Paeds · neurology-neurodisability-and-neuromuscular
Spinal muscular atrophy
Also known as SMA · Werdnig-Hoffmann disease · Type 1 SMA · Dubowitz disease · Type 2 SMA · Kugelberg-Welander disease · Type 3 SMA · Proximal spinal muscular atrophy
Fellowship guide to spinal muscular atrophy in children. Covers the autosomal recessive SMN1 deletion on chromosome 5q13 with the inverse relationship to SMN2 copy number and the exon 7 splicing defect that depletes survival motor neuron protein and causes anterior horn cell degeneration, the five SMA types from the lethal type 0 and the Werdnig-Hoffmann type 1 to the milder type 3 and adult type 4, the clinical picture of severe symmetric proximal hypotonia and areflexia with tongue fasciculations and paradoxical breathing but preserved intellect, the genetic diagnosis by SMN1 deletion testing and SMN2 copy number, the three disease-modifying therapies nusinersen the intrathecal antisense oligonucleotide risdiplam the oral small molecule and onasemnogene abeparvovec the AAV9 gene therapy, the ENDEAR CHERISH FIREFISH SUNFISH NURTURE and STR1VE trial evidence, newborn screening and presymptomatic treatment, and the multidisciplinary respiratory nutritional and orthopaedic care.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
Overview & Definition
A floppy infant whose limbs lie limp and splayed, whose reflexes have vanished, but whose eyes track and follow and whose face is bright and alert, is presenting one of the most important diagnoses in paediatric neurology. Spinal muscular atrophy is an autosomal recessive disorder in which loss of the survival motor neuron 1 gene, or SMN1, on chromosome 5q13 depletes the survival motor neuron protein and causes progressive degeneration of the anterior horn cells of the spinal cord and the motor nuclei of the brainstem. The result is severe, symmetric, proximal muscle weakness and atrophy, with the legs more affected than the arms and the trunk and breathing muscles progressively involved. It is the commonest genetic cause of infant death. [1]
The reason this disease is now central to the paediatric exam, and to clinical practice, is that the prognosis has been transformed. For decades type 1 spinal muscular atrophy was a relentless disease in which an infant never sat, lost the ability to swallow and breathe, and died before two years of age. Three disease-modifying therapies have changed that trajectory. Nusinersen, an antisense oligonucleotide given into the cerebrospinal fluid, risdiplam, an oral small molecule, and onasemnogene abeparvovec, a single-infusion gene therapy, each raise the functional survival motor neuron protein and preserve motor neurons. The cardinal lesson of the past decade is that motor neurons lost before treatment do not grow back, so the earlier the therapy the better the outcome, and presymptomatic treatment begun through newborn screening is now the standard of care. [9][11]
The framing concept for the paediatrician is the floppy infant, and within it the spinal cord cause of hypotonia. The same bedside picture hides cerebral causes such as hypoxic injury, muscle disease such as congenital myopathy, and neuromuscular junction disorders such as congenital myasthenia. The discriminating clue is the pattern of the weakness and the preservation of the intellect. Spinal muscular atrophy gives symmetric, proximal, areflexic weakness with a bright and engaged child, and the diagnosis is confirmed by a single genetic test for the homozygous SMN1 deletion. [1]
Classification
Spinal muscular atrophy is sorted by severity, and the severity sorts by the SMN2 copy number, so the two are read together. The clinical classification spans five types, from the lethal type 0 to the mild adult type 4, and it predicts the motor milestone a child can reach and the lifespan they can expect without treatment. [1]
Type 1, also called Werdnig-Hoffmann disease, is the commonest and most severe form that survives the neonatal period, accounting for around half of all cases. Onset is before six months of age. The infant never achieves the ability to sit independently, develops profound weakness of the legs then the arms and the breathing muscles, and without treatment dies from respiratory failure before two years of age. Type 0 is the rarest and most severe form, with onset before birth, severe weakness and respiratory failure at delivery, joint contractures, and death within weeks. [3]
Type 2, or intermediate spinal muscular atrophy, has onset between six and eighteen months. The child achieves the ability to sit independently but never walks. Most survive into adulthood with appropriate respiratory and orthopaedic care. Type 3, or Kugelberg-Welander disease, has onset after eighteen months, and the child walks independently at some stage, though many later lose that ability. Lifespan is near normal. Type 4 is the rare adult-onset form with mild proximal weakness and a normal lifespan. [1]
