasive testing for everyone else. [3][5]
The clinical habit to carry is to order the genomic test early, to test the child and both parents together so that de novo variants are captured, to interpret every variant through the standard framework rather than by intuition, and to return to the data for reanalysis rather than accepting a negative result as final. [4][1]
Classification
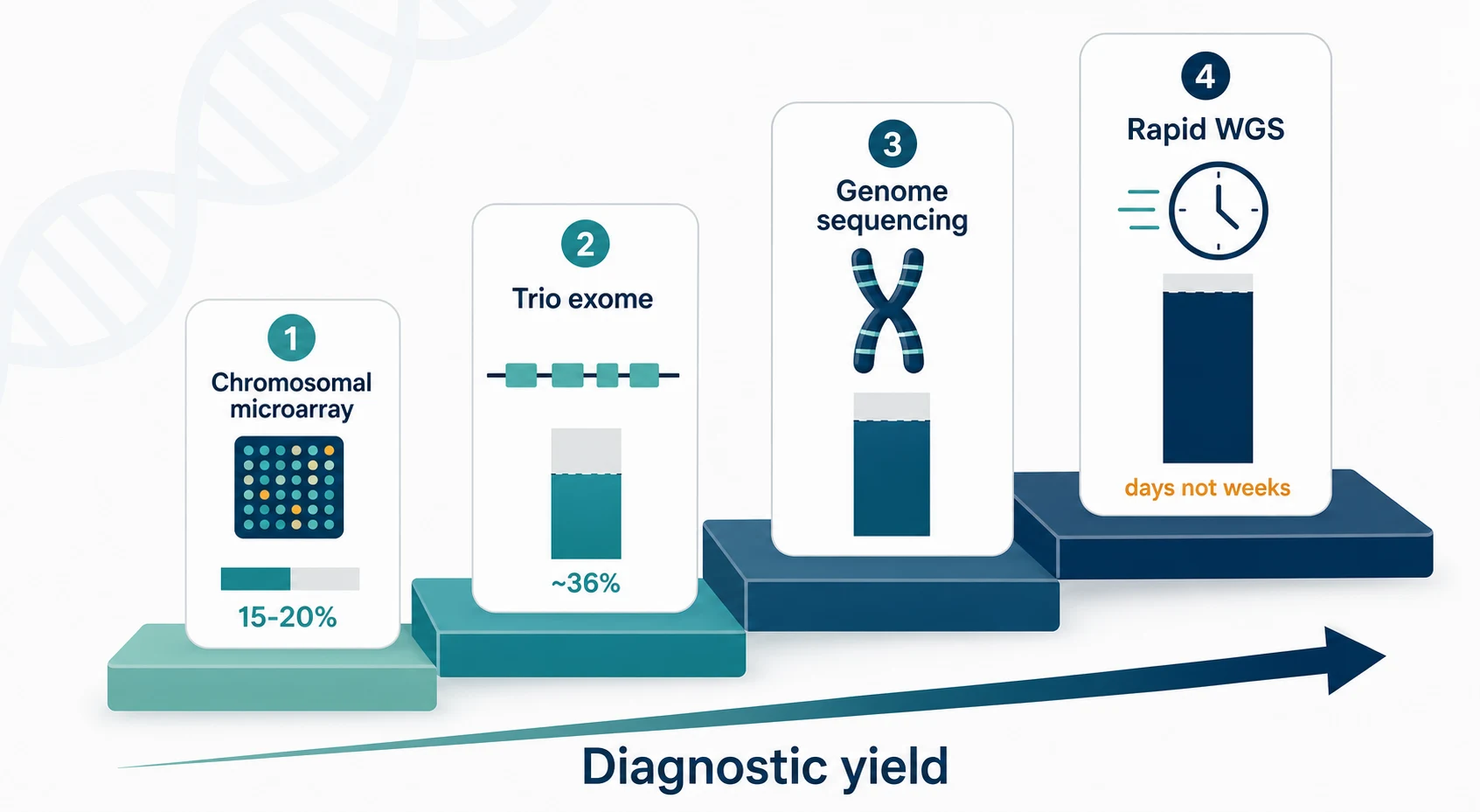
The practical classification of genomic testing follows the type of variant each technique detects and the order in which the tests are deployed. The clinician chooses the test by the phenotype and the acuity, and by what has already been done, because each tier captures a different class of variant and a negative tier is not a negative diagnosis. [4][3]
- First-tier for copy-number variants (deletions, duplications)
- Yield around fifteen to twenty per cent in developmental disability
- Detects neither balanced rearrangements nor single-nucleotide variants
- First-tier for single-nucleotide and coding variants
- Yield around thirty-six per cent in neurodevelopmental disorders
- Trio (proband and both parents) captures de novo change
- Whole sequence including non-coding and structural change
- Resolves repeat expansions and complex rearrangements
- Preferred when exome is negative and the picture is strongly genetic
- For the acutely ill infant in days, not weeks
- Changes acute management and reduces cost
- Established in neonatal and paediatric intensive care
The second axis separates phenotype-driven testing, where the clinical picture selects the gene or panel first, from genome-first testing, where an unselected genome is read and the phenotype is then reinterpreted against the finding in a process called reverse phenotyping. In practice the two converge, because a careful phenotype always raises the yield of any genome read, and a genome-first finding always sends the clinician back to re-examine the child for features they had missed. [9][4]
Epidemiology & Risk Factors
Rare disease is common in aggregate, and most rare diseases affect children and have a genetic basis. Roughly half of all rare diseases present in childhood, and the majority are neurological or neurodevelopmental, so the undiagnosed child in a paediatric neurology or developmental clinic is the core population for precision diagnosis. The diagnostic odyssey before the genomic era ran for years, involved repeated specialist visits and serial single-gene tests, and often ended without an answer. [9][3]
The risk factors that raise the prior probability of a monogenic cause, and therefore the yield of testing, cluster into a recognisable pattern. Severe global developmental delay or intellectual disability, developmental regression, early-onset or drug-resistant epilepsy, congenital anomalies alongside the neurological features, a positive family history, and consanguinity each raise the yield. Sporadic severe disease in a child of unaffected parents carries a particularly high prior of a de novo dominant variant, which is why trio sequencing outperforms proband-only testing. [5][6]
The burden of the odyssey is not only medical: it includes repeated anaesthetics for investigations, parental anxiety and lost income, and missed opportunities for therapy and reproductive counselling. Early genomic testing has been shown to shorten the odyssey, reduce cost, and improve outcomes, which is why it has moved from last resort to first tier. [8][9]
Pathophysiology
