Paeds · neurology-neurodisability-and-neuromuscular
Peripheral neuropathies
Also known as Charcot-Marie-Tooth disease · CMT · Hereditary motor and sensory neuropathy · HMSN · Chronic inflammatory demyelinating polyneuropathy · CIDP · Hereditary neuropathy with liability to pressure palsies · HNPP
Fellowship guide to peripheral neuropathies in children covering the inherited and acquired polyneuropathies. Details Charcot-Marie-Tooth disease subtypes CMT1A with PMP22 duplication, CMT1B with MPZ, CMT2A with MFN2, CMTX1 with GJB1 connexin 32, and HNPP with PMP22 deletion, the clinical phenotype of distal wasting with pes cavus and foot drop, the nerve conduction study distinction between demyelinating under thirty-eight metres per second and axonal patterns, the paediatric CMT clinical practice guideline of Yiu 2022, the acquired neuropathies including chronic inflammatory demyelinating polyradiculoneuropathy with its greater than eight weeks criterion and treatment with intravenous immunoglobulin and corticosteroids, diabetic neuropathy in youth, chemotherapy-induced and vincristine neuropathy that can unmask Charcot-Marie-Tooth disease, and the multidisciplinary management with orthotics, physiotherapy, foot surgery, pain control, and genetic counselling.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
Overview & Definition
A child who walks on the outer edges of high-arched feet, trips over unseen kerbs, and cannot feel the vibration of a tuning fork is showing one of the most recognisable patterns in paediatric neurology. Peripheral neuropathy is a disorder of the peripheral nerves, the motor, sensory, and autonomic fibres that connect the spinal cord to the limbs and the trunk, and in childhood it divides cleanly into the inherited and the acquired. The inherited family is dominated by Charcot-Marie-Tooth disease, the commonest inherited neuromuscular disorder of humans, with a prevalence of around one in two thousand five hundred. The acquired family is dominated by the treatable immune neuropathies, the toxic and metabolic neuropathies, and diabetic neuropathy in youth. [1]
The reason this topic matters above all is that the two families demand opposite management. The inherited neuropathies progress slowly over years, have no disease-modifying therapy, and are managed with orthotics, physiotherapy, foot surgery, pain control, and genetic counselling. The acquired neuropathies can progress over weeks and are treatable with immunoglobulin, corticosteroids, or removal of the offending drug or toxin. Confusing the two wastes the child's time and the family's hope, because a treatable CIDP mislabelled as Charcot-Marie-Tooth disease goes untreated while an incurable CMT given immunoglobulin gains nothing. [1][8]
The single concept that frames the whole topic is the nerve conduction study, which tells you whether the problem is the myelin or the axon. A motor conduction velocity under thirty-eight metres per second with uniform slowing across all nerves points to an inherited demyelinating disease, the CMT1 family. A velocity above thirty-eight with reduced amplitudes points to axonal disease, the CMT2 family. Patchy, non-uniform slowing with conduction block points to an acquired demyelinating process, the CIDP family. That one number, thirty-eight metres per second, sorts the disease at the bedside and drives the entire workup. [1]
Classification
Peripheral neuropathies are sorted along two axes that both change the workup and the management. The first sort is hereditary versus acquired, which sets whether the disease is treatable. The second sort is demyelinating versus axonal, which is read off the nerve conduction study and sets the likely gene and the prognosis. [1]
The hereditary family is dominated by Charcot-Marie-Tooth disease, historically called hereditary motor and sensory neuropathy. The disease sorts first by conduction velocity into the demyelinating CMT1, the axonal CMT2, and the dominant intermediate CMTDI. CMT1A, caused by a duplication of the PMP22 gene on chromosome seventeen, accounts for around half of all Charcot-Marie-Tooth disease and is the single most common form worldwide. CMT1B carries mutations in MPZ, the gene for myelin protein zero. CMT2A, caused by mutations in MFN2 encoding mitofusin 2, is the commonest axonal form and can include optic atrophy. CMTX1, from variants in GJB1 encoding connexin 32, is X-linked and affects males more severely while carrier females can be mildly affected. Hereditary neuropathy with liability to pressure palsies, caused by a deletion of the same PMP22 gene, produces recurrent focal mononeuropathies rather than a length-dependent polyneuropathy. [1][3][5]
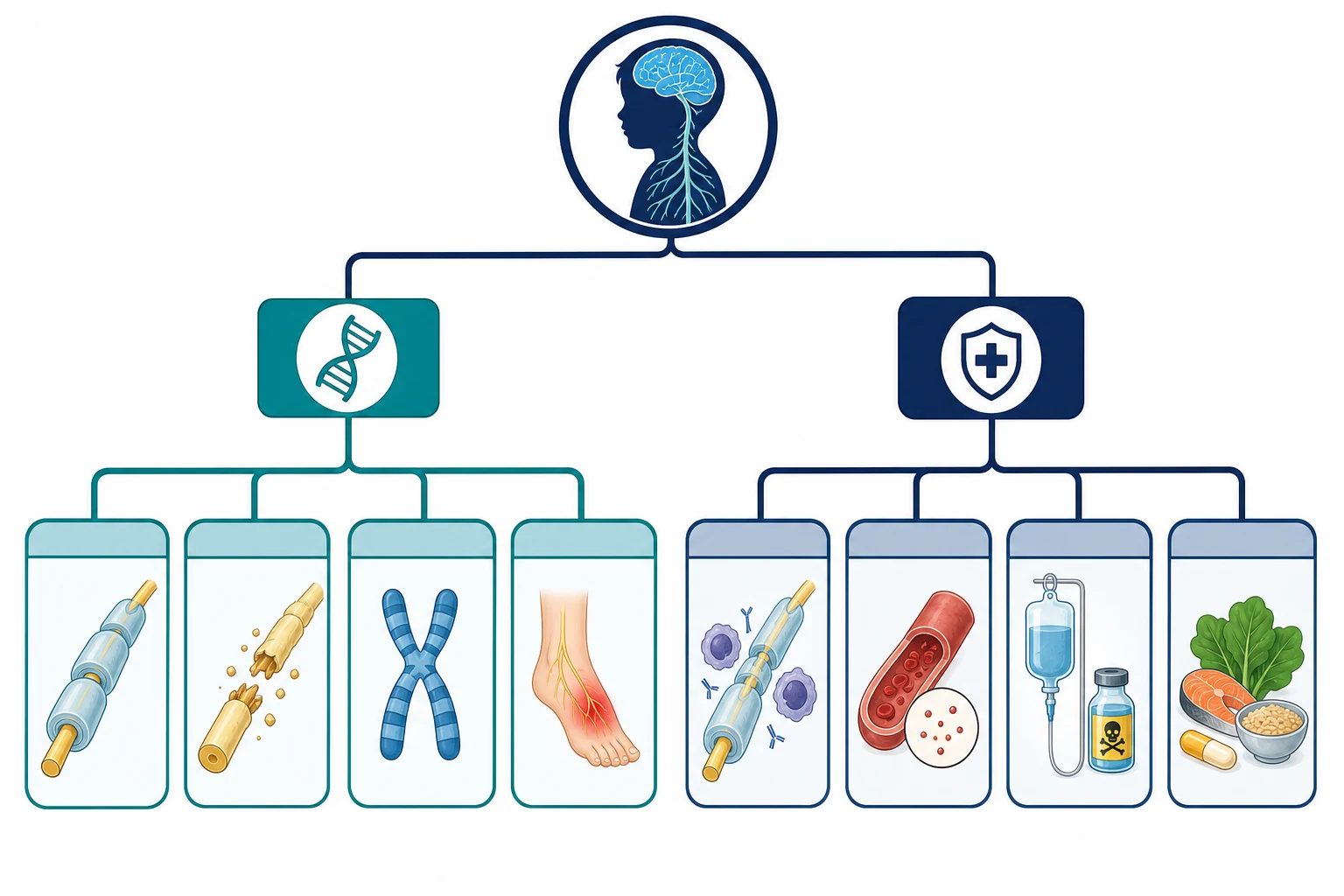
The acquired family divides into the immune, the metabolic and nutritional, the toxic, and the infectious. Chronic inflammatory demyelinating polyradiculoneuropathy is the immune neuropathy that matters most, because it is common and treatable, and it is defined by progression over more than eight weeks, which separates it from Guillain-Barre syndrome. Diabetic neuropathy, once a disease of adults, now affects youth with type 2 diabetes and a significant minority of those with long-standing type 1 diabetes. Chemotherapy-induced peripheral neuropathy, especially from vincristine, platinums, and taxanes, is the commonest toxic neuropathy in paediatric oncology. [8][10][11]
