Paeds · ophthalmology
Leukocoria and retinoblastoma
Also known as White pupillary reflex · Cat-eye reflex · Retinoblastoma · RB1 tumour suppressor gene · Two-hit hypothesis · International Intraocular Retinoblastoma Classification · Trilateral retinoblastoma · Ophthalmic artery chemosurgery
Fellowship guide to leukocoria and retinoblastoma in children. Covers the white pupillary reflex as the red-flag sign, the differential diagnosis of leukocoria from the retinoblastoma through the congenital cataract, the Coats disease, the retinopathy of prematurity and the toxocariasis, the RB1 tumour suppressor gene on chromosome thirteen and the Knudson two-hit hypothesis, the heritable and the non-heritable forms, the International Intraocular Retinoblastoma Classification of groups A through E, the imaging with ultrasound and magnetic resonance imaging, the urgent referral to the ocular oncology centre, the risk-adapted treatment from the focal laser and cryotherapy through the ophthalmic artery chemosurgery and the systemic chemotherapy to the enucleation, the trilateral retinoblastoma and its poor prognosis, and the survivorship surveillance for the second malignancy and the late effects.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
Overview & Definition
A parent sees a white glow in the pupil of their infant in the flash of a photograph, and the family is unsettled by something they cannot name. A general practitioner performs the red reflex at the six-week check and finds a yellow-white reflex where there should be a warm orange-red. Both have met leukocoria, the white pupillary reflex, and the most important single sign in paediatric ophthalmology, because behind it may sit retinoblastoma, the commonest primary intraocular malignancy of childhood. The clinician who sees the white pupil and moves to the urgent referral without delay is the clinician who saves the eye and the life, because the early retinoblastoma is one of the most curable of all childhood cancers and the late retinoblastoma is one of the most lethal. [1][2]
Retinoblastoma is a tumour of the developing retina that arises from the immature cone-precursor cell, and it is driven by the loss of both copies of the RB1 tumour suppressor gene on the long arm of chromosome thirteen. The disease sits at the intersection of the paediatrics, the ophthalmology, the oncology and the clinical genetics, because it is at once a cancer, a genetic disease and a model of the tumour suppressor gene. The Knudson two-hit hypothesis, proposed by Alfred Knudson in nineteen-seventy-one to explain the age and the laterality of retinoblastoma, gave the world the concept of the tumour suppressor gene, and it is one of the most consequential ideas in all of oncology. The candidate who can hold the biology, the classification, the staging and the treatment in one frame holds the whole topic. [1][5][6]
The clinical gravity of the disease is concentrated in the speed of the referral and in the resource of the country. In the high-income setting, where the red reflex is checked at the newborn examination and the well-child visits, the retinoblastoma presents with the leukocoria or the strabismus, the disease is confined to the eye, and the survival is above ninety-five percent. In the low-income setting, where the red reflex is not checked and the specialist is far, the retinoblastoma presents with the proptosis, the orbital cellulitis or the intracranial spread, and the survival is below forty percent. The Global Retinoblastoma Study of the JAMA Oncology, which analysed over four thousand children across the world, showed that the single greatest determinant of the outcome of retinoblastoma is the national income, and it is the fact that the boards reward the candidate who can name. [2][3]
