Paeds · ophthalmology
Colour vision deficiency and inherited retinal disease
Also known as Colour blindness · Red-green colour deficiency · Protanopia · Deuteranopia · Tritanopia · Achromatopsia · Retinitis pigmentosa · Stargardt disease · Leber congenital amaurosis · Choroideremia · X-linked retinoschisis
Fellowship guide to colour vision deficiency and inherited retinal disease in children. Covers the inherited and the congenital colour vision defects from the red-green dichromacy through the achromatopsia, the Ishihara and the Farnsworth colour testing, the inherited retinal dystrophies from the retinitis pigmentosa through the Stargardt disease, the Leber congenital amaurosis, the choroideremia and the X-linked retinoschisis, the molecular genetics with the RPE65 and the ABCA4 genes, the electroretinography and the autofluorescence imaging, the molecular genetic testing, the supportive management with the low-vision aids and the genetic counselling, and the voretigene neparvovec gene therapy for the RPE65 Leber congenital amaurosis.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
Overview & Definition
A teacher notices that a boy in the first year of school mistakes the orange and the green of the reading-group charts, and the family is reassured that this is the common and the harmless red-green colour blindness, passed from the mother to the son. A different child, a toddler, is brought in because he does not see in the dusk and he bumps into the furniture in the dim hallway. The family learns that behind this lies a progressive inherited retinal dystrophy that will narrow his world to a tunnel over the years. Both children have met the inherited disorders of the retinal function. The clinician who holds the two ends of the spectrum — the benign and the common colour vision deficiency at one end, the serious and the progressive inherited retinal disease at the other — is the clinician who reassures correctly and refers promptly. [6][1]
Colour vision deficiency, the old colour blindness, is the reduced ability to distinguish certain colours. The great majority of the congenital cases are the inherited red-green defect, an X-linked recessive trait that affects roughly one in twelve males of the Northern European ancestry and a far smaller fraction of the females. The condition is stable from the birth, it does not progress, and it carries no threat to the sight or to the health, so its management is the explanation, the reassurance and the thoughtful vocational advice. The smaller group of the congenital colour defects, the tritan and the achromatopsia, is far rarer and it carries the photophobia and the reduced acuity that change the management. The distinction between the benign common defect and the serious rare one is the first task of the paediatric assessment. [6][7]
Inherited retinal disease is the larger and the more serious territory, the family of the genetic dystrophies of the retina that includes the retinitis pigmentosa, the Stargardt disease, the Leber congenital amaurosis, the choroideremia and the X-linked retinoschisis. These conditions are driven by the single-gene mutations of the phototransduction and the visual cycle, and they share the progressive loss of the photoreceptors and the sight. The retinitis pigmentosa is the commonest of the inherited retinal dystrophies, the Stargardt disease is the commonest of the macular dystrophies, and the Leber congenital amaurosis is the severe congenital form that may sit behind the nystagmus and the poor vision of the infant. The landmark of the field is the voretigene neparvovec, the gene therapy for the RPE65 form of the Leber congenital amaurosis, the first gene therapy ever approved for an inherited disease, and the proof that the molecular diagnosis can now open a molecular treatment. [1][3][11]
Classification
A father who is colour-blind and a sister who sees normally ask why the boy cannot tell the red from the green, and the answer is the inheritance. The classification of the colour vision defects follows the inheritance first and the cone second. The common red-green defect is the X-linked recessive trait of the long- and the medium-wave opsin genes, and it splits into the anomalous trichromacy of the weakened cone, the protanomaly and the deuteranomaly, and the dichromacy of the absent cone. The protanopia and the deuteranopia are the dichromat forms, and the deuteranomaly is the single commonest form. The tritan defect, the blue-yellow, is far rarer as a congenital trait and far more often the acquired effect of the optic nerve or the retinal disease, and the achromatopsia is the rare autosomal recessive absence of all the cone function with the severe visual impairment. [6][2]