Epidemiology & Risk Factors
Charcot-Marie-Tooth disease has a prevalence of around one in two thousand five hundred in the general population, which makes it the commonest inherited neuromuscular disorder. The Inherited Neuropathies Consortium natural history study of over a thousand patients confirmed that CMT1A is the single largest subtype, accounting for around half of all cases, and that the disease burden is driven by foot and hand weakness, sensory loss, and pain. Boys and girls are affected equally in autosomal forms, while the X-linked CMTX1 affects males more severely. [1][7]
The strongest risk factor for an inherited neuropathy is a family history, and it is the single most useful piece of information at the first consultation. The inheritance patterns are autosomal dominant for most CMT1A and CMT2A, autosomal recessive for less common and often more severe childhood-onset forms, and X-linked dominant for CMTX1. A new variant accounts for around ten percent of CMT1A cases and explains the child with no family history. The age at onset varies by subtype, with severe recessive forms presenting in infancy and the common CMT1A presenting in the first or second decade with foot deformity and clumsy gait. [1][5]
For the acquired neuropathies, the risk factors are different and treatable. Chronic inflammatory demyelinating polyradiculoneuropathy is less common in children than in adults but carries a better prognosis, with most children responding to intravenous immunoglobulin. Diabetic peripheral neuropathy affects around seven percent of youth with type 1 diabetes and around twenty-two percent of those with type 2 diabetes in the Jaiswal SEARCH study, driven by glycaemic control and duration. Chemotherapy-induced peripheral neuropathy affects a substantial proportion of children treated with vincristine for acute lymphoblastic leukaemia and sarcomas. [8][10][11]
Pathophysiology
The mechanism of the inherited neuropathies is a structural failure of either the myelin sheath or the axon, and the gene tells you which. The concept that holds the family together is that a single duplicated or deleted gene produces a lifelong, slowly progressive length-dependent degeneration of the longest peripheral nerves, which is why the symptoms begin in the feet. [1]
In CMT1A the problem is a one-point-four-megabase duplication on chromosome seventeen that contains the PMP22 gene, discovered by Lupski in 1991. The duplication causes the Schwann cell to overexpress peripheral myelin protein twenty-two, and the excess protein is misfolded and unstable, so the myelin sheath is over-produced, laid down abnormally, and then breaks down. The result is demyelination with abnormally thick irregular myelin on onion-bulb formations on biopsy, and the nerve conduction velocity falls to under thirty-eight metres per second uniformly across all nerves because every Schwann cell carries the same genetic dose. The gene dose matters: a duplication causes CMT1A and a deletion of the same gene causes hereditary neuropathy with liability to pressure palsies, because too much or too little of the same protein destabilises the myelin. [2][3]
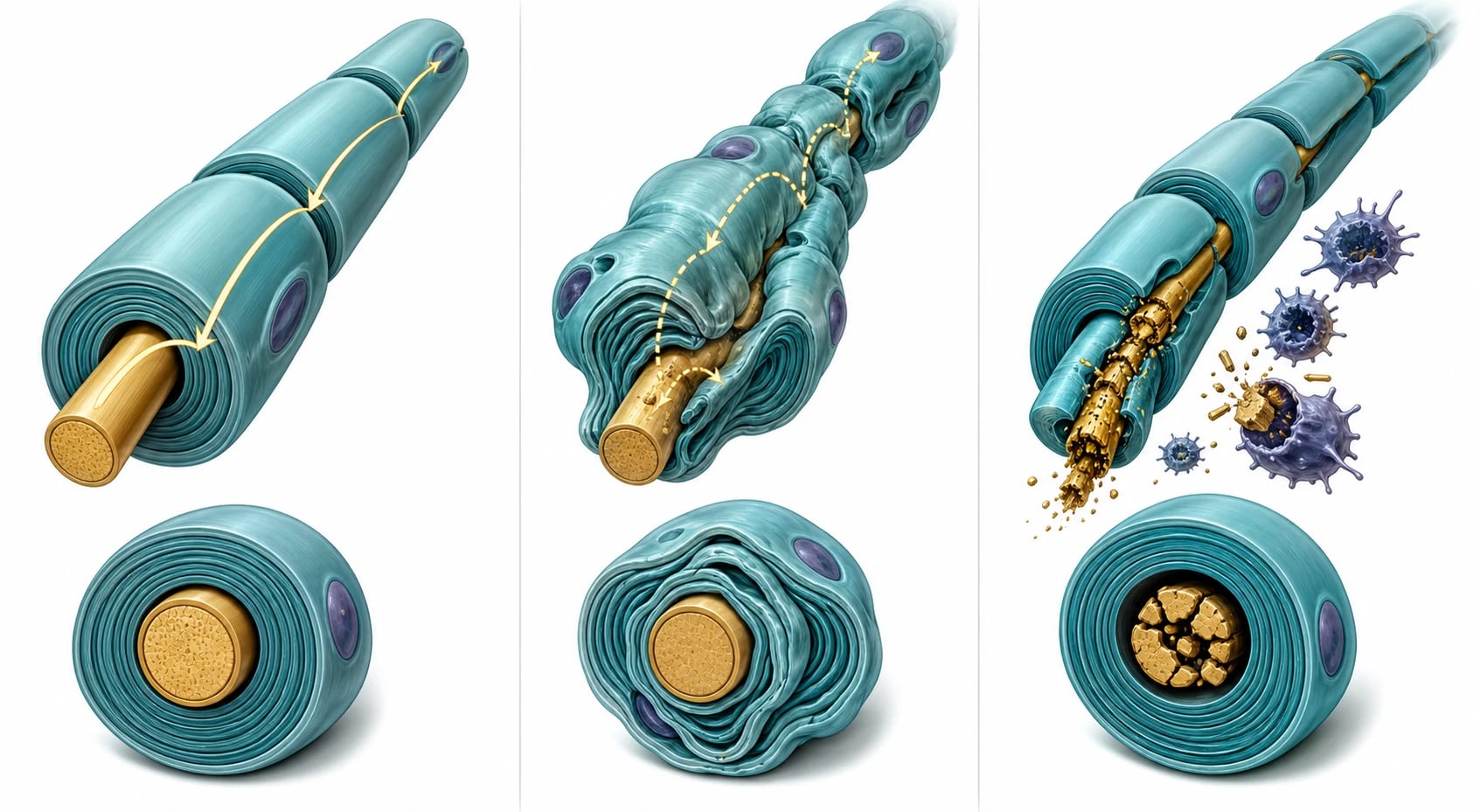
In the axonal forms the myelin is relatively preserved and the axon itself degenerates. CMT2A is caused by mutations in MFN2, which encodes mitofusin 2, a protein that tethers the endoplasmic reticulum to the mitochondria and governs mitochondrial transport along the nerve. When mitofusin 2 fails, the distal axon cannot maintain its energy supply and undergoes length-dependent Wallerian degeneration, which is why CMT2A presents with muscle wasting, reduced compound muscle action potential amplitudes, and relatively preserved conduction velocity. CMTX1 is caused by variants in GJB1 encoding connexin 32, a gap-junction protein expressed in Schwann cells that allows nutrient exchange across the myelin, and it produces an intermediate picture with features of both demyelination and axonal loss. [4][5]
Chronic inflammatory demyelinating polyradiculoneuropathy is driven by a T-cell and antibody-mediated attack on the myelin and the nerve roots, similar to Guillain-Barre syndrome but chronic and relapsing rather than monophasic. The macrophages strip the myelin in a patchy, multifocal pattern, which is why the nerve conduction study shows non-uniform conduction block and temporal dispersion rather than the uniform slowing of CMT1. The disease responds to immunomodulation because the immune attack is ongoing, which is the key difference from the static genetic diseases. [8]
Clinical Presentation
The child with an inherited neuropathy presents with a pattern that is so characteristic that the diagnosis is often made from the end of the bed. The weakness and wasting are distal and length-dependent, beginning in the intrinsic foot muscles and the peroneal nerve, and climbing to the calves, the hands, and the forearms over years. The legs take on the classic inverted-champagne-bottle contour, with normal thighs and wasted lower legs, and the hands develop the same pattern later. The foot deformity is the sign that brings most children to attention: pes cavus with a high medial arch, clawed hammer toes, and a varus heel that rolls the ankle outward and makes the child walk on the outer edge of the foot. [1]