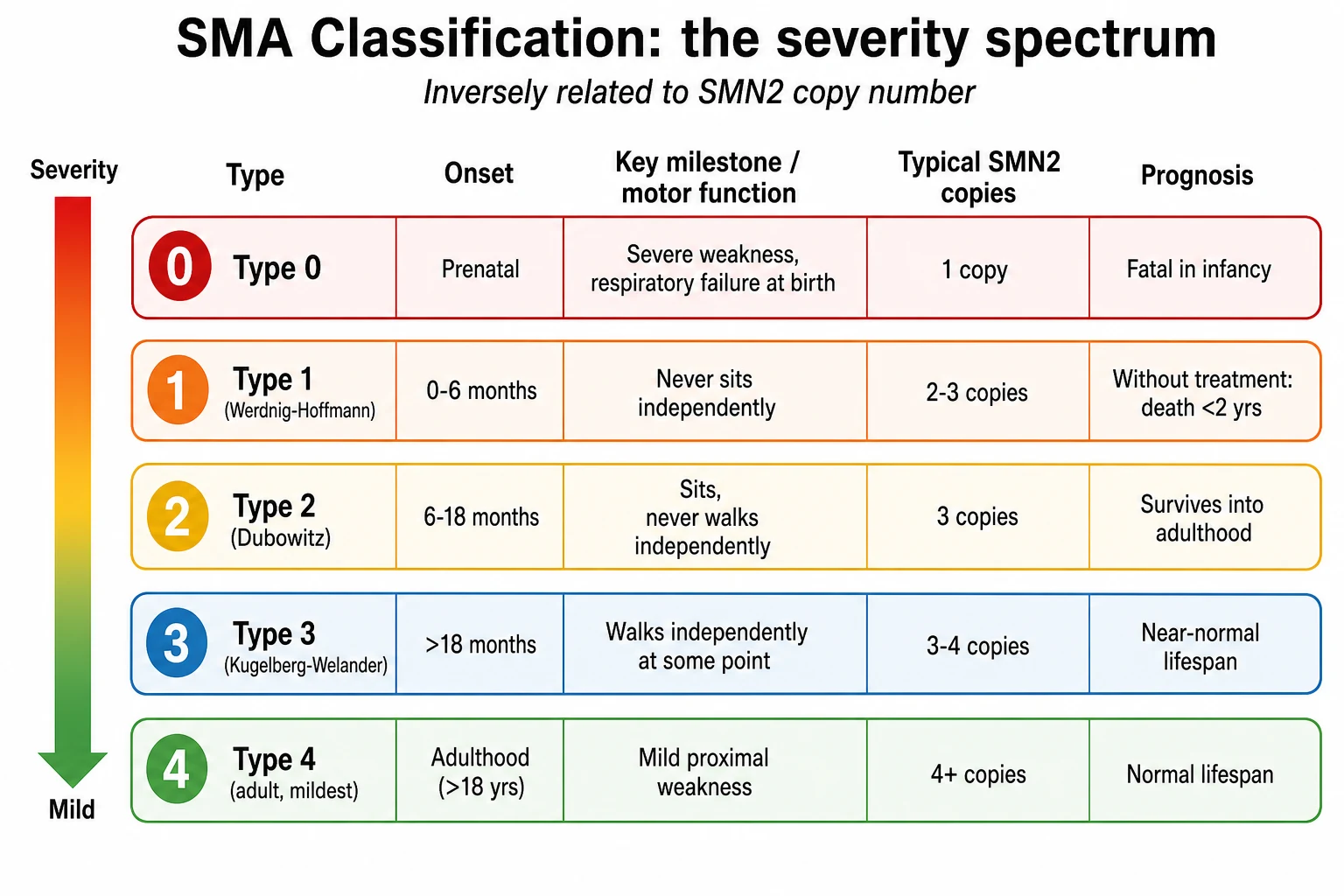
The genetic modifier that sets where a child sits on this spectrum is the SMN2 copy number. SMN2 is a nearly identical backup gene that sits next to SMN1 on chromosome 5. Most of its transcripts skip exon 7 and produce a short, unstable protein, so each copy makes only a small amount of functional survival motor neuron protein. A child with one SMN2 copy usually has type 0 or severe type 1 disease, two copies give classic type 1, three copies give type 2 or type 3, and four or more copies give the milder type 3 or type 4 disease. The copy number is the single best predictor of severity, though it is not a perfect one, and a few exceptional cases break the rule. [1]
Epidemiology & Risk Factors
Spinal muscular atrophy is one of the commonest autosomal recessive disorders and the leading genetic cause of infant mortality. The disease frequency sits around one in ten thousand live births, and the carrier frequency of a single SMN1 deletion in the general population is around one in fifty to one in sixty. Because it is autosomal recessive, each child of two carrier parents has a one in four chance of being affected, a one in two chance of being a carrier, and a one in four chance of inheriting neither deletion. [1]
The disease is found in every population, with carrier frequencies that vary a little by ancestry but remain high everywhere, which is why population carrier screening and newborn screening are now international priorities. Around one to two percent of cases arise from a de novo mutation rather than from two inherited alleles, and a small number come from a mild SMN1 point mutation on one chromosome paired with a deletion on the other. [1]
There is no environmental risk factor and no trigger. The disease is set at conception by the inheritance of two faulty SMN1 alleles, and the whole of the subsequent clinical course is shaped by the SMN2 copy number and the age at which treatment begins. This pure genetic determinism is what makes newborn screening so powerful, because identifying the genotype at birth allows treatment before the motor neurons are lost. [12]
Pathophysiology
The single sentence to hold is that the child inherits two broken copies of the SMN1 gene, the backup SMN2 gene cannot make enough of the survival motor neuron protein, and without that protein the motor neurons die. [1]
The survival motor neuron protein is a housekeeping protein that every cell needs to assemble the small nuclear ribonucleoprotein complexes that process messenger RNA. It is essential everywhere, but it is needed most by the motor neurons, which are the largest cells with the longest axons and the greatest demand for RNA processing. When the protein falls below a critical threshold, the motor neurons of the anterior horn of the spinal cord and the motor nuclei of the brainstem degenerate, their axons withdraw from the muscle, and the denervated muscle atrophies. The sensory neurons and the intellect are spared, which is why sensation is preserved and the child remains bright. [1]
The key to the disease and to all three treatments lies in the difference between SMN1 and SMN2. The two genes are nearly identical, but a single silent change in SMN2 disrupts a splicing enhancer so that most of its messenger RNA skips exon 7. The resulting protein lacks a critical piece, is unstable, and is rapidly degraded, so each SMN2 copy produces only about ten to fifteen percent of the functional protein that an SMN1 copy makes. This is why more SMN2 copies buffer the disease, and why the SMN2 copy number predicts the type. [1]
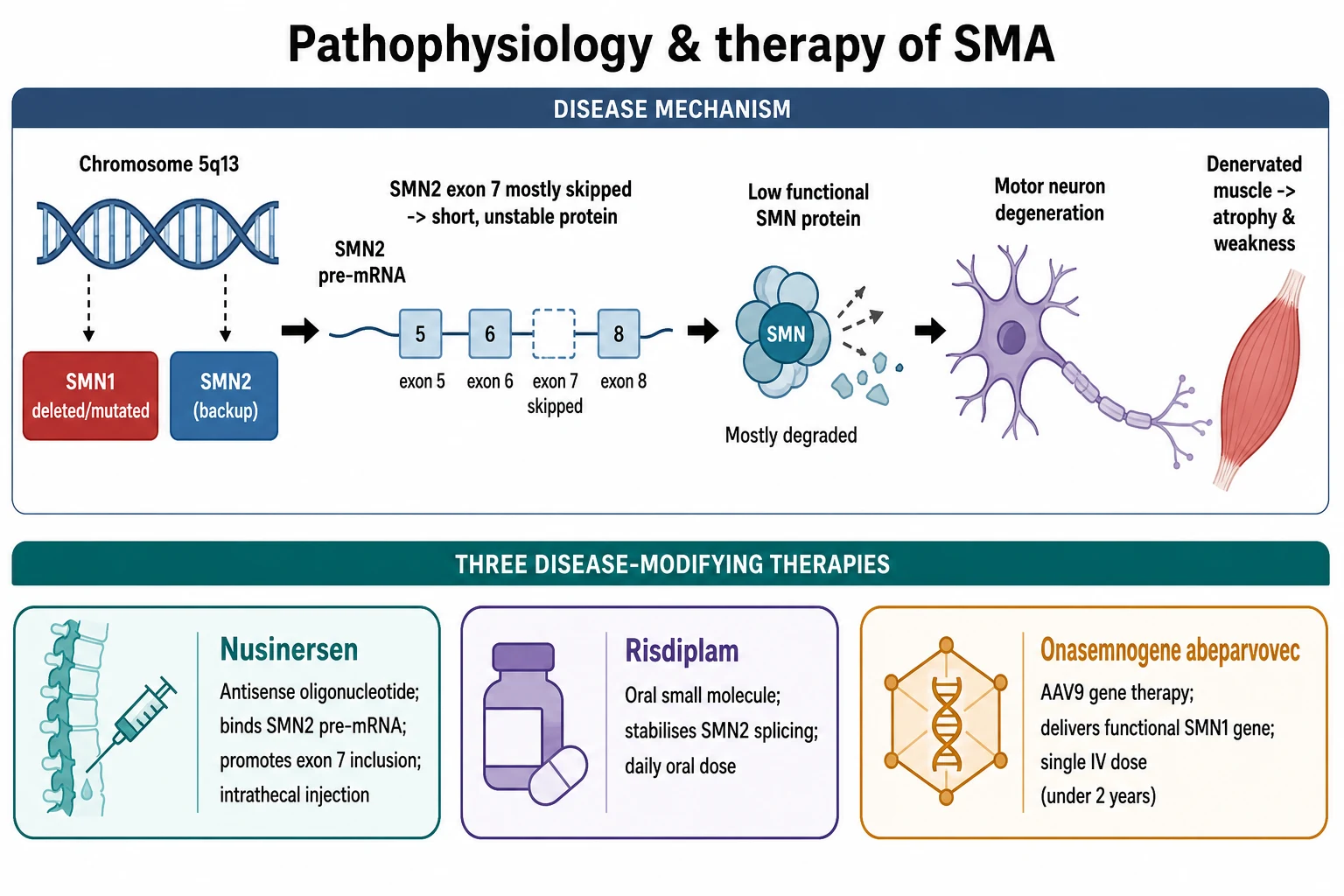
The three disease-modifying therapies each attack the same protein deficit by a different route. Nusinersen is an antisense oligonucleotide that binds a silencing sequence in SMN2 and forces the splicing machinery to include exon 7, raising the amount of functional protein. Risdiplam is a small molecule taken by mouth every day that stabilises the same splicing step. Onasemnogene abeparvovec is a gene therapy that uses an adeno-associated virus type 9, or AAV9, vector to deliver a working copy of the SMN1 gene into the motor neurons in a single intravenous infusion, so the cell makes its own full-length protein. All three work only if motor neurons remain to be saved, which is why timing is everything. [4][6][8]
Clinical Presentation
The presentation bends to the type, and the type bends to the SMN2 copy number, but the core picture is the same: symmetric, proximal weakness with areflexia in a bright and engaged child. The paediatrician who holds this picture in mind will rarely miss the diagnosis. [1]