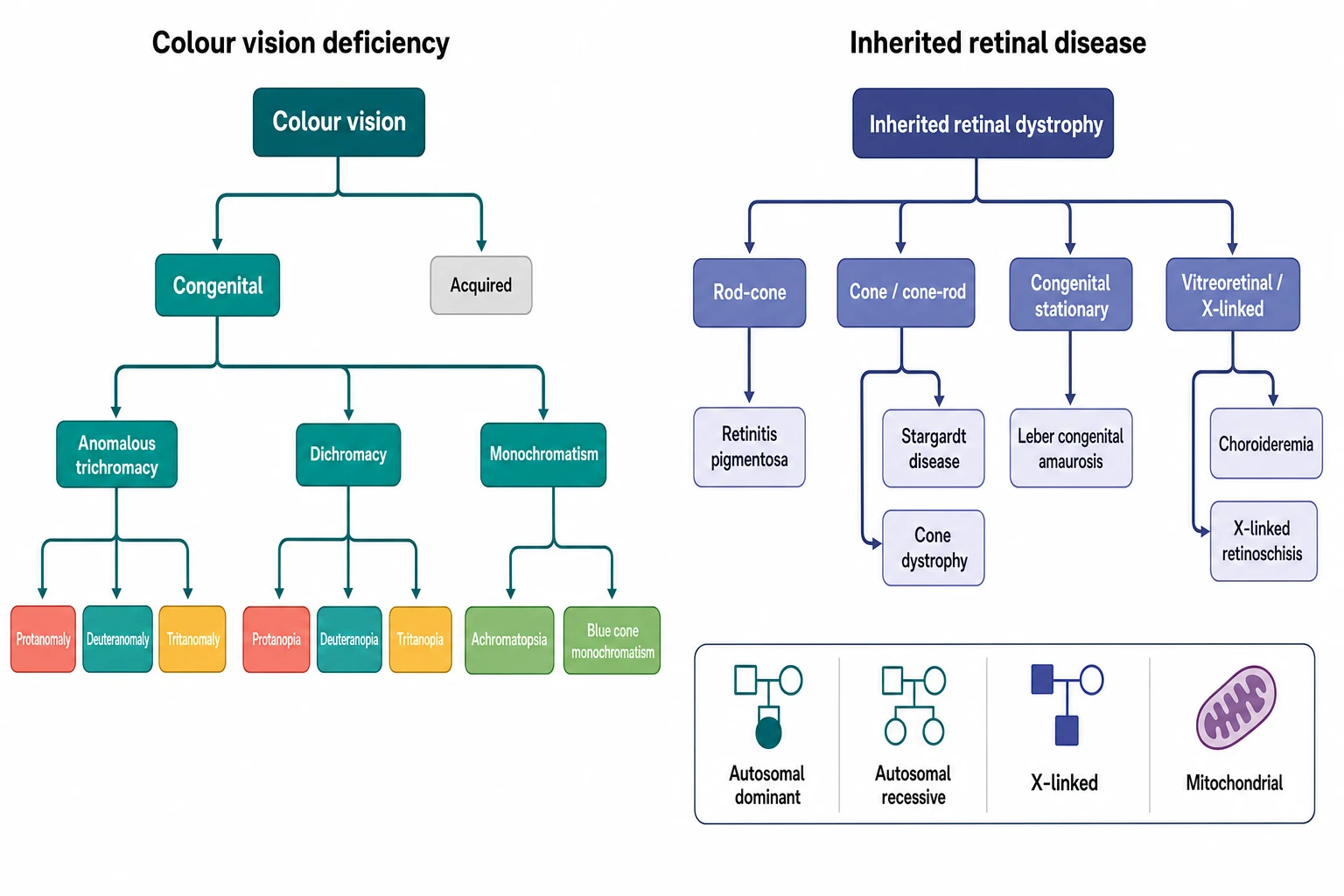
The inherited retinal disease carries its own classification, and it rests on the photoreceptor first affected and on the inheritance. The rod-cone dystrophy, the retinitis pigmentosa, begins with the rods and the night blindness and the field loss, and it is the commonest form. The cone and the cone-rod dystrophy begin with the cones and the central acuity and the colour, and the Stargardt disease is the commonest of the macular dystrophies in this group. The congenital stationary forms, the Leber congenital amaurosis and the congenital stationary night blindness, present in the infancy with the nystagmus and the poor vision. The vitreoretinal and the X-linked forms, the choroideremia and the X-linked retinoschisis, carry the specific inheritance that guides the counselling of the mother and the sisters. The molecular genetics now sorts the diseases by the gene, and the RPE65, the ABCA4, the RPGR and the CHM genes carry the names the boards reward. [1][8]
Red-green CVD
common, benign
- X-linked recessive, one in twelve males
- Stable from birth, no threat to sight
- Deuteranomaly the commonest form
- Reassurance and vocational advice
Retinitis pigmentosa
rod-cone
- Commonest inherited retinal dystrophy
- Night blindness and field constriction
- RHO, USH2A, RPGR genes
- Low-vision support, gene therapy trials
Stargardt disease
macular
- Commonest macular dystrophy
- Autosomal recessive ABCA4
- Central acuity and colour loss in adolescence
- No cure, low-vision and genetic counselling
Leber congenital amaurosis
congenital, treatable
- Severe vision loss from infancy
- Nystagmus, roving eye movements
- RPE65 form is treatable
- Voretigene neparvovec gene therapy
Epidemiology & Risk Factors
The epidemiology of the colour vision deficiency is the epidemiology of the X chromosome, and the boards reward the candidate who holds it. The red-green colour deficiency affects roughly eight percent of the males and well under one percent of the females of the Northern European ancestry. The global review of Birch showed that the prevalence is fairly constant across the populations, with the deuteranomaly as the commonest form and the protan defects as the next. The 2025 meta-analysis of the global prevalence in the children and the adolescents confirmed the figure and showed that the prevalence sits highest in the European and the Middle Eastern populations and lowest in some of the African and the Indigenous populations. The reason is the founder effect and the genetic drift. [6][7]
The inherited retinal disease is far rarer but far graver, and the retinitis pigmentosa is the commonest single entity with the prevalence of roughly one in four thousand, the figure that the boards reward. The Stargardt disease is the commonest macular dystrophy, with the prevalence of roughly one in ten thousand, and the Leber congenital amaurosis accounts for a smaller fraction but a disproportionate share of the congenital blindness. The choroideremia and the X-linked retinoschisis are the rare X-linked forms that cluster in the families and that carry the counselling of the carrier females. The single greatest risk factor across the whole field is the family history, and the consanguinity and the founder effect raise the prevalence of the autosomal recessive forms in the closed and the isolated populations. [1][2]
The access to the diagnosis and the care varies by the geography and the socioeconomic status, and it is the equity issue that the boards reward. The molecular genetic testing, the specialist electroretinography and the gene therapy are concentrated in the tertiary centres of the high-income setting. The child of the rural and the remote community, the Indigenous community and the migrant family is the child most at risk of the delayed diagnosis and the missed treatment. The candidate who links the family history and the equity to the access to the genetic testing and the gene therapy demonstrates the breadth the examiner rewards. [9][11]
Pathophysiology