A monogenic neurogenetic disease arises from a variant that disrupts a gene whose function is required for the developing or functioning nervous system. The variant types run from single-nucleotide variants and small insertions or deletions, through larger copy-number variants of whole exons or genes, to repeat expansions, structural rearrangements, and regulatory change in the non-coding genome. No single test detects all of them, which is why the ladder exists: the microarray sees the copy-number variants, the exome sees the coding single-nucleotide variants, and the genome sees the rest. [4][2]
The inheritance patterns drive the pre-test counselling and the choice of a trio. Autosomal dominant disorders may be inherited from an affected parent or arise de novo, and de novo dominant change is the dominant mechanism in sporadic severe neurodevelopmental disease. Autosomal recessive disorders cluster in consanguineous families and founder populations. X-linked disorders affect males disproportionately but carrier females can be variably affected, and mitochondrial disorders show maternal inheritance with variable heteroplasmy. Reading the trio at the variant level distinguishes a de novo change from an inherited one and is what allows a confident pathogenic call in a sporadic case. [1][9]
Not every variant in a disease-associated gene causes disease. Penetrance is incomplete for many genes, expressivity is variable, and the same gene can cause different phenotypes in different people, which is why the genotype must always be read back against the phenotype. The variant of uncertain significance is the honest expression of this uncertainty, and it is the single most common source of error when it is over-interpreted as pathogenic. [1][9]
Clinical Presentation
The child reaches the genomic testing pathway through a handful of presenting problems, and recognising them triggers the test rather than another round of single-gene assays. Global developmental delay or intellectual disability of uncertain cause is the commonest entry point, followed by developmental regression, early-onset or drug-resistant epilepsy, neonatal hypotonia, a progressive movement disorder, and a combination of congenital anomalies with neurological features. Each of these, when unexplained after initial workup, meets the threshold for tiered genomic testing. [3][2]
The features that raise the yield further are a positive family history of a similar disorder, consanguinity, older paternal age, a dysmorphic gestalt, and a multi-system picture that joins neurological to cardiac, skeletal, renal, or ophthalmic findings. A regression after a period of normal development points toward a neurodegenerative or metabolic process and raises the prior of a treatable condition, which makes the speed of diagnosis matter. The acutely ill infant with an unexplained encephalopathy or refractory seizures is the time-critical presentation that justifies rapid whole-genome sequencing in days. [6][7]
The phenotype is not a label to attach after the test; it is the input that raises the yield of the test itself. Deep phenotyping with structured Human Phenotype Ontology terms, a careful dysmorphology examination, and targeted biochemistry and imaging allows the laboratory to prioritise the right genes and the clinician to reinterpret a genome-first finding against the child in front of them. [9][5]
Differential Diagnosis
The first differential is whether the undiagnosed neurological presentation is genetic at all, because that decision determines whether genomic testing is the right next step. An acquired cause such as hypoxic-ischaemic encephalopathy, a perinatal or postnatal infection, an autoimmune encephalitis, a structural lesion, or a nutritional or toxic exposure must be considered and excluded where appropriate, although a genetic cause can coexist with an acquired insult and the two are not mutually exclusive. [9][3]