The gait changes in ways that the family reports before the child does. A foot drop from tibialis anterior weakness produces a slapping or steppage gait in which the knee is lifted high to clear the dropped toes, and the child trips, sprains ankles, and fatigues over distance. The sensory loss is for the large fibres, so vibration and joint position sense are lost early while pain and temperature are relatively spared, which is why the child may develop painless foot ulcers or trophic skin changes. The deep tendon reflexes are reduced or absent, beginning at the ankles, and their absence in a child with foot deformity is one of the most valuable bedside signs. The hands are affected later, with weakness of grip, fine motor difficulty, and intrinsic hand muscle wasting. [1][6]
The tempo is the feature that separates the inherited from the acquired, and it is measured in years rather than weeks. The child with CMT1A is often labelled clumsy or flat-footed for years before the diagnosis, and a careful history reveals that the foot deformity and the clumsiness have been present since the child first walked. The child with chronic inflammatory demyelinating polyradiculoneuropathy, by contrast, develops progressive weakness over weeks to months, often with proximal and distal involvement, sensory symptoms, and a large-fibre sensory loss. The child with HNPP presents not with a polyneuropathy but with recurrent painless focal palsies, such as a wrist drop after leaning on a desk, a foot drop after squatting, or a brachial plexus palsy after carrying a heavy backpack. [3][8]
NEUROPATHY
Differential Diagnosis
The differential divides into the neuropathies that look like CMT and the diseases of the spinal cord, the muscle, and the anterior horn cell that look like a neuropathy from a distance. The discriminating features are the distribution of the weakness, the reflex pattern, the sensory involvement, and the family history. [1]
The first fork is between an inherited neuropathy and a treatable acquired one. Chronic inflammatory demyelinating polyradiculoneuropathy can present with foot deformity and areflexia that mimic Charcot-Marie-Tooth disease, and the Fernandez-Garcia study showed that a significant fraction of children referred for genetic neuropathy testing have an acquired demyelinating process or a genetically undefined picture. The discriminating features are the tempo of weeks to months, the presence of proximal weakness, and the patchy non-uniform conduction block on nerve conduction studies. The importance of this fork is that CIDP responds to immunotherapy and CMT does not, so missing a treatable CIDP is the classic pitfall. [8][9]
The second fork is between a neuropathy and a disorder of the motor unit elsewhere. Spinal muscular atrophy produces proximal weakness and hypotonia, not distal wasting, and it is excluded by the pattern and by genetic testing for the SMN1 deletion. Duchenne muscular dystrophy produces proximal weakness with a Gowers sign and pseudohypertrophy, not distal wasting and sensory loss. Hereditary spastic paraplegia produces a spastic rather than a flaccid gait, with brisk reflexes and extensor plantar responses. Friedreich ataxia combines neuropathy with ataxia, cardiomyopathy, and diabetes, and it carries a GAA trinucleotide repeat in the FXN gene. [1]
Clinical & Bedside Assessment
The bedside assessment of a child with suspected neuropathy follows a sequence that begins with the feet and ends with the family tree. The child should be examined barefoot and in shorts, and the first look is at the foot deformity and the muscle bulk. Look for pes cavus with a high arch that does not flatten on weight-bearing, clawed hammer toes, a varus heel, and wasting of the intrinsic foot muscles and the peroneal compartment that produces the inverted-champagne-bottle leg. Inspect the hands for intrinsic muscle wasting, then watch the child walk. [1][6]
The gait examination is where the foot drop becomes visible. Ask the child to walk on the heels, which is impossible with a tibialis anterior weakness below Medical Research Council grade four, and to walk on the toes, which tests the gastrocnemius and the tibialis posterior. The steppage gait, with excessive hip and knee flexion to clear the dropped foot, is the sign that brings most families to the doctor. Look for ankle spraining, tripping, and the inability to run. Examine the strength of ankle dorsiflexion, eversion, plantar flexion, and the intrinsic hand muscles, and grade each using the Medical Research Council scale. [6]
The sensory and reflex examination completes the bedside picture. Test vibration sense at the great toe with a one-hundred-and-twenty-eight-hertz tuning fork, joint position sense at the great toe, and light touch and pinprick in a stocking distribution. The large fibres are lost first in Charcot-Marie-Tooth disease, so vibration and proprioception are impaired while pain and temperature are relatively spared. Check the deep tendon reflexes at the knees and the ankles, and note that the ankle jerks are the first to disappear. Then take the family history, which is the most powerful diagnostic tool in the inherited neuropathies, and ask specifically about high arches, foot drop, abnormal gait, ankle sprains, and orthopaedic foot surgery in parents, siblings, and grandparents. [1][6]