Type 1, the Werdnig-Hoffmann form, presents in the first six months. The mother often reports reduced fetal movements in retrospect. The infant is floppy, lies in a frog-leg posture with the limbs abducted and externally rotated, and cannot lift the limbs against gravity. The legs are weaker than the arms, so the severe limb weakness with relatively preserved facial and eye movement is a defining pattern. The deep tendon reflexes are absent. The tongue shows fasciculations, a fine rippling movement that is one of the most specific signs in paediatric neurology. The intercostal muscles are weak while the diaphragm is relatively spared, so the chest sinks in as the abdomen balloons out with each breath, the paradoxical or belly breathing that produces the bell-shaped chest, and a weak cough predicts respiratory failure. [3]
The intellect is always spared. A profoundly weak, floppy infant who fixes and follows, smiles, and tracks the examiner is the signature picture of type 1 disease, and the contrast between the alert face and the limp body is the single most valuable bedside clue. Swallowing becomes weak, secretions pool, and poor weight gain and aspiration follow. Without treatment the weakness progresses to respiratory failure, and death occurs before two years of age, most often from pneumonia and ventilatory failure. [3]
Type 2 presents between six and eighteen months, usually after the child has learned to sit but before or as they attempt to stand. The weakness is again proximal and symmetric, the reflexes are absent or reduced, and a fine tremor of the outstretched hands is common and characteristic. Scoliosis, hip dislocation, and joint contractures develop over time and dominate the long-term management. Type 3 presents after eighteen months in a child who has walked, with a waddling gait, difficulty climbing stairs, and progressive proximal weakness. The course is slower, lifespan is near normal, and disability accrues over years to decades. [1]
MOTOR
Differential Diagnosis
The differential is the floppy infant, and the task is to separate the spinal cord cause from the cerebral, the muscle, and the neuromuscular junction causes. The discriminating features are the pattern of the weakness, the reflexes, the presence of fasciculations, and the level of the intellect and the social interaction. [1]
The first fork is between a spinal cause and a cerebral cause of hypotonia. A cerebral cause, such as hypoxic-ischaemic encephalopathy, chromosomal disorder, or metabolic brain disease, tends to give diffuse hypotonia with the weakness less prominent than the tone change, often with brisk reflexes, seizures, developmental delay across all domains, and an infant who is not socially engaged. Spinal muscular atrophy gives pure motor weakness with absent reflexes, a bright and engaged child, and no sensory or cognitive loss. The spinal muscular atrophy infant is weak but socially normal, which is the discriminating clue. [1]
The second fork is between spinal muscular atrophy and other lower motor neuron or neuromuscular disorders. Congenital myopathies such as central core disease and congenital muscular dystrophies such as merosin-deficient disease give hypotonia and weakness from birth but usually with facial involvement, calf hypertrophy, or raised creatine kinase. Congenital myasthenic syndromes give fatigable ptosis and ophthalmoplegia. Guillain-Barre syndrome is acute and acquired, not chronic and congenital. Spinal cord injury from birth trauma gives an asymmetric and often improving picture. The tongue fasciculations, the symmetric proximal pattern, the areflexia, and the preserved intellect together point to spinal muscular atrophy, and the diagnosis is then confirmed by the SMN1 deletion test. [1]
The remaining mimics each carry a pitfall. Prader-Willi syndrome gives severe neonatal hypotonia with poor feeding but later hyperphagia and hypogonadism, and the methylation test settles it. Spinal cord birth injury and transverse myelitis are acute rather than chronic. The rare X-linked myotubular myopathy gives severe neonatal weakness with a macrocephalic, hypotonic infant. In every case the combination of the symmetric proximal areflexic weakness, the tongue fasciculations, the spared intellect, and a positive SMN1 deletion test is diagnostic. [1]
Clinical & Bedside Assessment
The bedside assessment confirms the pattern and quantifies the threat, because respiratory failure is the immediate danger in the severe infantile forms. Watch the child breathe first. Paradoxical breathing, where the chest retracts and the abdomen protrudes on inspiration, signals intercostal muscle weakness, and a weak cough with pooled secretions signals bulbar weakness and impending ventilatory failure. Measure the oxygen saturation, but remember that carbon dioxide retention precedes hypoxaemia in neuromuscular respiratory failure, so a normal oxygen level does not exclude ventilatory failure. [2]