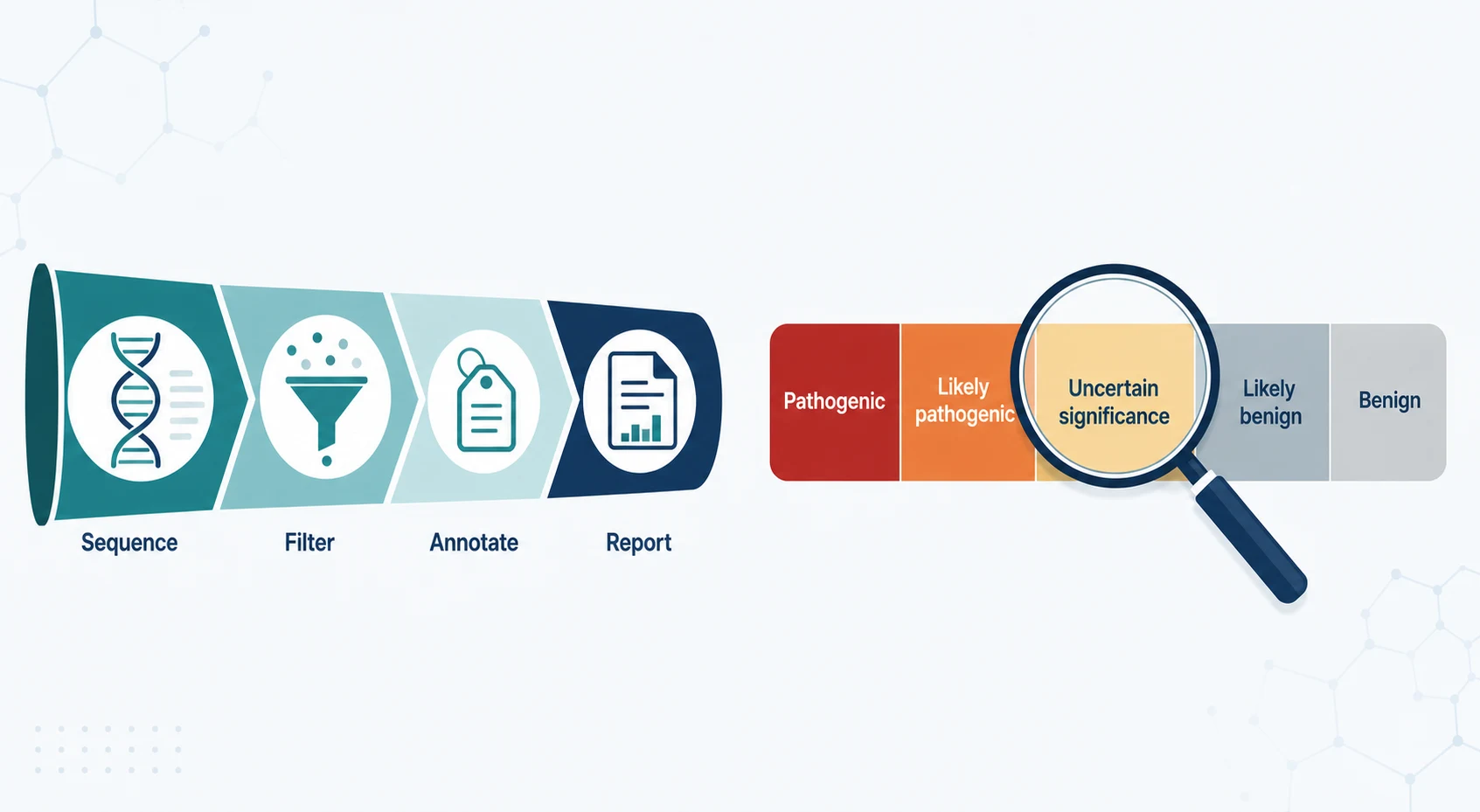
The second differential runs across the genomic test results themselves, because the laboratory returns several kinds of finding and each is managed differently. A pathogenic or likely pathogenic variant in a gene that matches the phenotype is a diagnostic result, a variant of uncertain significance is an honest admission of insufficient evidence, a pathogenic variant in a gene that does not match the phenotype may be incidental, and a secondary finding in a recommended actionable gene may be returned regardless of the presenting problem. [1][4]
- Diagnostic result
- Directs therapy and counselling
- Ends further diagnostic testing
- Insufficient evidence to call
- Manage by the phenotype, not the variant
- Reanalyse over time
- Variant not related to the presentation
- Return only if actionable and consented
- Do not anchor the diagnosis to it
- Not the end of the investigation
- Reanalyse periodically
- Move to genome sequencing if not done
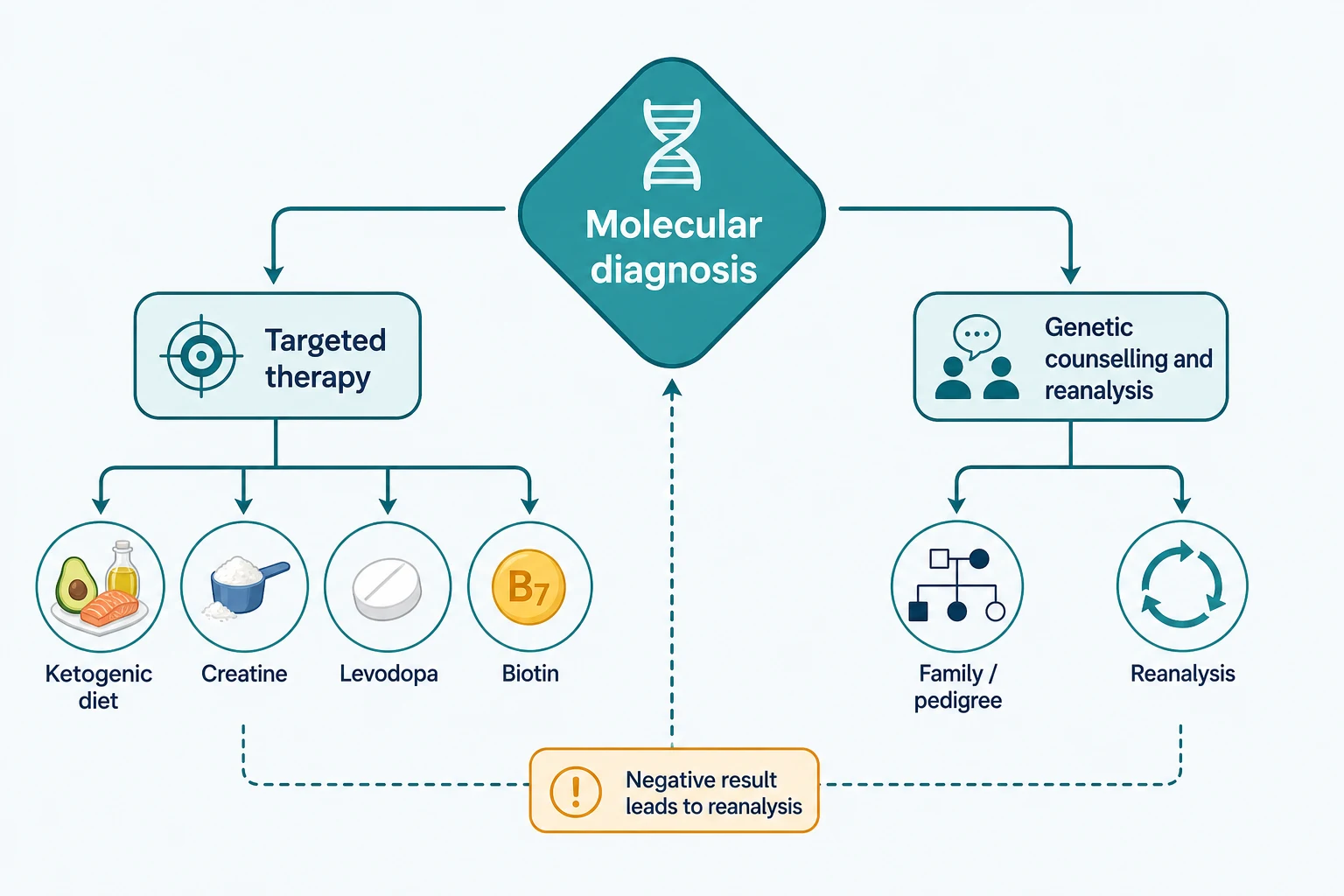
The treatable conditions form a differential all of their own, because each is a diagnosis that doubles as a treatment trigger. Glucose transporter one deficiency presents with epilepsy and a movement disorder and responds to the ketogenic diet, creatine transporter deficiency presents with intellectual disability and seizures and responds to creatine supplementation, dopa-responsive dystonia presents with a fluctuating dystonia and responds to levodopa, and biotinidase deficiency presents with seizures and skin changes and responds to biotin. Recognising the phenotype and confirming the gene turns a chronic disability into a treated condition. [11][10]
Clinical & Bedside Assessment
The bedside assessment is deep phenotyping, and its quality sets the yield of every downstream test. The history captures a three-generation pedigree with consanguinity, parental ages, prior losses and stillbirths, the pregnancy and perinatal period, the developmental trajectory with any regression, the seizure semiology, and the response to any trials of therapy. The examination looks for a dysmorphic gestalt, cutaneous stigmata of a phakomatosis, neurocutaneous markers, organomegaly, and the pattern of tone, power, reflexes, and movement disorder. [9][3]
TRIOS
The pre-test counselling sets expectations before the blood is drawn. The family is told the likely yield, the chance of a variant of uncertain significance, the possibility of an incidental or secondary finding in an actionable gene, and the option to opt in or out of receiving such findings. Consent is taken for the storage and periodic reanalysis of the data, and the family is told that a negative result today may become a positive result in a year as new genes are discovered. [4][1]
The investigations that complete the phenotyping run alongside the genomic test rather than after it. A metabolic and biochemical screen, neurophysiology, and neuroimaging define the phenotype and occasionally yield a treatable diagnosis in their own right, such as a low cerebrospinal fluid glucose consistent with glucose transporter one deficiency or a depleted creatine peak on magnetic resonance spectroscopy consistent with a creatine disorder. [11][10]
Investigations