Classification
The classification of the child with a white pupil rests on two axes, and the examination rewards the candidate who holds both. The first is the differential diagnosis of leukocoria itself, the long list of the conditions that can produce the white pupillary reflex, from the retinoblastoma through the congenital cataract, the Coats disease, the retinopathy of prematurity, the persistent fetal vasculature and the toxocariasis. The second is the classification of the retinoblastoma once it is diagnosed, which is the International Intraocular Retinoblastoma Classification that grades the intraocular tumour by the size, the location and the seeding, and that drives the treatment from the focal therapy to the enucleation. [1][3]
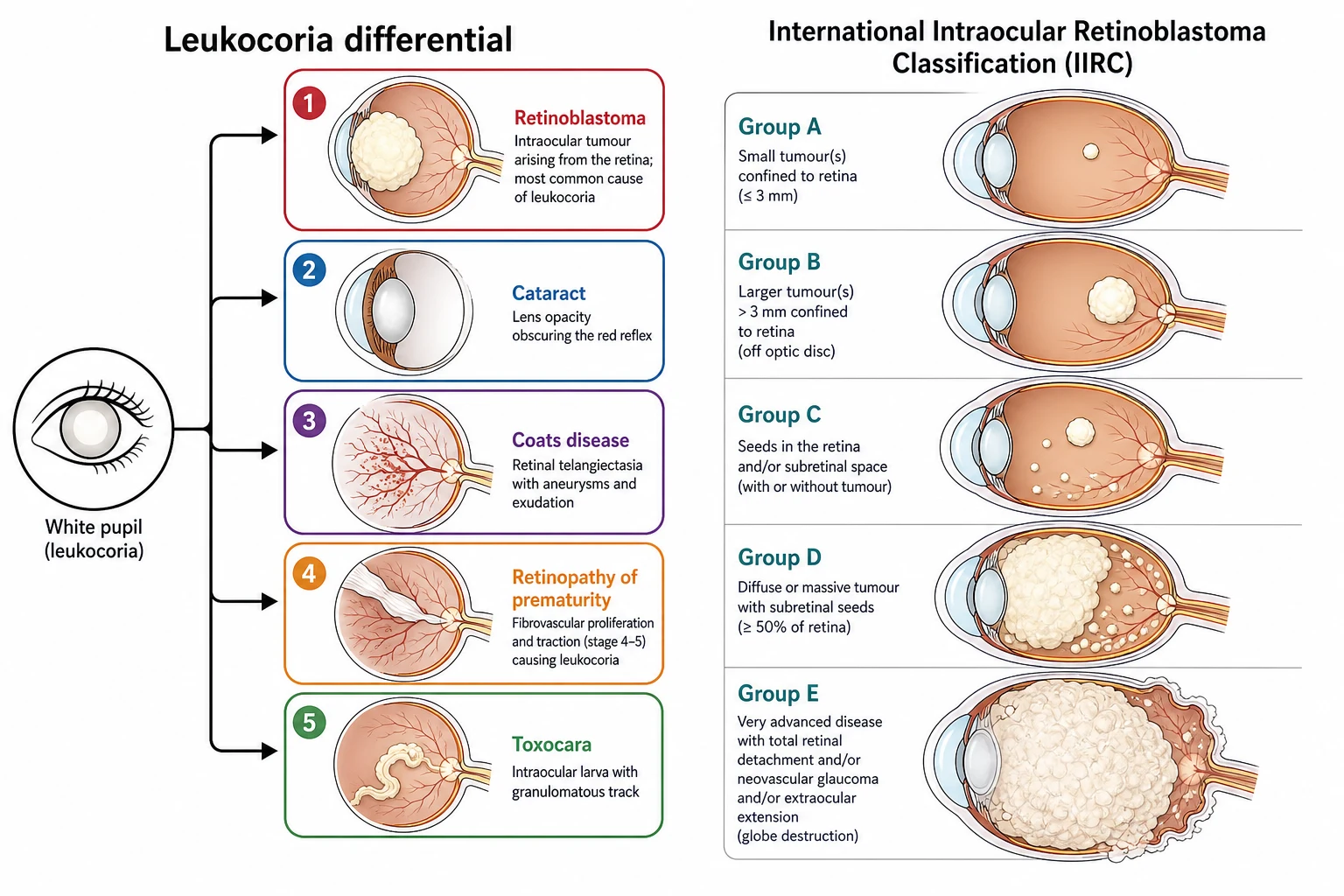
The differential diagnosis of leukocoria is the long list that the boards probe, and the candidate who can walk through it from the most dangerous to the commonest demonstrates the breadth the examiner rewards. The retinoblastoma is the most dangerous and the diagnosis that must never be missed, and it presents as the cream-white mass behind the lens with the dilated vessels and the possible strabismus. The congenital cataract is the commonest cause of the unilateral leukocoria in the high-income setting, and it presents as the opacity within the lens itself, visible without the ophthalmoscope. The Coats disease is the retinal telangiectasia with the exudative retinal detachment of the young boy, and it produces the yellow-white reflex behind the lens. The retinopathy of prematurity is the fibrovascular proliferation of the premature infant, and the persistent fetal vasculature is the remnants of the hyaloid vessel with the microphthalmia and the cataract. The toxocariasis is the nematode larval granuloma of the child exposed to the puppy, and the retinal coloboma is the sectoral defect of the inferior retina. [1][3]
The International Intraocular Retinoblastoma Classification, developed by Linn Murphree and the international consortium to replace the older Reese-Ellsworth classification, grades the intraocular disease by the features that predict the response to the chemotherapy and the focal therapy. The group A is the small tumour, three millimetres or less in the greatest dimension, confined to the retina and away from the optic disc and the foveola, and it carries an excellent prognosis with the focal therapy alone. The group B is the larger tumour or any tumour involving the macula or the disc, or any tumour with a subretinal fluid less than three millimetres from the margin, and it is treated with the chemotherapy and the focal consolidation. The group C is the tumour with the focal subretinal or vitreous seeds within three millimetres of the tumour, and the group D is the diffuse seeds or the massive tumour with the retinal detachment. The group E is the unsalvageable eye, defined by the massive tumour, the neovascular glaucoma, the invasion of the anterior chamber or the optic nerve, or the destruction of the globe, and it is treated by the enucleation. [1][3]
Group A
small, confined
- Tumour three millimetres or less
- Confined to the retina
- Away from the disc and the foveola
- Focal therapy alone, excellent prognosis
Group B
macula or disc
- Larger tumour or involvement of the macula or disc
- Subretinal fluid close to the margin
- Chemotherapy with focal consolidation
- Good globe salvage and vision
Group C
focal seeds
- Focal subretinal or vitreous seeds
- Within three millimetres of the tumour
- Chemotherapy plus focal and the chemosurgery
- Moderate globe salvage
Group D
diffuse seeds