The focused examination looks for the pattern that confirms the diagnosis and for the features that argue for a mimic. Lay the infant supine and note the frog-leg posture and the head lag. Test the limbs against gravity and confirm the symmetric proximal weakness that is worse in the legs. Check the deep tendon reflexes and confirm that they are absent. Look at the tongue at rest for the fine fasciculations, and watch the face and eyes to confirm that social interaction, fixing and following, and facial strength are preserved. Examine the spine for scoliosis and the hips for dislocation in the older child. Take a careful family history of neuromuscular disease or unexplained infant death and a birth history of reduced fetal movements. [1]
The motor examination is best quantified with a standard scale, because the baseline and the response to treatment are tracked numerically. The Children's Hospital of Philadelphia Infant Test of Neuromuscular Disorders, or CHOP-INTEND, scores motor function in the severely affected infant, and the Hammersmith Infant Neurological Examination and the Hammersmith Functional Rating Scale track infants and toddlers. The older ambulant child is scored with the six-minute walk test and the Hammersmith Functional Motor Scale Expanded. The value of the score is that it sets the baseline before treatment and measures the response, because the examiners and the families need to see the numbers. [1]
In ANZ practice, spinal muscular atrophy is part of the newborn bloodspot screen, so most new diagnoses are presymptomatic infants referred from screening rather than the decompensating infant on the ward. The peripheral or rural clinician who meets a floppy infant confirms the airway and breathing, takes the genetic test, and arranges urgent retrieval to a tertiary paediatric neuromuscular centre where the treatment decision is made, because the window to preserve motor neurons closes with every week of delay. [12]
Investigations
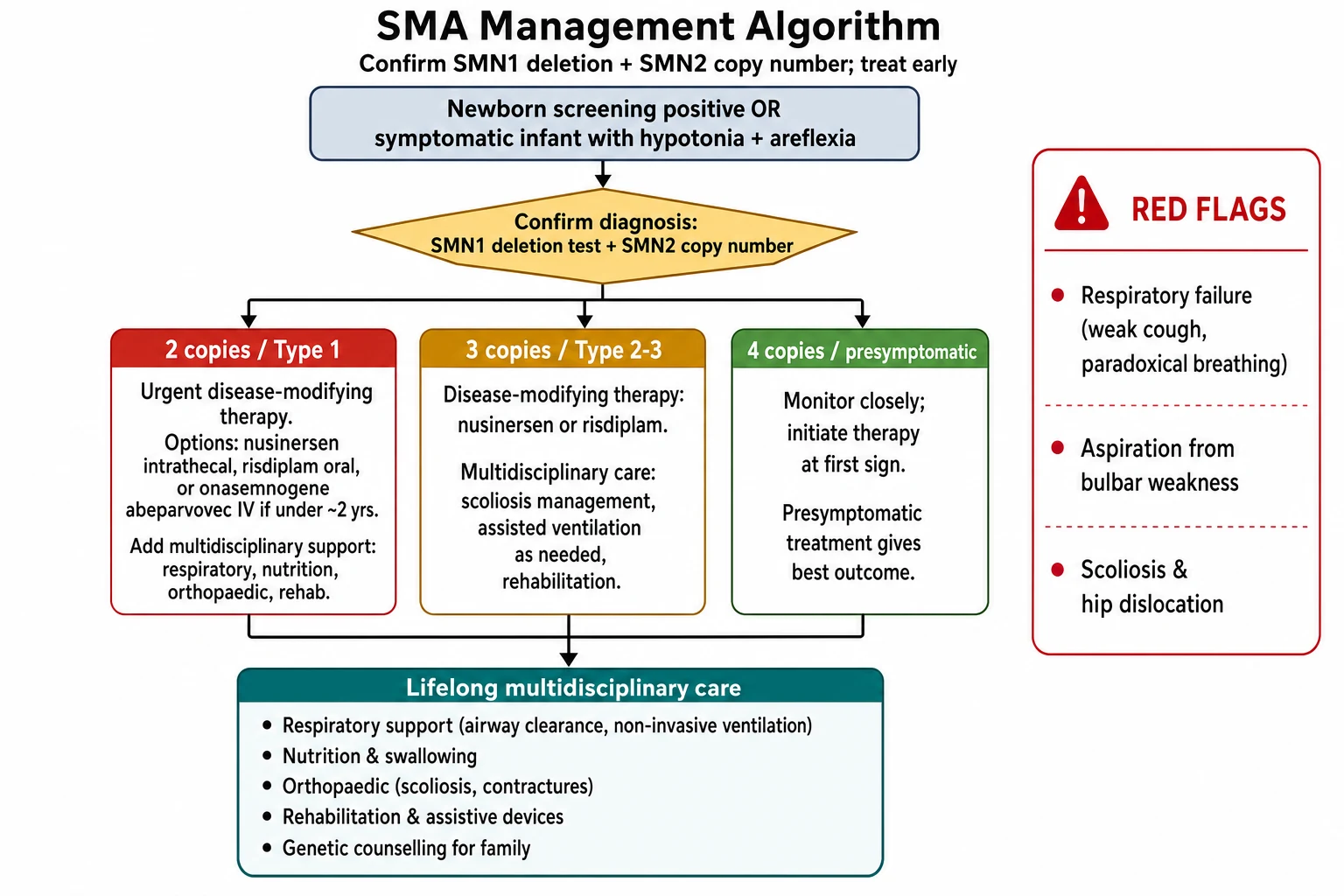
The diagnosis of spinal muscular atrophy is genetic and is made by a single blood test, and the era of muscle biopsy and electromyography as first-line tools is over for the typical case. Send a targeted mutation analysis or multiplex ligation-dependent probe amplification for the homozygous deletion of SMN1 exon 7, which is present in more than ninety-five percent of affected individuals. A positive result in the right clinical picture confirms the diagnosis without further testing. [1]
The next test, and the one that sets the type and the treatment plan, is the SMN2 copy number. Quantitative testing gives the number of SMN2 copies, and that number predicts the likely severity and drives the urgency and the choice of therapy. A presymptomatic infant found on newborn screening with two SMN2 copies has classic type 1 risk and is treated urgently, while four copies allow a watch-and-treat approach. The SMN2 copy number is also the number the family remembers and asks about, so it must be explained clearly. [10]
The remaining investigations support the clinical picture and exclude mimics when the genetic picture is atypical. The creatine kinase is normal or only mildly raised, which helps separate spinal muscular atrophy from a muscular dystrophy. Electromyography shows denervation with fibrillation and large motor units, but it is rarely needed when the deletion test is positive. Muscle biopsy shows grouped atrophy of type 1 and type 2 fibres, again a second-line test. The rare child with a single SMN1 deletion and an atypical picture needs sequencing of the remaining SMN1 allele for a point mutation. Pulmonary function testing and a sleep study quantify the respiratory reserve and guide the timing of respiratory support, and a swallowing assessment guides nutritional management. [1][2]
[2]Management — Resuscitation
Resuscitation protects the airway and the breathing in the severely affected infant while the genetic diagnosis is confirmed and the treatment is planned. A type 1 infant with paradoxical breathing, a weak cough, and pooled secretions is heading toward respiratory failure, and the carbon dioxide can rise while the oxygen saturation still reads normally. Involve the paediatric intensive care team early, and the threshold to support the breathing is driven by the respiratory effort, the cough, and the carbon dioxide, not by the oxygen saturation alone. [2]