The investigations form a tiered ladder in which each test detects a different class of variant and a negative tier is followed by the next tier rather than accepted as a final answer. Chromosomal microarray is the first-tier test for copy-number variants and reaches a yield of around fifteen to twenty per cent in unexplained developmental disability, which is why it remains the starting point where a copy-number burden is suspected. [2][3]
Exome sequencing is the first-tier test for neurodevelopmental disorders because it reads the coding single-nucleotide and small insertion-deletion variants that account for most monogenic disease, and a trio of the proband and both parents captures the de novo variants that dominate sporadic severe disease. Genome sequencing reads the whole sequence and resolves repeat expansions, complex rearrangements, and non-coding regulatory change that the exome misses, and it is the next step when a strongly genetic picture has a negative exome. [3][4]
In the acutely ill infant, rapid whole-genome sequencing compresses the timeline from weeks to days and changes acute management. Studies in the neonatal and paediatric intensive care unit report diagnostic yields in the order of a quarter to forty per cent, with a meaningful proportion of diagnoses leading to a change in therapy and to the withdrawal of invasive testing, and a reduction in the cost and length of hospitalisation. [6][7][8]
- Copy-number variants
- Yield around fifteen to twenty per cent
- First-tier where a CNV is suspected
- Coding single-nucleotide variants
- Yield around thirty-six per cent
- Captures de novo change
- Non-coding and structural change
- Resolves repeat expansions
- Next step after a negative exome
- Days rather than weeks
- For the acutely ill infant
- Changes acute management
A negative result triggers periodic reanalysis of the existing data, because new gene-disease associations are published continuously and a variant that was uninterpretable one year may be reclassified the next. The laboratory should store the data and offer scheduled reanalysis, and the clinician should return the patient to the pathway at intervals rather than discharging them as undiagnosed. [3][9]
Management — Resuscitation
The resuscitation equivalent in precision diagnosis is the time-critical molecular workup of the acutely ill infant, in whom an unexplained encephalopathy or refractory seizures carry a real risk of death or irreversible brain injury and a treatable cause may be hidden in the genome. Rapid whole-genome sequencing is ordered the moment a monogenic cause is plausible, in parallel with the metabolic and infective workup, because a diagnosis within days rather than weeks changes what the team does at the bedside. [6][8]
A rapid diagnosis changes management in two directions. It directs therapy toward a treatable cause, such as starting a ketogenic diet for glucose transporter one deficiency, biotin for biotinidase deficiency, or pyridoxine for pyridoxine-dependent epilepsy, each of which can halt seizures or encephalopathy that no standard anticonvulsant would control. It also directs the withdrawal of non-contributory invasive testing, the de-escalation of intensive support where a progressive fatal disorder is confirmed, and the redirection of care toward comfort and family-centred goals. [7][11]
The team does not wait for the genome to manage the acute problem: seizures are treated, the airway and circulation are supported, and empirical treatable-condition therapy such as biotin and pyridoxine is given on suspicion where the phenotype fits, because these are safe and potentially disease-modifying while the genome is pending. [11][10]
Management — Definitive & Stepwise