In ANZ practice, children with suspected inherited neuropathy are managed through regional paediatric neurology services with telehealth support for rural and remote centres. The nerve conduction studies and the genetic testing are centralised, and the paediatric Charcot-Marie-Tooth clinic coordinates orthotics, physiotherapy, podiatry, and orthopaedic surgery. The threshold for referral to a neurologist is any child with pes cavus, unexplained foot drop, or a family history of inherited neuropathy, because early orthotic intervention preserves mobility and prevents contractures. [6]
Investigations
The investigations confirm the neuropathy, sort it into demyelinating or axonal, and then identify the gene or the acquired cause. The nerve conduction study is the gatekeeper test, because it sets the direction of the genetic workup and separates the inherited from the acquired. [1]
Nerve conduction studies measure the compound muscle action potential amplitude, the distal motor latency, the conduction velocity, and the F-wave latency in the motor nerves, and the sensory nerve action potential amplitude and velocity in the sensory nerves. The single most important number is the median or ulnar motor conduction velocity. A uniform slowing to under thirty-eight metres per second across multiple nerves, with the slowing proportional to the nerve length and without conduction block, is the signature of CMT1 and points to a PMP22 duplication test. A velocity above thirty-eight metres per second with reduced compound muscle action potential amplitudes points to the axonal CMT2 family and an MFN2 test. Non-uniform slowing with conduction block and temporal dispersion points to an acquired demyelinating neuropathy, most often CIDP, and argues against a uniform genetic disease. The sensory nerve action potentials are reduced or absent in axonal disease and relatively preserved in early demyelinating disease. [1][8]
Genetic testing is targeted by the nerve conduction study and the family history, and it has moved from single-gene sequential testing to next-generation sequencing panels. The first test for a child with uniform demyelinating slowing is a PMP22 duplication and deletion analysis by multiplex ligation-dependent probe amplification, which diagnoses CMT1A and HNPP. If that is negative, a hereditary neuropathy panel covering MPZ, GJB1, MFN2, and dozens of other genes is the next step. The Fernandez-Garcia study reminds us that not every child with a neuropathy phenotype has a genetic cause, and that a treatable acquired CIDP must be excluded before the child is labelled with an incurable hereditary disease. [5][9]
[6]The investigations for acquired neuropathy are different and treatable. The cerebrospinal fluid in CIDP shows albuminocytological dissociation, a raised protein with a normal cell count, similar to Guillain-Barre syndrome but persistent. The glycosylated haemoglobin and the oral glucose tolerance test screen for diabetes. The vitamin B12 and the copper levels screen for nutritional neuropathy. The autoimmune and paraprotein screens look for an underlying cause of CIDP. The chemotherapy history identifies the toxic culprit. MRI of the nerve roots may show hypertrophy in CMT1 or enhancement in CIDP. [8][10][11]
Management — Resuscitation
Most peripheral neuropathies in children are chronic and do not present as emergencies, but two scenarios demand immediate action. The first is a child on vincristine who develops severe acute weakness, which may unmask an undiagnosed Charcot-Marie-Tooth disease and can progress to irreversible quadriparesis and respiratory failure. The vincristine must be stopped immediately, the child assessed for foot deformity and family history, and the nerve conduction studies and genetic testing arranged urgently. The second is a child with rapidly progressive CIDP who develops respiratory or bulbar weakness, which is managed on the same intensive care ladder as Guillain-Barre syndrome with bedside spirometry and early intubation. [8][11]
The child with established Charcot-Marie-Tooth disease does not need resuscitation but does need anticipatory care. Assess the respiratory function if the vital capacity is reduced, which can occur in severe forms. Assess the scoliosis, which can accompany the foot deformity and the trunk weakness. Assess the pain, which affects a significant minority of children with CMT and is often neuropathic and undertreated. Assess the mobility and the fall risk, because ankle instability and foot drop make falls a daily burden and a source of secondary injury. [6]
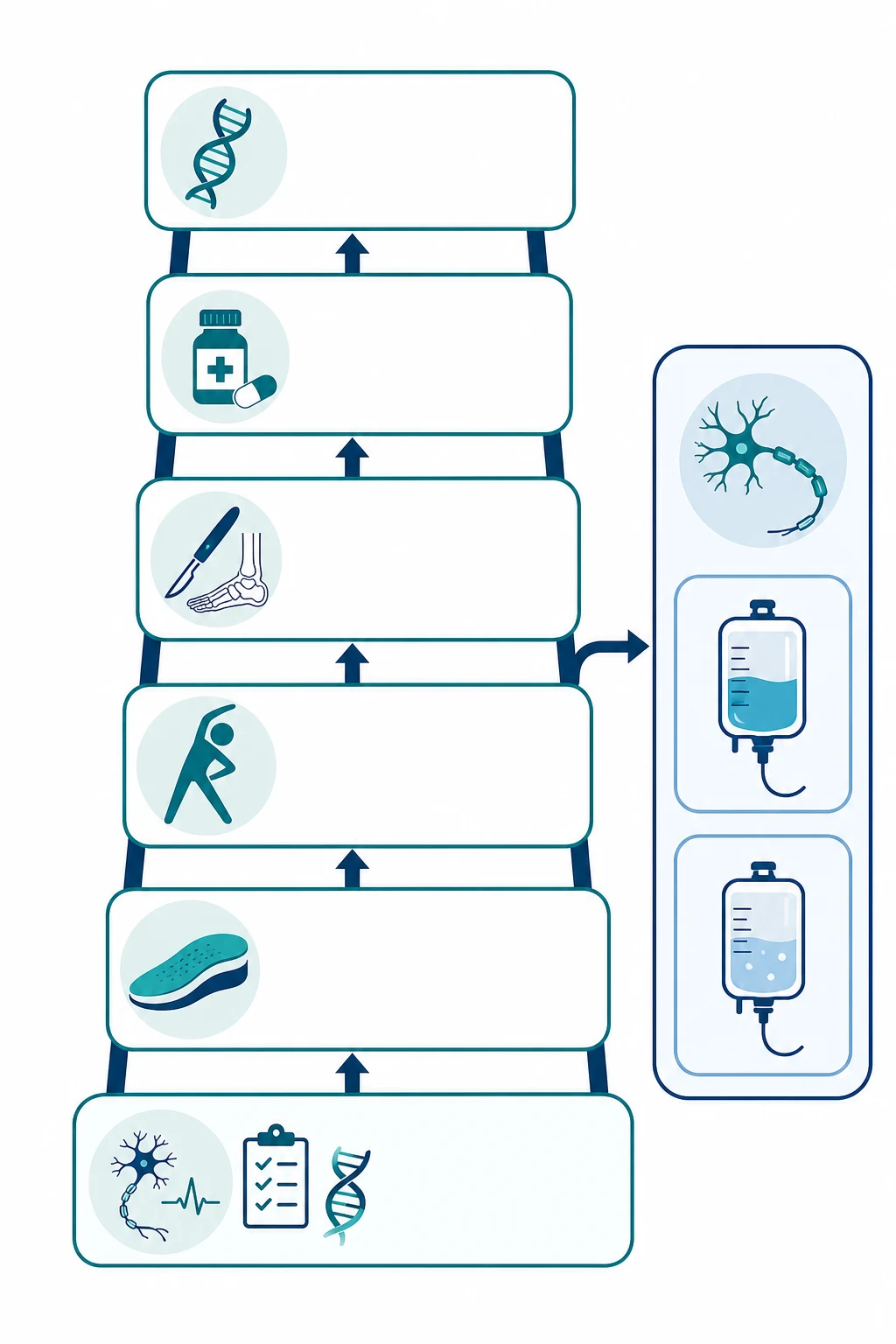
Management — Definitive & Stepwise