The pathophysiology of the colour vision begins with the three cone photoreceptors and the three opsins, and the candidate who holds the phototransduction holds the disease. The normal colour vision is the trichromacy of the three cones, the short-wave blue cone of the OPN1SW gene on the chromosome seven, the medium-wave green cone of the OPN1MW gene on the X chromosome, and the long-wave red cone of the OPN1LW gene, also on the X chromosome. The red-green colour deficiency arises from the rearrangement and the unequal crossing-over of the adjacent OPN1LW and OPN1MW genes on the X chromosome, which produces the hybrid gene and the shifted or the absent cone, and the X-linked recessive inheritance follows directly from the location of the genes. The tritan defect arises from the OPN1SW gene, and the achromatopsia arises from the loss of all the cone function through the CNGA3, the CNGB3 and the GNAT2 genes. [6][8]
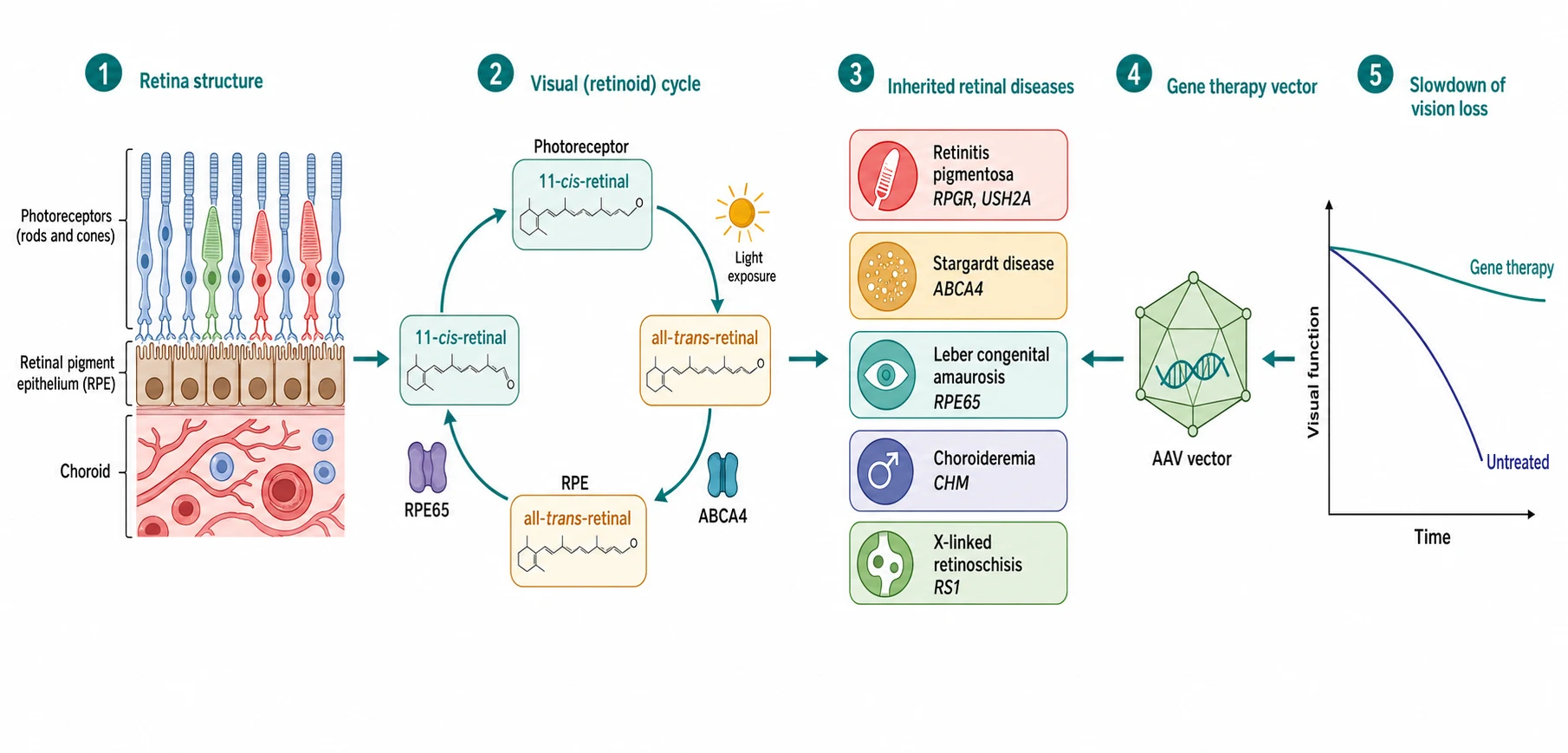
The inherited retinal disease is the single-gene disorder of the photoreceptor and the visual cycle, and the molecular defect drives the cell that dies first. The retinitis pigmentosa begins with the rod, the photoreceptor of the dim light and the periphery, and the mutation of the RHO, the USH2A or the RPGR gene triggers the apoptosis of the rod and the night blindness and the ring scotoma that widen into the tunnel. The Stargardt disease is the defect of the ABCA4 gene, the transporter of the retinoid in the visual cycle, and the build-up of the toxic lipofuscin in the retinal pigment epithelium destroys the cone-rich macula and takes the central acuity. The Leber congenital amaurosis is the severe defect of the visual cycle, and the RPE65 gene is the one that blocks the regeneration of the eleven-cis-retinal, which is why the restoration of the RPE65 gene by the vector can restore the vision. [1][8][3]
The choroideremia is the defect of the CHM gene that encodes the Rab escort protein. The loss of the protein causes the progressive degeneration of the retinal pigment epithelium, the photoreceptors and the choriocapillaris in the male, beginning in the periphery and sparing the macula until the late stage. The X-linked retinoschisis is the defect of the RS1 gene that encodes the retinoschisin protein of the retinal cell adhesion, and the loss splits the retinal layers and produces the foveal schisis of the young boy. The unifying idea is that the molecular diagnosis now predicts the natural history and, in the RPE65 form, it opens the treatment, which is why the genetic testing is no longer the academic exercise of the past but the gateway to the gene therapy and the clinical trial. [5][11]
Clinical Presentation
The child with the colour vision deficiency presents through the school or the family, and the presentation is the quiet one that the vigilant teacher or the colour-blind parent catches. The common red-green defect is noticed when the child mistakes the red and the green of the teaching materials, the coloured pencils or the traffic-light game, or when the colour-blind father sees his own pattern in the son. The acuity is normal, the fields are full and the fundus is healthy, and the child has no complaint of the vision, because the defect is stable from the birth and the child has never known the other way. The rarer congenital defects present differently: the achromatopsia presents with the photophobia, the nystagmus and the poor acuity, and the tritan defect presents with the blue-yellow confusion that may point to the acquired rather than the congenital cause. [6][2]
The inherited retinal disease presents through several doors, and each carries its own clue. The retinitis pigmentosa presents with the night blindness, the difficulty in the dim light and the playground, the bumping into the objects and the gradual constriction of the field that the child describes as the narrowing world. The Stargardt disease presents with the central reading difficulty and the reduced acuity in the late childhood or the adolescence, with the slow decline that the child hides for the months before the family seeks the help. The Leber congenital amaurosis presents in the infancy with the nystagmus, the roving eye movements, the poor fixation, the eye-pressing or the poking, and the severely reduced vision that the family and the health visitor notice in the first months. [1][12]
The boards probe the presentations that distinguish the serious from the benign, and the candidate who holds them earns the marks. The photophobia and the nystagmus point to the cone dysfunction and the achromatopsia, while the night blindness points to the rod-cone dystrophy. The central acuity loss points to the macular dystrophy, while the peripheral field loss points to the retinitis pigmentosa. The symmetrical and the slowly progressive course points to the inherited disease, while the sudden or the asymmetric loss points to the acquired or the inflammatory cause that demands the urgent referral. The family history of the childhood blindness, the night blindness or the registered sight impairment is the clue that drives the pedigree and the genetic testing. [1][2]