Airway protection and secretion management are the immediate priorities. Suction the secretions, position the child, and assess the swallow, because aspiration from bulbar weakness is a leading cause of pneumonia and death in untreated type 1 disease. Where the cough is weak and the secretions are unmanageable, non-invasive ventilation and cough-assist devices are introduced early and reduce the hospitalisation rate. A nasogastric or gastrostomy tube secures nutrition and hydration when the swallow is unsafe, and the decision about long-term gastrostomy is part of the multidisciplinary plan, not an emergency measure. [2]
The genetic confirmation is sent as soon as the diagnosis is suspected, because the whole of the modern management depends on it. The SMN1 deletion test and the SMN2 copy number are ordered together, and the paediatric neuromuscular service is contacted while the result is awaited, because the disease-modifying therapy should begin as soon as the diagnosis is confirmed and not be delayed for the older, slower workup. [1]
Management — Definitive & Stepwise
The definitive treatment is disease-modifying therapy, and the principle that governs every decision is to start early, because motor neurons that have died do not recover. Three therapies are now licensed, and the choice turns on the age, the SMN2 copy number, the symptom status, and the local availability and funding, which varies by region. [4]
Nusinersen is an antisense oligonucleotide that binds the intronic splicing silencer in SMN2 and forces the inclusion of exon 7, raising the amount of functional survival motor neuron protein. It is given by intrathecal injection, because it does not cross the blood-brain barrier, as a loading course on days one, fifteen, twenty-nine, and sixty-four, then as a maintenance dose every four months, often lifelong. The ENDEAR trial in infantile-onset disease showed that nusinersen improved motor function and reduced the risk of death or permanent ventilation compared with sham, and the CHERISH trial in later-onset disease showed a significant gain in motor function. The NURTURE study of presymptomatic infants given nusinersen showed the best outcomes of all, with the great majority sitting and most walking, results impossible in untreated type 1 disease. [4][5][9]
Disease-modifying therapies for spinal muscular atrophy (paediatric)
Risdiplam is a small molecule taken by mouth every day that stabilises the SMN2 splicing machinery and includes exon 7. Its great advantage over nusinersen is the oral route and the systemic distribution, which reaches the peripheral as well as the central nervous system. The FIREFISH trial in type 1 disease showed that risdiplam improved survival and motor function, with a substantial proportion of infants sitting without support, an outcome rarely seen in untreated type 1 disease. The SUNFISH trial confirmed benefit in type 2 and type 3 disease. Risdiplam is now the first agent to treat every type of spinal muscular atrophy and is especially valuable where intrathecal access is difficult, such as in severe scoliosis. [8]
Onasemnogene abeparvovec is a gene therapy that uses an AAV9 vector to deliver a working copy of the SMN1 gene, given as a single intravenous infusion. The pivotal Mendell 2017 trial of a high dose in type 1 infants showed a dramatic improvement, with infants achieving motor milestones never before seen in the disease, and the STR1VE trial confirmed the benefit in symptomatic infants with two SMN2 copies. The therapy is approved for children under two years of age, because the immune response to the viral vector and the disease biology make it most effective and safest in the youngest patients, and the SPR1NT study of presymptomatic infants showed that treatment before symptoms gives the best motor outcomes of any therapy to date. [6][7][11]
The treatment pathway, week by week
Week 0: Confirm the SMN1 deletion and SMN2 copy number; refer urgently to the paediatric neuromuscular service; assess airway, breathing, swallow, and nutrition
Week 0 to 1: Choose disease-modifying therapy by age, copy number, and symptom status; treat presymptomatic infants immediately after a screen-positive result
Throughout: Layer on respiratory support with cough-assist and non-invasive ventilation, nutritional support with gastrostomy where needed, and orthopaedic surveillance for scoliosis and contractures
Months to years: Continue therapy and multidisciplinary care; track motor function with CHOP-INTEND or the Hammersmith scales; support development, education, and family wellbeing
Lifelong: Coordinate transition to adult neuromuscular care; arrange genetic counselling for the family and carrier testing for siblings
Specific Subtypes & Scenarios
The type 1 infant is the scenario that drives most of the exam and most of the acute practice, and the modern principle is to treat early and to treat the whole child. A symptomatic type 1 infant under two years meets the criteria for all three therapies, and the choice turns on the centre, the family, and the funding, with the shared goal of preserving the surviving motor neurons and preventing the respiratory failure and the aspiration that killed untreated infants. The NURTURE data make the case that no type 1 infant should be left untreated, because even the most severely affected gain function and survival. [9][11]