The definitive management of Charcot-Marie-Tooth disease is supportive, multidisciplinary, and lifelong, and the Yiu 2022 clinical practice guideline for the management of paediatric Charcot-Marie-Tooth disease is the evidence-based standard. No disease-modifying therapy has proven effective for CMT1A. The ascorbic acid trials, based on the rationale that reducing PMP22 overexpression might slow the disease, showed no benefit, and the progesterone antagonists and the neurotrophin 3 gene therapy remain experimental. The management is built around preserving mobility, preventing contractures, controlling pain, and supporting participation. [6]
Management of paediatric Charcot-Marie-Tooth disease
The orthotic management is the backbone of treatment and the intervention that preserves mobility the longest. An ankle-foot orthosis corrects the foot drop, stabilises the ankle, reduces falls, and improves the steppage gait, and the Yiu guideline recommends it for any child with functional impairment from dorsiflexion weakness. The orthosis should be custom-moulded, lightweight, and reviewed regularly as the child grows. Intrinsic hand splints and adaptive equipment address the later hand weakness. Podiatry manages the foot deformity, the calluses, and the nail care, and the footwear must accommodate the orthosis and the high arch. [6]
The management ladder, year by year
At diagnosis: Confirm with nerve conduction studies and genetic testing; establish the subtype and the inheritance pattern; refer to the multidisciplinary clinic
First years: Fit ankle-foot orthoses for foot drop; start physiotherapy for stretching and strengthening; arrange podiatry and footwear modification
Throughout childhood: Monitor gait, strength, sensation, and pain annually; treat neuropathic pain with gabapentin or pregabalin; screen for scoliosis and respiratory involvement
Adolescence: Consider orthopaedic surgery for fixed foot deformity, such as tendon transfer, calcaneal osteotomy, or triple arthrodesis; address hand weakness with occupational therapy
Transition: Hand over to adult neurology and rehabilitation; provide genetic counselling for reproductive planning; screen for pregnancy-related and anaesthetic risks
The definitive management of the acquired neuropathies is treatable and time-sensitive. Chronic inflammatory demyelinating polyradiculoneuropathy responds to intravenous immunoglobulin two grams per kilogram as a loading dose over two to five days, to corticosteroids, and to plasma exchange, and the choice depends on the severity, the side-effect profile, and the maintenance strategy. The Bunschoten 2019 Lancet Neurology review established that first-line therapy is intravenous immunoglobulin or corticosteroids, that plasma exchange is effective for refractory disease, and that maintenance therapy is needed because most patients relapse when treatment is withdrawn. Diabetic neuropathy is managed by optimising glycaemic control, because tight control reduces the progression in type 1 diabetes, and by neuropathic pain agents. Chemotherapy-induced neuropathy is managed by dose reduction or cessation of the offending agent, which must be balanced against the oncological benefit, and by rehabilitation. [8][10][11]
Specific Subtypes & Scenarios
CMT1A is the commonest and the prototype of the demyelinating family. It is caused by a duplication of the PMP22 gene on chromosome seventeen, it is inherited in an autosomal dominant pattern, and it presents in the first or second decade with pes cavus, foot drop, distal weakness, and areflexia. The nerve conduction velocity is uniformly slowed to under thirty-eight metres per second. The disease progresses slowly over decades, and most patients remain ambulant into late adulthood with orthotic support. [1][2]
CMT2A is the commonest axonal form, caused by mutations in MFN2 encoding mitofusin 2. It presents in childhood with a more severe phenotype than CMT1A in many cases, with distal wasting, weakness, and sensory loss, and a subset develops optic atrophy that can progress to visual impairment. The nerve conduction velocities are preserved above thirty-eight metres per second, and the compound muscle action potential amplitudes are reduced, reflecting axonal loss. The natural history study of Pipis 2020 showed that CMT2A causes significant disability and that the rate of progression varies with the specific MFN2 variant. [4]
CMTX1 is caused by variants in GJB1 encoding connexin 32, and it is inherited in an X-linked dominant pattern. Males are affected with a typical neuropathy phenotype, while carrier females can be mildly affected or asymptomatic. The Record 2023 natural history study confirmed that CMTX1 produces an intermediate neuropathy with features of both demyelination and axonal loss, that males are more severely affected than females, and that a minority of males develop transient central nervous system symptoms such as stroke-like episodes, which reflect the expression of connexin 32 in central myelin. [5]
Hereditary neuropathy with liability to pressure palsies is caused by a deletion of the PMP22 gene, the mirror image of the CMT1A duplication, and it was characterised by Chance in 1993. It presents not as a polyneuropathy but as recurrent, painless focal mononeuropathies at common compression sites, such as a peroneal palsy at the fibular head, an ulnar palsy at the elbow, or a radial palsy at the spiral groove. The episodes resolve over days to weeks, and the nerve conduction studies show focal slowing at compression sites with mild background demyelination. The management is avoidance of prolonged compression and protective padding, and most patients do well with conservative care. [3]