Differential Diagnosis
The differential diagnosis rests on the cause of the colour change and the vision loss, and the boards probe the candidate who can walk through it. The congenital red-green defect is the common and the benign cause, and it is distinguished by the stable course, the normal acuity and the positive family history. The acquired colour defect, by contrast, is the new and the progressive change in the older child or the adult, and it points to the optic neuropathy, the macular disease or the drug toxicity, with the tritan blue-yellow defect as the common acquired pattern. The photophobia and the nystagmus raise the achromatopsia and the albinism, and the poor vision from the infancy raises the Leber congenital amaurosis, the cortical visual impairment and the congenital stationary night blindness. [6][2]
The progressive vision loss raises the long list of the inherited retinal dystrophies, and the history sorts them. The night blindness and the field loss point to the retinitis pigmentosa, the central acuity loss to the Stargardt disease, the chorioretinal atrophy in the male to the choroideremia, and the foveal schisis in the young boy to the X-linked retinoschisis. The non-inherited causes must be excluded, and they include the vitamin A deficiency of the malnutrition and the malabsorption, which causes the night blindness and the dry eye, and the drug toxicity of the hydroxychloroquine, the ethambutol and the vigabatrin, which cause the acquired field and the colour defects. The candidate who names the reversible cause of the night blindness, the vitamin A deficiency, alongside the inherited ones demonstrates the breadth the examiner rewards. [1][8]
Clinical & Bedside Assessment
The bedside assessment begins with the colour vision test, and the Ishihara plate test is the one the boards reward. The test is performed in the good daylight or the standard room light, with the child wearing the glasses if they use them, and the plates held at the comfortable reading distance of about seventy-five centimetres. The child names the number on each plate, and the normal child reads twelve or more of the fourteen screening plates correctly, while the child with the red-green defect misreads the plates in the pattern that distinguishes the protan from the deutan. The Ishihara is the excellent screen for the red-green defect, and it is the poor test for the blue-yellow tritan defect, which the Hardy-Rand-Rittler plates and the Farnsworth D15 detect better, and the gold standard is the anomaloscope that measures the matching range. [6][2]
The rest of the assessment is the full ophthalmic and the paediatric examination. The acuity is measured with the age-appropriate test, the preferential looking in the infant and the logMAR letter in the older child, and the colour test is paired with the visual field by the confrontation, the pupil reactions and the fundus. The fundus shows the healthy retina of the colour vision deficiency, the bone-spicule pigmentation, the attenuated arterioles and the waxy pallor of the disc in the retinitis pigmentosa, the beaten-bronze macula and the pisciform flecks of the Stargardt disease, and the chorioretinal atrophy of the choroideremia. The family history is taken as the three-generation pedigree, and the drawing of the affected males and the carrier females of the X-linked pedigree is the skill the boards reward. [1][12]
Investigations
The investigation of the inherited retinal disease rests on the electrophysiology, the imaging and the genetics, and the candidate who holds the three earns the marks. The electroretinography is the functional test that separates the rod-cone from the cone and the stationary from the progressive, and the full-field electroretinography measures the rod and the cone response across the whole retina, while the pattern electroretinography measures the ganglion-cell response of the macular pathway. The extinguished rod response with the reduced cone response confirms the retinitis pigmentosa, the cone-dominated reduction confirms the cone dystrophy, and the near-absent rod and cone response confirms the Leber congenital amaurosis. [1][2]