The presymptomatic infant found on newborn screening is the scenario that defines the modern standard of care, and the principle is that the SMN2 copy number sets the urgency. An infant with two copies has classic type 1 risk and is treated urgently, ideally before six weeks of age, because the motor neurons are lost from the first weeks of life. An infant with three copies is treated early, because the natural history shows that many develop type 2 disease. An infant with four copies is monitored closely, with most guidelines recommending treatment at the first clinical or neurophysiological sign, because the risk of progression is real and presymptomatic treatment gives the best outcome. [10][11]
The child with established scoliosis and the technical difficulty of intrathecal access is a common practical problem, because nusinersen requires repeated lumbar punctures that become impossible as the spine curves. The solutions are image-guided intrathecal access, an implanted intrathecal port, or a switch to oral risdiplam, which avoids the spine altogether. The child on long-term ventilation is another scenario, because the tracheostomy and the ventilator do not preclude treatment, and the motor gains from therapy can allow weaning in some. The young adult in transition is the final scenario, because the treated children are now surviving into adulthood, and the systems for adult neuromuscular care, reproductive counselling, and independent living are still being built. [1]
Complications & Pitfalls
The life-threatening complications are the respiratory and the nutritional. Ventilatory failure from diaphragmatic and intercostal weakness, and aspiration pneumonia from bulbar weakness and pooled secretions, are the leading causes of death in untreated type 1 disease. Scoliosis progresses with growth and compounds the respiratory failure by restricting lung capacity, and hip dislocation and joint contractures limit mobility and care. The complications are now preventable or modifiable with early therapy and systematic multidisciplinary care, which is why the untreated natural history no longer defines the prognosis. [2]
The classic pitfalls are the ones the examiners reward a candidate for naming. The first is attributing a floppy infant to immaturity or cerebral causes and missing the absent reflexes and the tongue fasciculations. The second is delaying the SMN1 deletion test while ordering electromyography and muscle biopsy that the genetic test makes unnecessary. The third is treating the oxygen saturation rather than the carbon dioxide in neuromuscular respiratory failure. The fourth is waiting for symptoms to begin treatment in a screen-positive infant, when the motor neurons are already being lost. The fifth is failing to arrange the multidisciplinary respiratory, nutritional, and orthopaedic care that determines the long-term function as much as the drug. [1]
The long-term complications dominate the lived burden of the disease for the treated child and family. Scoliosis and restrictive lung disease, joint contractures, the burden of repeated intrathecal injections, the stress of a chronic and technology-dependent childhood, and the educational and psychosocial impact of a visible disability all require active management. A treated child who gains motor function still needs the physiotherapist, the orthotist, the respiratory nurse, the surgeon, and the school, and the multidisciplinary clinic is the model of care. [1]
Prognosis & Disposition
The prognosis of spinal muscular atrophy has been transformed, and the single fact to give the family is that early treatment changes everything. Untreated type 1 disease was universally fatal before two years of age. With early nusinersen in the NURTURE study, the great majority of presymptomatic infants survived without permanent ventilation, most sat, and many walked, outcomes that were impossible in the natural history. The presymptomatic gene therapy data show even greater motor gains. The prognosis is now set by the age and the symptom status at the start of treatment, the SMN2 copy number, and the quality of the multidisciplinary care. [9][11]
The predictors of a better outcome are an earlier age at the start of therapy, a presymptomatic or early-symptomatic presentation, a higher SMN2 copy number, a higher baseline motor function, and the systematic delivery of respiratory and nutritional support. The predictors of a poorer outcome are a late presentation after substantial motor neuron loss, a low SMN2 copy number, established respiratory failure, and a failure to deliver the multidisciplinary care. The motor outcome is best tracked with the CHOP-INTEND in the infant and the Hammersmith scales in the older child, and the numbers are the honest measure of the response. [1]