The definitive pathway is a stepwise ladder that moves from phenotyping through the right tier of test to variant interpretation, return of results, and the action the diagnosis unlocks. Each step has an explicit decision, and the team never stops at a single negative result. [4][3]
Deep phenotyping with a three-generation pedigree, dysmorphology, Human Phenotype Ontology coding, and targeted biochemistry and imaging
Chromosomal microarray as the first-tier test for copy-number variants where a CNV burden is suspected
Trio exome or genome sequencing as first-tier for neurodevelopmental disorders, capturing de novo change
Rapid whole-genome sequencing in the acutely ill infant to return a diagnosis in days
Variant interpretation through the American College of Medical Genetics five-tier framework, with a multidisciplinary review of uncertain calls
Return of results with genetic counselling, including uncertain and secondary findings, and a documented plan
Action the diagnosis: start treatable-condition therapy, stop non-contributory testing, counsel on recurrence, and enrol in trials
Periodic reanalysis of negative or uncertain results as new gene-disease associations are discovered
The variant interpretation is the step where error most often enters, and it is governed by the standard five-tier framework that combines population frequency, computational prediction, segregation in the family, and functional evidence into a call of pathogenic, likely pathogenic, uncertain significance, likely benign, or benign. A variant of uncertain significance is managed by the phenotype and reanalysed, never reported to the family as the cause of the disease, and a pathogenic call requires the variant to match the clinical picture. [1][9]
The return of results is a clinical act, not a letter in the post. The clinician and the genetic counsellor sit with the family, explain the diagnosis in plain language, set honest expectations about prognosis and therapy, discuss recurrence risk and reproductive options, and arrange the practical next steps. Where the diagnosis is treatable, the therapy is started; where it is not, the family is linked to a support network and to a natural-history or trial programme. [9][11]
Specific Subtypes & Scenarios
The treatable neurogenetic conditions are the heart of precision diagnosis, because in each a confirmed molecular diagnosis doubles as a treatment trigger that converts a chronic disability into a managed disease. Glucose transporter one deficiency, caused by SLC2A1 variants, presents with early-onset epilepsy, a paroxysmal movement disorder, and developmental delay, and responds to a ketogenic diet that supplies the brain with ketone bodies as an alternative fuel, with the diagnosis supported by a low cerebrospinal fluid glucose. [11][9]
Creatine transporter deficiency, caused by SLC6A8 variants, presents with intellectual disability, seizures, and behavioural change in males, shows a depleted creatine peak on magnetic resonance spectroscopy, and responds to oral creatine supplementation with adjunctive arginine and glycine to support replenishment, although the response is partial. Dopa-responsive dystonia, caused most often by GCH1 variants, presents with a childhood-onset fluctuating dystonia that worsens through the day and responds dramatically to low-dose levodopa, and it is the treatable mimic of cerebral palsy that every clinician should exclude. [10][9]
- Epilepsy and paroxysmal movement disorder
- Low cerebrospinal fluid glucose
- Ketogenic diet
- Intellectual disability and seizures in males
- Depleted creatine on spectroscopy
- Oral creatine supplementation
- Fluctuating diurnal dystonia