[2] [9]The imaging adds the structural detail that the function cannot. The fundus autofluorescence shows the lipofuscin of the retinal pigment epithelium, and the ring of the increased autofluorescence at the vascular arcades of the retinitis pigmentosa and the flecks and the atrophy of the Stargardt disease are the patterns the boards reward. The optical coherence tomography shows the thinning of the outer retina and the loss of the ellipsoid zone, and it measures the macula that the gene therapy demands to be viable. The molecular genetic testing is the definitive investigation, and the next-generation sequencing panel of the inherited retinal disease genes, now covering over a hundred and seventy genes, identifies the pathogenic variant in the majority of the children and it opens the door to the gene therapy and the trial. The candidate who names the panel and the RPE65 result as the gateway to the voretigene neparvovec demonstrates the reasoning the boards reward. [9][11]
Management — Resuscitation
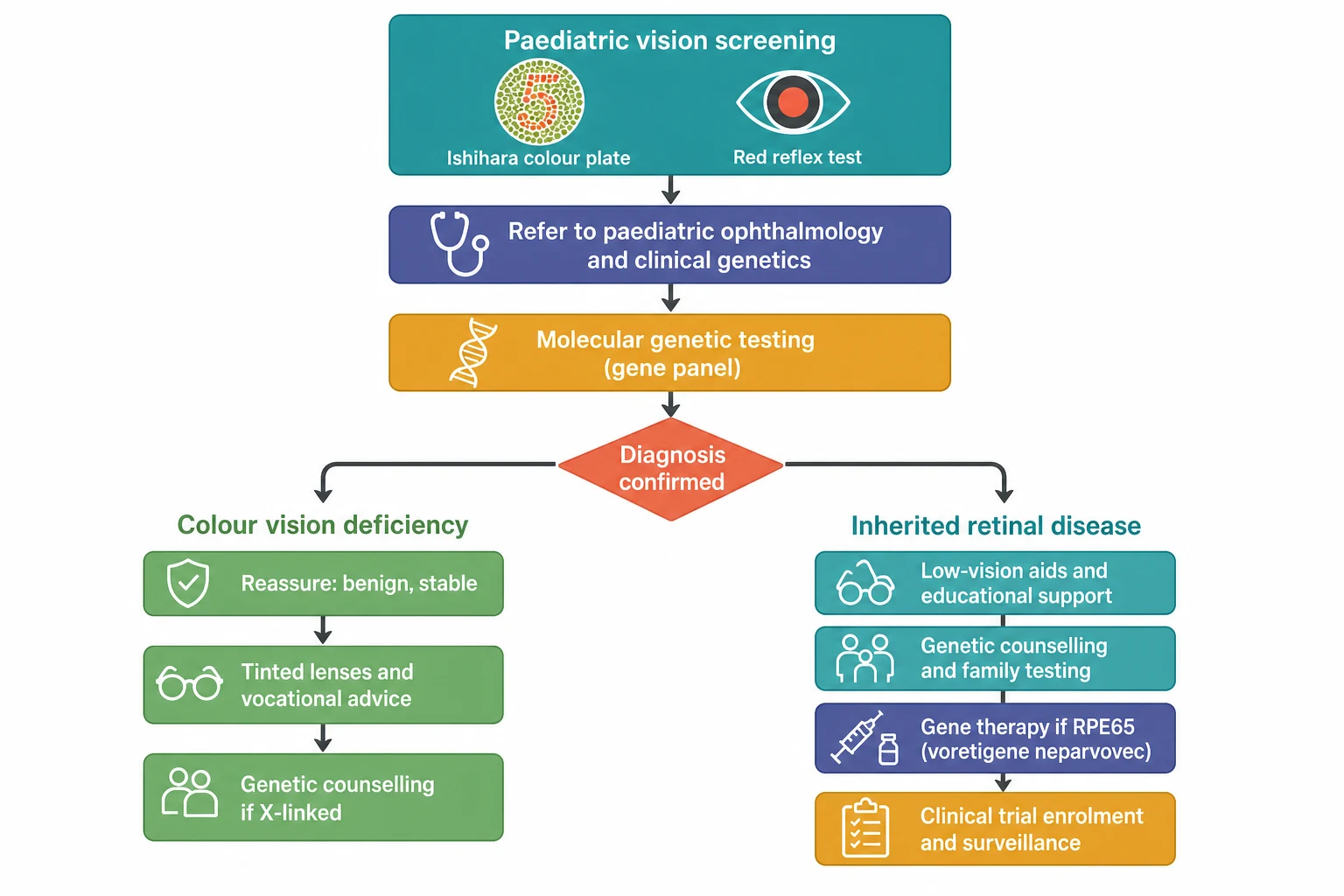
The resuscitation of the child with the inherited retinal disease is the urgent referral and the safety, because there is no acute collapse but there is the acute danger of the missed treatable cause. The infant with the nystagmus, the photophobia and the poor vision is referred promptly to the paediatric ophthalmology service for the electroretinography and the genetic testing, because the treatable RPE65 Leber congenital amaurosis must not be missed. The child with the sudden or the rapidly progressive vision loss is referred the same day, because the sudden loss is never a stable inherited disease and it points to the inflammatory, the vascular or the neoplastic cause that the time-sensitive treatment may save. [1][3]
The safety extends to the family and the school. The child with the field loss of the retinitis pigmentosa is assessed for the safe mobility and the traffic, and the low-vision service and the educational support are engaged early. The family is offered the genetic counselling and the testing of the siblings and the carrier females, and the psychosocial support of the diagnosis is addressed from the first visit, because the inherited retinal disease carries the burden that the family carries for the years. [10][11]
Management — Definitive & Stepwise
The definitive management splits at the diagnosis, and the colour vision deficiency and the inherited retinal disease travel the two different paths. The colour vision deficiency has no treatment and it needs none, because the condition is stable and it carries no threat to the sight. The management is the honest explanation that the defect is common, inherited and permanent, the reassurance that the acuity and the field are normal. The thoughtful vocational advice guides the child away from the careers that demand the perfect colour discrimination, such as the electrical work, the aviation and the railway signalling. The tinted lenses and the contact lenses may aid the discrimination in the selected case, but they do not correct the defect, and the candidate who says so earns the marks. [6][2]
The inherited retinal disease has the stepwise management that builds from the support to the gene therapy and the trial. The first step is the low-vision support, with the magnifiers, the electronic aids and the educational adaptation that keep the child in the mainstream school. The second step is the genetic counselling, with the testing of the siblings and the carrier females, the recurrence-risk advice and the prenatal and the preimplantation options for the severe autosomal recessive and the X-linked forms. The third step is the gene therapy for the eligible child, and the voretigene neparvovec is the approved gene therapy for the biallelic RPE65 mutation with the viable retinal cells, delivered as the single subretinal injection of the adeno-associated virus vector carrying the normal RPE65 gene into each eye. [3][11]
Voretigene neparvovec (Luxturna) for the RPE65 Leber congenital amaurosis
Dose
The approved dose per the regulatory label, a single treatment per eye, administered once in the lifetime. The candidate should know the mechanism and the indication rather than the exact vector-genome number, and should state that the treatment is reserved for the child with the viable retinal cells confirmed on the optical coherence tomography.