outcome trajectory with early treatment
Best outcomes: most sit, many walk; near-normal survival in NURTURE and SPR1NT data
Disposition follows the type and the respiratory status. A type 1 infant in respiratory failure goes to the paediatric intensive care unit for ventilatory support while the genetic diagnosis and the treatment plan are confirmed. A presymptomatic infant from newborn screening is managed in the neuromuscular clinic with urgent treatment and outpatient respiratory and nutritional surveillance. A treated type 2 or type 3 child is managed in the multidisciplinary clinic with regular respiratory, orthopaedic, and rehabilitation review, and a planned transition to adult neuromuscular care in the late teenage years. [1]
Special Populations
The presymptomatic infant found on newborn screening is the population that defines modern care, and the principle is that the SMN2 copy number sets the timing. The infant with two copies is treated within weeks, because the motor neurons are lost from the first weeks of life and the NURTURE data show that early treatment lets most sit and many walk. The infant with three copies is treated early, and the infant with four copies is monitored closely with treatment at the first sign, because presymptomatic treatment gives the best outcome of any approach. [10][11]
The Aboriginal and Torres Strait Islander child and the child from a remote setting may present with established disease rather than through screening, because of distance, access, and the variable rollout of newborn screening. The threshold to test and to retrieve is low, and the equity of access to the funded therapies is a real issue that the clinician advocates for, because the untreated natural history is still the reality where access fails. The child from a refugee or migrant family needs an early interpreter and genetic counselling in language, and the family needs to understand the one in four recurrence risk and the options for carrier testing and prenatal diagnosis in future pregnancies. [12]
The technology-dependent child on long-term ventilation and gastrostomy feeds, and the child in out-of-home care, are populations for whom the coordination of the multidisciplinary care is as important as the drug. The family of a newly diagnosed child carries a heavy emotional and financial burden, and the genetic counselling, the peer support, and the social work input are part of treatment, not an afterthought. The cardinal rule across all these populations is that access to early treatment should not depend on geography, wealth, or background, because the disease does not discriminate and neither should the care. [1]
Evidence, Guidelines & Regional Differences
The diagnostic and care framework rests on the 2018 consensus standard of care, published in two parts by Mercuri and Finkel, which set the recommendations for diagnosis, rehabilitation, orthopaedic and nutritional care in part one and pulmonary, acute, and ethical care in part two. The Mercuri 2020 critical review of the type 1 natural history remains the reference for the untreated course against which the therapies are measured, and it confirms the universal fatality before two years of age in untreated type 1 disease. [1][2][3]
The treatment evidence is dominated by five landmark trials. The ENDEAR trial of nusinersen versus sham in infantile-onset disease showed improved motor function and a reduced risk of death or permanent ventilation, and the CHERISH trial showed a significant motor gain in later-onset disease. The Mendell 2017 trial of a high-dose AAV9 gene therapy in type 1 infants showed dramatic motor gains and established onasemnogene abeparvovec, and the STR1VE trial confirmed the benefit in symptomatic infants with two SMN2 copies. The FIREFISH trial of the oral risdiplam in type 1 disease showed improved survival and motor function. The NURTURE study of presymptomatic nusinersen showed the best outcomes of all. [4][5][6][7][8][9]
ENDEAR 2017 — nusinersen in infantile-onset SMA
Randomised, double-blind, sham-controlled trial of intrathecal nusinersen versus sham procedure in 121 infants with type 1 spinal muscular atrophy.
Key finding
Nusinersen improved the attainment of motor milestones and significantly reduced the risk of death or the need for permanent ventilation compared with sham control.
Practice change
Nusinersen became the first disease-modifying therapy for spinal muscular atrophy and established that early antisense-mediated SMN2 splicing modification changes the natural history of type 1 disease.