Chronic inflammatory demyelinating polyradiculoneuropathy is the acquired neuropathy that the paediatrician must not miss. It is defined by progressive symmetric weakness over more than eight weeks, which separates it from the monophasic Guillain-Barre syndrome that peaks within four weeks. It involves proximal and distal muscles, it produces a sensory loss and areflexia, and the nerve conduction studies show patchy non-uniform demyelination with conduction block. It responds to intravenous immunoglobulin, corticosteroids, and plasma exchange, and the response itself is diagnostic, because the hereditary neuropathies do not respond to immunotherapy. [8][9]
Diabetic peripheral neuropathy in youth was quantified by the Jaiswal SEARCH study, which found a prevalence of around seven percent in type 1 diabetes and around twenty-two percent in type 2 diabetes, driven by glycaemic control, diabetes duration, and lipid profiles. It presents as a distal symmetric sensory neuropathy with numbness, tingling, and pain in the feet, and it is managed by optimising glycaemic control and treating neuropathic pain. The rising prevalence of youth-onset type 2 diabetes makes this an increasingly important acquired neuropathy in adolescents. [10]
Chemotherapy-induced peripheral neuropathy is the commonest toxic neuropathy in paediatric oncology, and vincristine is the leading culprit. It produces a length-dependent sensorimotor neuropathy with foot drop, sensory loss, constipation, and jaw pain, and it is dose-dependent and often partially reversible. The critical scenario is the child with undiagnosed Charcot-Marie-Tooth disease who receives vincristine, because the drug can unmask the genetic disease and cause severe, irreversible neuropathy, as reviewed by Bjornard in the Lancet Child and Adolescent Health. The lesson is to screen for foot deformity and family history before vincristine and to use dose reduction or alternative agents in affected children. [11]
Complications & Pitfalls
The complications of Charcot-Marie-Tooth disease are the accumulated consequences of a lifelong, slowly progressive neuropathy. Falls and ankle sprains from foot drop and ankle instability are the most common daily burden, and they cause secondary injury and fear of walking. Foot deformity progresses from flexible to fixed, and the calluses, the pressure areas, and the occasional painless ulcer from sensory loss add to the morbidity. Hand weakness limits fine motor tasks, writing, and independence in dressing and eating. Scoliosis accompanies the trunk weakness in some children. Neuropathic pain affects a significant minority and is undertreated. Fatigue is pervasive and limits school and social participation. [1][6]
The classic pitfalls are the ones that examiners reward a candidate for naming. The first is labelling a treatable CIDP as an incurable Charcot-Marie-Tooth disease, which deprives the child of immunotherapy. The Fernandez-Garcia study showed that this happens in real practice, and the guard is the nerve conduction study, which shows patchy non-uniform slowing in CIDP versus uniform slowing in CMT1. The second is failing to take the family history, which is the single most powerful diagnostic tool in the inherited neuropathies and which identifies the inheritance pattern and guides the genetic testing. The third is performing a single-gene test in the wrong order, such as testing MFN2 before PMP22 duplication in a child with uniform demyelinating slowing. The fourth is giving vincristine to a child with undiagnosed CMT, which can cause irreversible neuropathy. The fifth is under-treating the pain and the foot deformity, which are the lived burden of the disease. [1][9][11]
The anaesthetic and surgical pitfalls matter for children who need procedures. Charcot-Marie-Tooth disease increases the risk of positioning injuries and pressure palsies under anaesthesia, because the nerves are already vulnerable to compression, and succinylcholine and volatile agents should be used with caution because of the theoretical risk of malignant hyperthermia-like reactions, although modern evidence suggests that total intravenous anaesthesia is safe. The HNPP precaution is the same but more acute, because the deleted PMP22 gene makes the nerves exquisitely sensitive to compression, and careful padding and positioning are essential. [1][3]
Prognosis & Disposition
The prognosis of Charcot-Marie-Tooth disease is one of slow progression over decades with preservation of ambulation, and that is the message to give the family. Most children with CMT1A remain ambulant into late adulthood with orthotic support, and the life expectancy is normal. The strongest predictor of the disease severity is the subtype, with CMT2A and the recessive forms causing more disability than CMT1A, and the Fridman natural history study of the Inherited Neuropathies Consortium confirmed that the disease burden is driven by foot and hand weakness, sensory loss, and pain rather than by life-threatening complications. [1][7]