- Dramatic response to levodopa
- Treatable mimic of cerebral palsy
- Seizures, hypotonia, skin changes
- Detected on newborn screening
- Biotin supplementation
Pyridoxine-dependent epilepsy, caused by ALDH7A1 variants, presents with refractory neonatal seizures that respond to pyridoxine, and biotinidase deficiency presents with seizures, hypotonia, and skin changes that respond to biotin and is often detected on newborn screening. Wilson disease, caused by ATP7B variants, presents with a movement disorder or psychiatric change alongside liver disease and responds to chelation and zinc. The gene-modifying therapies for spinal muscular atrophy and Duchenne muscular dystrophy are mutation-specific, so a confirmed molecular diagnosis is the prerequisite for treatment with nusinersen, risdiplam, or onasemnogene for spinal muscular atrophy and exon-skipping or gene therapy for Duchenne. [9][11]
The acutely ill infant is the scenario where speed is itself the therapy. Rapid whole-genome sequencing in the neonatal or paediatric intensive care unit returns a diagnosis in days, and the studies show that a meaningful proportion of these diagnoses change management, from starting a treatable-condition therapy to redirecting care in a progressive fatal disorder. The undiagnosed school-age child is the scenario of the long odyssey, where reanalysis and the move from exome to genome recover diagnoses years after the first negative test. [7][8][3]
Complications & Pitfalls
The most common and most dangerous pitfall is the over-interpretation of a variant of uncertain significance as the cause of the disease. A variant of uncertain significance is a statement that the evidence is insufficient to call, and treating it as pathogenic leads to a wrong diagnosis, wrong counselling about recurrence, and wrong therapy. The correct approach is to manage the child by their phenotype, to segregate the variant through the family if possible, and to reanalyse it as evidence accrues, while never reporting it to the family as the answer. [1][4]
The second pitfall is ending the workup at a single negative test. A negative exome is not a negative diagnosis, because the yield accrues with trio sequencing, with the move to genome sequencing, and with periodic reanalysis as new gene-disease associations are discovered. Discharging a child as undiagnosed after one test, with no plan to reanalyse, closes a door that would have opened a year later. [3][9]
The third pitfall is missing a treatable condition because the test was not ordered or was ordered too late. Glucose transporter one deficiency, creatine transporter deficiency, dopa-responsive dystonia, pyridoxine-dependent epilepsy, and biotinidase deficiency are each treatable, and a delay in diagnosis is a delay in therapy that the child does not get back. The equity pitfall is that genomic testing is unevenly available, and children in remote, indigenous, and disadvantaged communities face longer odysseys, which is why access and data sovereignty are part of good practice. [11][10][9]
Prognosis & Disposition
The prognosis is set by the underlying gene, but the act of diagnosis itself improves outcomes regardless of the specific prognosis. A confirmed molecular diagnosis stops the diagnostic odyssey, withdraws non-contributory and invasive testing, enables accurate recurrence-risk and reproductive counselling, and links the family to a community, to a natural-history study, and to a clinical trial where one exists. For the treatable conditions the prognosis is transformed by therapy, and for the progressive disorders the prognosis is at least made honest and plannable. [9][3]