The fourth step is the clinical trial and the surveillance, because the gene therapy is the RPE65 success and the candidate must know that the rest of the field is in the trial. The choroideremia gene therapy of MacLaren showed the safety and the early functional gain, and the trials of the X-linked retinoschisis, the Stargardt disease and the achromatopsia are the ongoing frontier of the field. The retinitis pigmentosa has the emerging therapies of the neuroprotection, the optogenetics and the retinal prosthesis. The vitamin A palmitate, by contrast, is the controversial adult treatment that is not used in the child because of the hepatotoxicity and the limited evidence, and it is avoided in the ABCA4 Stargardt disease because it may accelerate the atrophy. The child with the inherited retinal disease is enrolled in the specialist surveillance and the register, so that the eligible trial and the approved therapy reach the child as they emerge. [5][10]
Colour vision deficiency
Inherited retinal disease
Specific Subtypes & Scenarios
The retinitis pigmentosa is the commonest and the prototype of the inherited retinal dystrophy, and the boards probe it deeply. The disease presents with the night blindness in the childhood or the adolescence, it progresses to the ring scotoma and the tunnel vision over the decades, and it spares the central acuity until the late stage. The inheritance follows the autosomal dominant RHO form and the autosomal recessive USH2A form. The severe RPGR form is the X-linked disease of the young male, and the syndromic forms include the Usher syndrome with the deafness and the Bardet-Biedl syndrome with the obesity, the polydactyly and the learning difficulty. The candidate who names the Usher and the Bardet-Biedl syndrome alongside the isolated retinitis pigmentosa demonstrates the syndromic breadth the examiner rewards. [1][10]
The Stargardt disease is the commonest macular dystrophy, and it presents with the central reading difficulty and the acuity loss in the late childhood or the adolescence. The autosomal recessive ABCA4 defect causes the build-up of the lipofuscin and the beaten-bronze macula with the pisciform flecks, and the early pattern of the macular degeneration in the ABCA4 retinopathy is the finding the imaging reveals. The acuity often falls to the six over eighteen to the six over sixty range, and the disease spares the peripheral field, so the child keeps the mobility but loses the reading, which makes the low-vision support and the educational adaptation the core of the management. [12][2]
The Leber congenital amaurosis is the severe congenital form, and it is the treatable one. The disease presents in the infancy with the nystagmus, the roving eye movements, the poor fixation and the eye-pressing, and the electroretinography shows the near-absent response. The RPE65 form is the one the gene therapy treats, and the voretigene neparvovec phase three trial of Russell showed the improvement of the functional vision on the multi-luminance mobility test, with the durable gain at the one-year and the longer follow-up of Maguire. The candidate who holds the RPE65 link, the vector and the trial earns the marks, and the candidate who knows that the treatment is the RPE65 form and not the whole disease demonstrates the precision the boards reward. [3][4]
The choroideremia and the X-linked retinoschisis are the X-linked forms that carry the counselling of the family. The choroideremia presents in the boy and the young man with the night blindness and the progressive chorioretinal atrophy of the mid-periphery, sparing the macula until the late stage, and the CHM gene therapy trial of MacLaren showed the safety and the early gain. The X-linked retinoschisis presents in the young boy with the reduced acuity and the foveal schisis on the imaging, and the split of the retinal layers by the RS1 defect may cause the vitreous haemorrhage or the retinal detachment in the severe case. The mother and the sisters are the carrier females, and the counselling and the testing of the family is the management that the boards reward. [5][8]
Complications & Pitfalls
The complications of the inherited retinal disease are the loss of the sight, the loss of the independence and the burden on the family. The retinitis pigmentosa progresses to the severe field loss and the registered sight impairment over the decades, the Stargardt disease to the loss of the reading vision, and the Leber congenital amaurosis to the profound vision loss from the infancy. The X-linked retinoschisis carries the acute risk of the vitreous haemorrhage and the retinal detachment, and the syndromic forms carry the hearing loss of the Usher syndrome and the multisystem burden of the Bardet-Biedl syndrome. The complications of the gene therapy are the surgical risk of the subretinal injection, the inflammation and the cataract, and the candidate who balances the gain against the risk earns the marks. [1][4]