The prognosis of chronic inflammatory demyelinating polyradiculoneuropathy is more favourable in children than in adults, with most children responding to intravenous immunoglobulin and achieving remission. The Bunschoten review established that first-line therapy with intravenous immunoglobulin or corticosteroids is effective, that maintenance therapy is often needed to prevent relapse, and that the long-term outcome in children is good with early and sustained treatment. The acquired neuropathies from diabetes and chemotherapy are more variable, and they depend on the control of the underlying disease and the cessation of the offending agent. [8][10][11]
functional trajectory
Consider foot surgery for fixed deformity; address hand weakness; provide genetic counselling
Disposition follows the subtype and the treatability. A child with suspected CMT is managed in the outpatient paediatric neurology and rehabilitation clinic, with orthotic and physiotherapy input and annual review. A child with suspected CIDP is referred urgently to paediatric neurology for nerve conduction studies and immunotherapy, because the disease is treatable and the window for treatment is early. A child with chemotherapy-induced neuropathy is managed jointly with the oncology team, with dose reduction or cessation of the culprit and with rehabilitation. A child with HNPP is managed with compression avoidance and protective strategies. [6][8]
Special Populations
The young child often presents with delayed motor milestones, clumsy gait, or frequent falls rather than reported weakness, so a low threshold for examining the feet, the reflexes, and the gait is essential. Pes cavus that is present from the first steps, toe-walking, and ankle spraining in a toddler are the signs that bring the family to the doctor, and the diagnostic clue is the combination of the foot deformity with reduced reflexes and a family history. The child with an existing neurodisability is at particular risk of having the neuropathy attributed to the primary condition, and any new weakness or sensory loss needs the same workup. [1]
The adolescent faces the dual burden of the disease and the transition to adult care. The foot deformity may require orthopaedic surgery, the hand weakness may limit career choices, and the genetic counselling for reproductive planning is a key developmental task. The adolescent girl with a family history of CMT needs counselling about the inheritance, the risk to future children, and the pregnancy-related risks, which include a possible deterioration in the third trimester and the postpartum period. The transition to adult neurology should be planned and structured, with a handover of the genetic diagnosis and the management plan. [6]
The child from a refugee or migrant family may carry a recessive form of CMT that is rare in the host population, and consanguinity increases the risk, so a three-generation pedigree and an interpreter are essential. The Aboriginal and Torres Strait Islander child and the child from a remote setting may present late after years of unrecognised foot deformity, so the threshold for referral is low and telehealth support is central. The child receiving chemotherapy for cancer is the population at risk of vincristine-induced neuropathy, and the vincristine precaution in suspected CMT is a cross-cutting safety rule that protects every child with foot deformity who enters the oncology pathway. [11]
Evidence, Guidelines & Regional Differences
The diagnostic and genetic framework rests on the discovery by Lupski in 1991 that CMT1A is caused by a PMP22 duplication, and on the parallel discovery by Chance in 1993 that HNPP is caused by a deletion of the same gene. These two papers established the concept of gene dosage in the peripheral nerve and remain the foundation of the genetic classification. The comprehensive Pareyson 2009 Lancet Neurology review set the modern synthesis of the diagnosis, the natural history, and the management of Charcot-Marie-Tooth disease, and it remains the standard overview. [1][2][3]
The natural history evidence rests on the Inherited Neuropathies Consortium cohort, reported by Fridman in 2015, which characterised the subtypes and the disease burden in over a thousand patients. The Pipis 2020 study established the natural history of CMT2A as the commonest axonal form, and the Record 2023 study established the genotype-phenotype correlation for CMTX1. These studies are the evidence base for the prognosis and the counselling that the clinician gives the family. [4][5][7]
The management evidence is anchored by the Yiu 2022 clinical practice guideline for the management of paediatric Charcot-Marie-Tooth disease, published in the Journal of Neurology Neurosurgery and Psychiatry, which provides evidence-based recommendations on assessment, orthotics, surgery, and rehabilitation for children. The ascorbic acid trials for CMT1A showed no benefit, and no disease-modifying therapy has yet proven effective, so the guideline endorses supportive multidisciplinary care. [6]
Ascorbic acid for CMT1A (Pareyson 2011)
Double-blind randomised placebo-controlled trials of ascorbic acid at varying doses in adults and children with CMT1A, testing the rationale that reducing PMP22 overexpression might slow demyelination.