Disposition is to a clinical genetics service working with neurology, metabolism, and the multidisciplinary team, with a documented plan for reanalysis and for the return of any updated result. The family is offered genetic counselling for recurrence risk and reproductive options including prenatal and preimplantation diagnosis, and the adolescent approaching transition is prepared for adult genetics and adult neurology services. The child with a treatable condition is monitored for response to therapy, and the child with an untreatable condition is supported with palliative and developmental services as the phenotype requires. [4][11]
The reanalysis schedule is part of the disposition, because a negative or uncertain result today may become diagnostic within one to two years as new genes are published, and the family should know that the door remains open. [3]
Special Populations
The neonate and young infant are the population in whom precision diagnosis is most time-critical, because an unexplained encephalopathy or refractory seizure may hide a treatable cause and because the developing brain is vulnerable to delay. Rapid whole-genome sequencing in the neonatal intensive care unit returns a diagnosis in days and changes acute management, and empirical treatable-condition therapy such as biotin and pyridoxine is given on suspicion while the genome is pending. [6][8]
Indigenous and remote communities face longer diagnostic odysseys because of distance, workforce, and access, and good practice combines telehealth outreach with attention to data sovereignty and to the cultural safety of genetic counselling. Migrant and consanguineous communities carry a higher burden of autosomal recessive disease through founder variants, and a three-generation pedigree that captures consanguinity raises the yield of testing and the accuracy of counselling. [9][3]
The adolescent approaching transition holds a molecular diagnosis made in childhood and faces the move to adult services, reproductive decisions, and the autonomy to decide on secondary findings and reanalysis for themselves. The child from a disadvantaged background faces the cost and the coordination barriers of a long odyssey, and the clinician advocates for equitable access to testing, counselling, and the therapies and trials that a diagnosis unlocks. [4][9]
Evidence, Guidelines & Regional Differences
The evidence base rests on a sequence of consensus statements and meta-analyses that moved genomic testing from last resort to first tier. The Miller consensus established chromosomal microarray as a first-tier test for unexplained developmental disability on the strength of its copy-number variant yield, and the Srivastava meta-analysis of more than thirty thousand individuals established exome as a first-tier test for neurodevelopmental disorders with a pooled diagnostic yield of around thirty-six per cent. The Manickam technical standard codifies the practice of exome and genome sequencing in children, and the Richards standards govern the interpretation of every variant. [2][3][4][1]
Srivastava et al, Genet Med 2019
Key finding
A systematic review and meta-analysis of exome sequencing in individuals with neurodevelopmental disorders pooled data from more than thirty thousand subjects and reported a diagnostic yield of around thirty-six per cent, with higher yields for trio sequencing and for reanalysis over time.
Practice change
Exome sequencing is a first-tier clinical diagnostic test for individuals with neurodevelopmental disorders, and the yield improves with trio design and periodic reanalysis.