The newborn screening framework is set by the Glascock 2020 recommendations for the treatment of infants found by screening, which stratify the urgency by SMN2 copy number, and by the Cooper 2024 systematic review of presymptomatic treatment, which confirms that earlier therapy gives better motor outcomes. The Dangouloff 2021 worldwide survey of newborn screening programs maps the global rollout and the remaining gaps in access. [10][11][12]
Regional differences are real and matter for the family. Newborn screening for spinal muscular atrophy is now standard in Australia and Aotearoa New Zealand, the United States, and much of Europe, but the funding and the availability of the three therapies vary by jurisdiction, and the cost of gene therapy is a barrier in many health systems. The threshold to treat and the choice of agent are broadly consistent across the RACP, RCPCH, ABP, and RCPSC contexts, but the local pathway and the funding must be confirmed with the neuromuscular service at the point of care, because the equity of access to early treatment is the unfinished business of the field. [12]
Exam Pearls
Spinal muscular atrophy is the commonest inherited cause of infant death and an autosomal recessive loss of the SMN1 gene on chromosome 5q13, with the severity set by the SMN2 copy number. The clinical picture is symmetric, proximal weakness with areflexia, tongue fasciculations, and paradoxical breathing, but the intellect is always spared. The single most valuable bedside clue is the bright, socially engaged, alert infant with a profoundly weak and floppy body, and the diagnosis is confirmed by the homozygous SMN1 deletion test. [1]
The SMN2 copy number is the chief modifier and the predictor of the type, with one copy predicting type 0 or severe type 1, two copies classic type 1, three copies type 2 or type 3, and four or more copies the milder type 3 or type 4. The exon 7 splicing defect, by which SMN2 produces mostly truncated unstable protein, is the key to the disease and to two of the three treatments. [1]
The three disease-modifying therapies are nusinersen, an intrathecal antisense oligonucleotide, risdiplam, a daily oral small molecule, and onasemnogene abeparvovec, a single-infusion AAV9 gene therapy approved for children under two years. The ENDEAR and CHERISH trials established nusinersen, the FIREFISH trial established risdiplam, and the Mendell 2017 and STR1VE trials established gene therapy. The principle that governs every decision is to treat early, because the NURTURE study showed that presymptomatic nusinersen lets most infants sit and many walk, outcomes impossible in untreated type 1 disease, and motor neurons lost before treatment do not recover. [4][9][6]
Newborn screening with presymptomatic treatment is the modern standard of care, and the SMN2 copy number sets the urgency: two copies is treated urgently, three copies early, and four copies is monitored closely with treatment at the first sign. The key mimic to separate is the cerebral cause of hypotonia, which gives developmental delay, brisk reflexes, and a poorly social infant, in contrast to the bright, areflexic, fasciculating child of spinal muscular atrophy. The two rules that protect the child are to send the SMN1 deletion test early and to treat as soon as the diagnosis is confirmed. [1][11]
References
- [1]Mercuri E, Finkel RS, Muntoni F, et al Diagnosis and management of spinal muscular atrophy: Part 1: Recommendations for diagnosis, rehabilitation, orthopedic and nutritional care. Neuromuscul Disord, 2018.PMID 29290580
- [2]Finkel RS, Mercuri E, Meyer OH, et al Diagnosis and management of spinal muscular atrophy: Part 2: Pulmonary and acute care; medications, supplements and immunizations; other organ systems; and ethics. Neuromuscul Disord, 2018.PMID 29305137
- [3]Mercuri E, Bertini E, Iannaccone ST Longitudinal natural history of type I spinal muscular atrophy: a critical review. Orphanet J Rare Dis, 2020.PMID 32248834
- [4]Finkel RS, Chiriboga CA, Vajsar J, et al, ENDEAR Study Group Nusinersen versus Sham Control in Infantile-Onset Spinal Muscular Atrophy. N Engl J Med, 2017.PMID 29091570
- [5]Mercuri E, Darras BT, Chiriboga CA, et al, CHERISH Study Group Nusinersen versus Sham Control in Later-Onset Spinal Muscular Atrophy. N Engl J Med, 2018.PMID 29443664
- [6]Mendell JR, Al-Zaidy S, Shell R, et al Single-Dose Gene-Replacement Therapy for Spinal Muscular Atrophy. N Engl J Med, 2017.PMID 29091557
- [7]Day JW, Chiriboga CA, Crawford TO, et al Onasemnogene abeparvovec gene therapy for symptomatic infantile-onset spinal muscular atrophy in patients with two copies of SMN2 (STR1VE): an open-label, single-arm, multicentre, phase 3 trial. Lancet Neurol, 2021.PMID 33743238
- [8]Baranello G, Darras BT, Chiriboga CA, et al, FIREFISH Working Group Risdiplam in Type 1 Spinal Muscular Atrophy. N Engl J Med, 2021.PMID 33626251
- [9]De Vivo DC, Bertini E, Swoboda KJ, et al, NURTURE Study Group Nusinersen initiated in infants during the presymptomatic stage of spinal muscular atrophy: Interim efficacy and safety results from the Phase 2 NURTURE study. Neuromuscul Disord, 2019.PMID 31704158
- [10]Glascock J, Sampson J, Haidet-Phillips A, et al Revised Recommendations for the Treatment of Infants Diagnosed with Spinal Muscular Atrophy Via Newborn Screening Who Have 4 Copies of SMN2. J Neuromuscul Dis, 2020.PMID 32007960
- [11]Cooper K, Prasad S, Bueser L, et al Systematic Review of Presymptomatic Treatment for Spinal Muscular Atrophy. Int J Neonatal Screen, 2024.PMID 39189228
- [12]Dangouloff T, Servais L, on behalf of the SMA NBS World Study Group Newborn screening programs for spinal muscular atrophy worldwide: Where we stand and where to go. Neuromuscul Disord, 2021.PMID 33985857