Key finding
No benefit was demonstrated for ascorbic acid over placebo on the primary outcome of the Charcot-Marie-Tooth Neuropathy Score at twelve months.
Practice change
No disease-modifying therapy has proven effective for CMT1A. Management remains supportive with orthotics, physiotherapy, surgery, and pain control, as endorsed by the Yiu 2022 paediatric guideline.
The acquired neuropathy evidence is anchored by the Bunschoten 2019 Lancet Neurology review of CIDP, which set the modern standard for the diagnosis and the treatment, and by the Jaiswal 2017 SEARCH study, which quantified the prevalence of diabetic neuropathy in youth. The Fernandez-Garcia 2021 study is the reminder that genetic neuropathies can present with CIDP-like features in childhood, and that the nerve conduction study pattern and the response to immunotherapy are the guards against mislabelling a treatable acquired neuropathy as an incurable hereditary disease. The Bjornard 2018 review established the framework for chemotherapy-induced peripheral neuropathy in children and the vincristine precaution in suspected CMT. [8][9][10][11]
Controversies remain. The place of next-generation sequencing panels versus sequential single-gene testing, the role of neurotrophin 3 gene therapy and progesterone antagonists for CMT1A, the threshold for orthopaedic surgery in the growing foot, and the optimal maintenance strategy for paediatric CIDP are all active areas. The evidence for new treatments is growing, but none has yet reached clinical practice, and the management of the inherited neuropathies remains supportive. [6][8]
Regional differences are small in principle because the genetic and the nerve conduction frameworks are international, but access matters in practice. Next-generation sequencing panels, paediatric neurology, and multidisciplinary Charcot-Marie-Tooth clinics are available in tertiary centres in ANZ, the UK, and North America, but the threshold for referral from rural and remote settings is higher, and telehealth support is central. The vincristine precaution in suspected CMT is consistent across the RACP, RCPCH, ABP, and RCPSC contexts, and the Yiu 2022 guideline is broadly endorsed internationally. [6][11]
Exam Pearls
Charcot-Marie-Tooth disease is the commonest inherited neuromuscular disorder, with a prevalence of around one in two thousand five hundred, and CMT1A from a PMP22 duplication accounts for around half of all cases. The phenotype is distal wasting, pes cavus, foot drop, steppage gait, areflexia, and large-fibre sensory loss, and the family history is the most powerful diagnostic tool. [1][2]
The nerve conduction velocity of thirty-eight metres per second is the number that sorts the disease. Uniform slowing under thirty-eight across all nerves is CMT1 demyelinating, and it points to a PMP22 duplication test. A velocity above thirty-eight with reduced amplitudes is CMT2 axonal, and it points to an MFN2 test. Patchy, non-uniform slowing with conduction block is an acquired demyelinating neuropathy, most often CIDP. [1][8]
Chronic inflammatory demyelinating polyradiculoneuropathy is defined by progression over more than eight weeks, and it responds to intravenous immunoglobulin two grams per kilogram, to corticosteroids, and to plasma exchange. It must not be confused with Guillain-Barre syndrome, which peaks within four weeks, or with Charcot-Marie-Tooth disease, which progresses over years and does not respond to immunotherapy. The acquired neuropathies are treatable, and the hereditary neuropathies are managed supportively. [8]
Vincristine can unmask or severely worsen Charcot-Marie-Tooth disease, and any child about to receive vincristine who has pes cavus, a family history of neuropathy, or unexplained weakness must be screened before the first dose. Hereditary neuropathy with liability to pressure palsies, from a PMP22 deletion, presents with recurrent painless focal mononeuropathies rather than a polyneuropathy. The two rules that protect the child are to examine the feet and the family at every consultation and to run the nerve conduction study before the genetic test. [1][3][11]
References
- [1]Pareyson D, Marchesi C Diagnosis, natural history, and management of Charcot-Marie-Tooth disease. Lancet Neurol, 2009.PMID 19539237
- [2]Lupski JR, de Oca-Luna RM, Slaugenhaupt S, et al DNA duplication associated with Charcot-Marie-Tooth disease type 1A. Cell, 1991.PMID 1677316
- [3]Chance PF, Alderson MK, Leppig KA, et al DNA deletion associated with hereditary neuropathy with liability to pressure palsies. Cell, 1993.PMID 8422677
- [4]Pipis M, Feely SME, Polke JM, et al Natural history of Charcot-Marie-Tooth disease type 2A: a large international multicentre study. Brain, 2020.PMID 33415332
- [5]Record CJ, Skorupinska M, Laura M, et al Genetic analysis and natural history of Charcot-Marie-Tooth disease CMTX1 due to GJB1 variants. Brain, 2023.PMID 37284795
- [6]Yiu EM, Bray P, Baets J, et al Clinical practice guideline for the management of paediatric Charcot-Marie-Tooth disease. J Neurol Neurosurg Psychiatry, 2022.PMID 35140138
- [7]Fridman V, Bundy B, Reilly MM, et al CMT subtypes and disease burden in patients enrolled in the Inherited Neuropathies Consortium natural history study: a cross-sectional analysis. J Neurol Neurosurg Psychiatry, 2015.PMID 25430934
- [8]Bunschoten C, Jacobs BC, Van den Bergh PYK, et al Progress in diagnosis and treatment of chronic inflammatory demyelinating polyradiculoneuropathy. Lancet Neurol, 2019.PMID 31076244
- [9]Fernandez-Garcia MA, Stettner GM, Kinali M, et al Genetic neuropathies presenting with CIDP-like features in childhood. Neuromuscul Disord, 2021.PMID 33386210
- [10]Jaiswal M, Divers J, Dabelea D, et al Prevalence of and risk factors for diabetic peripheral neuropathy in youth with type 1 and type 2 diabetes. Diabetes Care, 2017.PMID 28674076
- [11]Bjornard KL, Gilchrist LS, Inaba H, et al Peripheral neuropathy in children and adolescents treated for cancer. Lancet Child Adolesc Health, 2018.PMID 30236383