The rapid whole-genome sequencing studies in the acutely ill infant, from Willig and Meng through to Farnaes, show that a diagnosis in days changes management, reduces cost, and shortens hospitalisation, and they underpin the integration of rapid sequencing into neonatal and paediatric intensive care. Boycott framed the broader trajectory from gene discovery to translation, in which a molecular diagnosis is the gateway to therapy, counselling, and trial enrolment. [7][6][8][9]
Australian and New Zealand practice follows the tiered testing model with public access to chromosomal microarray and trio exome for paediatric neurodevelopmental disorders, funded genomic sequencing through state and national programmes, and rapid whole-genome sequencing established in the tertiary neonatal and paediatric intensive care units. Genetic counselling and reanalysis are coordinated through clinical genetics services, with attention to equitable access for rural, remote, and indigenous communities and to data sovereignty. [3][9]
Exam Pearls
The single most testable fact is that exome sequencing is a first-tier test for neurodevelopmental disorders with a pooled diagnostic yield of around thirty-six per cent, that chromosomal microarray remains first-tier for copy-number variants at around fifteen to twenty per cent, and that trio sequencing outperforms proband-only testing because it captures the de novo dominant variants that dominate sporadic severe disease. A negative result is never final: it is followed by genome sequencing and periodic reanalysis. [3][2]
The American College of Medical Genetics five-tier variant classification is pathogenic, likely pathogenic, uncertain significance, likely benign, and benign, and a variant of uncertain significance is managed by the phenotype and reanalysed, never reported as the diagnosis. The treatable conditions are the reward of precision diagnosis: glucose transporter one deficiency and the ketogenic diet, creatine transporter deficiency and creatine, dopa-responsive dystonia and levodopa, pyridoxine-dependent epilepsy and pyridoxine, and biotinidase deficiency and biotin. [1][11][10]
Rapid whole-genome sequencing in the acutely ill infant returns a diagnosis in days and changes acute management, from treatable-condition therapy to the redirection of care, and it reduces cost and length of stay. The gene-modifying therapies for spinal muscular atrophy and Duchenne muscular dystrophy are mutation-specific, so a confirmed molecular diagnosis is the prerequisite for treatment, and the same principle extends to every treatable condition unlocked by precision diagnosis. [6][8][9]
Self-test: what raises the yield of a trio exome above proband-only testing?
Sequencing the proband and both parents together allows the laboratory to distinguish a de novo variant from an inherited one, and de novo dominant change is the dominant mechanism in sporadic severe neurodevelopmental disease. Trio design also resolves autosomal recessive and X-linked inheritance at the variant level, supports segregation for uncertain calls, and shortens the time to a confident pathogenic interpretation. [5][1]
References
- [1]Richards S, Aziz N, Bale S, et al Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology Genet Med, 2015.PMID 25741868
- [2]Miller DT, Adam MP, Aradhya S, et al Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies Am J Hum Genet, 2010.PMID 20466091
- [3]Srivastava S, Love-Nichols JA, Dies KA, et al Meta-analysis and multidisciplinary consensus statement: exome sequencing is a first-tier clinical diagnostic test for individuals with neurodevelopmental disorders Genet Med, 2019.PMID 31182824
- [4]Manickam K, McClain MR, Demmer LA, et al Exome and genome sequencing for pediatric patients with congenital anomalies or intellectual disability: an evidence-based clinical guideline of the American College of Medical Genetics and Genomics (ACMG) Genet Med, 2021.PMID 34211152
- [5]Soden SE, Saunders CJ, Willig LK, et al Effectiveness of exome and genome sequencing guided by acuity of illness for diagnosis of neurodevelopmental disorders Sci Transl Med, 2014.PMID 25473036
- [6]Meng L, Pammi M, Saronwala A, et al Use of exome sequencing for infants in intensive care units: ascertainment of severe single-gene disorders and effect on medical management JAMA Pediatr, 2017.PMID 28973083
- [7]Willig LK, Petrikin JE, Smith LD, et al Whole-genome sequencing for identification of Mendelian disorders in critically ill newborns: a retrospective analysis of diagnostic and clinical findings Lancet Respir Med, 2015.PMID 25937001
- [8]Farnaes L, Hildreth A, Sweeney NM, et al Rapid whole-genome sequencing decreases infant morbidity and cost of hospitalization NPJ Genom Med, 2018.PMID 29644095
- [9]Boycott KM, Vanstone MR, Bulman DE, MacKenzie AE Rare-disease genetics in the era of next-generation sequencing: discovery to translation Nat Rev Genet, 2013.PMID 23999272
- [10]Dunbar M, Jaggumantri S, Sargent M, Stockler-Ipsiroglu S, van Karnebeek CD Treatment of X-linked creatine transporter (SLC6A8) deficiency: systematic review of the literature and three new cases Mol Genet Metab, 2014.PMID 24953403
- [11]Klepper J Glut1 deficiency syndrome: novel pathomechanisms, current concepts, and challenges J Inherit Metab Dis, 2025.PMID 40405536