The classic pitfalls are the two that the boards probe. The first is the attribution of the progressive vision loss to the benign colour vision deficiency, which delays the diagnosis of the retinitis pigmentosa and the macular dystrophy, because the clinician hears the colour complaint and stops the assessment short of the fields and the fundus. The second is the omission of the genetic testing, which leaves the family without the counselling and the child without the access to the gene therapy and the trial. The vitamin A palmitate is the third pitfall, prescribed in the hope of the slowing of the retinitis pigmentosa in the child, when the evidence is the modest adult benefit, the hepatotoxicity is the real risk, and the ABCA4 Stargardt disease may be accelerated by it. [10][12]
Prognosis & Disposition
The prognosis of the colour vision deficiency is excellent, because the condition is stable and it carries no threat to the sight or the health. The child keeps the normal acuity and the full field, and the only consequence is the career limitation that the vocational advice addresses. The prognosis of the inherited retinal disease is the variable one set by the gene, the cell and the stage, and the boards reward the candidate who holds the variation. The retinitis pigmentosa progresses over the decades to the severe field loss, with the central acuity spared until the late stage in many of the forms. The Stargardt disease stabilises at the moderate acuity loss with the spared peripheral field. The Leber congenital amaurosis carries the severe vision loss, and the RPE65 gene therapy now offers the functional gain that was absent a generation ago. [1][3]
The disposition is to the specialist centre and the multidisciplinary team. The child with the inherited retinal disease is referred to the paediatric ophthalmology service, the clinical genetics and the low-vision service, and the family is offered the genetic counselling and the psychosocial support from the diagnosis. The child with the RPE65 disease is referred to the specialist centre for the assessment of the gene therapy, and the child with the other gene is enrolled in the register and the surveillance, so that the emerging trial and the approved therapy reach the child. The equity of the access to the genetic testing and the gene therapy is the disposition issue the boards reward, because the tertiary service and the treatment are concentrated in the high-income setting and the urban centre. [9][11]
Special Populations
The Indigenous and the remote community child is the child at the greatest risk of the delayed diagnosis, because the specialist electrophysiology and the genetic testing are concentrated in the urban tertiary centre. The telehealth and the outreach ophthalmology service, the visiting electrophysiology and the mailed genetic panel are the models that close the gap, and the candidate who names them alongside the equity demonstrates the breadth the examiner rewards. The Indigenous populations also carry the founder-effect variation of the colour vision deficiency prevalence, and the global meta-analysis showed the lower prevalence in some of the populations, which is the fact the boards may probe. [7][9]
The migrant and the refugee family carry the consanguinity and the founder effect that raise the prevalence of the autosomal recessive dystrophies, and the language and the cultural barrier may delay the presentation. The child with the disability and the neurodiversity may not report the night blindness or the field loss, and the carer and the teacher are the ones who notice the bumping and the clumsiness. The socioeconomically disadvantaged family carries the barrier to the travel, the time off the work and the cost of the care, and the multidisciplinary plan must address the practical support as well as the clinical treatment. [10][11]
Evidence, Guidelines & Regional Differences
The evidence base of the field is the convergence of the molecular genetics, the electrophysiology and the gene therapy, and the boards reward the candidate who holds the landmark. The Hartong review of the retinitis pigmentosa in the Lancet laid out the rod-cone biology and the natural history, and it remains the reference the boards reward. The Russell phase three trial of the voretigene neparvovec in the Lancet showed the improvement of the functional vision on the multi-luminance mobility test, and it secured the first approval of a gene therapy for an inherited disease. The MacLaren trial of the choroideremia gene therapy opened the X-linked frontier, and the Georgiou review of the phenotyping and the genotyping of the inherited retinal diseases is the contemporary reference that holds the field. [1][3][5][2]
The regional differences are the access and the service model. The gene therapy is the high-cost treatment that the funding body must approve, and the access varies by the country and the insurer. The molecular genetic panel and the specialist electrophysiology are available in the tertiary centre of the high-income setting and absent in the low-resource setting, which is the equity gap that the global review of the gene therapy for the inherited retinal disease addressed. The candidate who links the molecular diagnosis to the access to the gene therapy and the trial, and who names the equity of the funding and the geography, demonstrates the regional reasoning the boards reward. [9][11]
[9] [11]Exam Pearls
The candidate who holds the following earns the marks the boards award. The common red-green colour deficiency is the X-linked recessive trait of the OPN1LW and the OPN1MW opsin genes on the X chromosome. It affects one in twelve males and well under one percent of the females, and the deuteranomaly is the commonest form, managed with the reassurance and the vocational advice. [6]
The Ishihara plate test is the excellent screen for the red-green defect and the poor test for the blue-yellow tritan defect, which the Hardy-Rand-Rittler and the Farnsworth tests detect and the anomaloscope measures as the gold standard. The acquired colour defect, especially the tritan, points to the optic neuropathy or the macular disease and demands the referral. [6][2]
The retinitis pigmentosa is the commonest inherited retinal dystrophy, with the prevalence of one in four thousand, the night blindness and the field constriction, the bone-spicule pigmentation, the attenuated vessels and the extinguished rod response on the electroretinography, and the Usher and the Bardet-Biedl syndrome as the syndromic forms. The Stargardt disease is the commonest macular dystrophy, the autosomal recessive ABCA4 defect, and the Leber congenital amaurosis is the severe congenital form. [1][12]
The RPE65 Leber congenital amaurosis is treated by the voretigene neparvovec, the first gene therapy approved for an inherited disease, a single subretinal injection of the adeno-associated virus vector per eye, with the gain on the multi-luminance mobility test and the durable follow-up, and the molecular genetic testing is the gateway to the treatment and the trial. [3][4]
References
- [1]Hartong DT, Berson EL, Dryja TP Retinitis pigmentosa. Lancet, 2006.PMID 17113430
- [2]Georgiou M, Kaiafa G, Larcher A, et al Phenotyping and genotyping inherited retinal diseases: Molecular genetics, clinical and imaging features, and therapeutics of macular dystrophies, cone and cone-rod dystrophies, rod-cone dystrophies, Leber congenital amaurosis, and cone dysfunction syndromes. Prog Retin Eye Res, 2024.PMID 38278208
- [3]Russell S, Bennett J, Wellman JA, et al Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial. Lancet, 2017.PMID 28712537
- [4]Maguire AM, Russell S, Wellman JA, et al Efficacy, Safety, and Durability of Voretigene Neparvovec-rzyl in RPE65 Mutation-Associated Inherited Retinal Dystrophy: Results of Phase 1 and 3 Trials. Ophthalmology, 2019.PMID 31443789
- [5]MacLaren RE, Groppe M, Barnard AR, et al Retinal gene therapy in patients with choroideremia: initial findings from a phase 1/2 clinical trial. Lancet, 2014.PMID 24439297
- [6]Birch J Worldwide prevalence of red-green color deficiency. J Opt Soc Am A Opt Image Sci Vis, 2012.PMID 22472762
- [7]Jeong YD, Al Owaifer AM, Kim YH, et al Global Prevalence of Congenital Color Vision Deficiency among Children and Adolescents, 1932-2022. Ophthalmology, 2025.PMID 40769301
- [8]Berger W, Kloeckener-Gruissem B, Neidhardt J The molecular basis of human retinal and vitreoretinal diseases. Prog Retin Eye Res, 2010.PMID 20362068
- [9]Sheck LHN, Bowdin SC, Skiadaresis T, et al Panel-based genetic testing for inherited retinal disease screening 176 genes. Mol Genet Genomic Med, 2021.PMID 33749171
- [10]Nguyen XT, Moysich SJ, Al-Barwani B, et al Retinitis Pigmentosa: Current Clinical Management and Emerging Therapies. Int J Mol Sci, 2023.PMID 37108642
- [11]Tan TE, Gasparini S, Ting DS, et al One down but many more to go: the state of gene therapy for inherited retinal disease. Regen Med, 2025.PMID 41054259
- [12]Khan KN, Kasujee I, Mahroo OA, et al Early Patterns of Macular Degeneration in ABCA4-Associated Retinopathy. Ophthalmology, 2018.PMID 29310964