- Diffuse subretinal or vitreous seeds
- Massive tumour or retinal detachment
- Intra-arterial chemotherapy or enucleation risk
- Reduced globe salvage
Group E
unsalvageable
- Massive tumour or globe destruction
- Neovascular glaucoma or anterior chamber invasion
- Enucleation is the treatment
- No hope of useful vision
Epidemiology & Risk Factors
Retinoblastoma is the commonest primary intraocular malignancy of childhood, and it accounts for roughly three percent of all childhood cancers. The incidence is estimated at one in fifteen thousand to one in twenty thousand live births, and it is consistent across the populations, with no strong racial or ethnic predilection. The disease is essentially confined to the young child, with the median age at diagnosis of about two years for the unilateral disease and under one year for the bilateral disease, and over ninety-five percent of cases present before the age of five years. The bilateral and the multifocal disease is the heritable germline form, and it accounts for roughly forty percent of the cases, while the unilateral disease is the non-heritable somatic form in the majority. [1][2]
The heritable and the non-heritable distinction is the central fact of the epidemiology and the genetic counselling, and it is the fact that the boards reward. The heritable retinoblastoma, which carries the germline mutation of the RB1 gene, presents earlier, with the bilateral or the multifocal disease, and it is passed with the near-complete penetrance of ninety percent or more. A parent with the heritable retinoblastoma has a fifty-percent chance of passing the mutated RB1 gene to each child, and the child who inherits the mutation will almost certainly develop the retinoblastoma, often in the first months of life. The non-heritable retinoblastoma, which carries the two somatic hits in a single retinal cell, presents later, with the unilateral unifocal disease, and it is not passed to the offspring. The candidate who can draw the family tree and explain the offspring risk is demonstrating the reasoning the boards reward. [1][7]
The most powerful risk factor is the family history, and the child with the affected parent or sibling demands the genetic testing and the early surveillance. Roughly ten percent of the retinoblastoma cases are familial, with the known germline RB1 mutation, and these children are enrolled in the surveillance protocol from the birth, with the regular examinations under anaesthesia that detect the tumour before the leukocoria and that salvage the vision. The environmental risk factors are weak and inconsistent, and no strong environmental cause has been identified for the somatic retinoblastoma. The access to the specialist care, the red reflex screening and the genetic testing varies by the geography and the socioeconomic status, and it is the equity issue that drives the global disparity in the outcome. [2][4]
Pathophysiology
The pathophysiology of retinoblastoma begins with the developing retina, because the tumour is a developmental disease of the immature retinal cell. The normal retina develops from the neuroblastic layer of the optic cup, and the retinoblastoma arises from the immature cone-precursor cell that retains the capacity to proliferate. The tumour appears in the young child because the retinal cell is still dividing and susceptible to the loss of the cell-cycle control, and it disappears as a risk after the retina matures and the cell-cycle control is established. The origin from the cone-precursor cell explains the characteristic features of the retinoblastoma, including the Flexner-Wintersteiner rosettes and the fleurettes on the histology, which are the attempts at the retinal differentiation. [1][3]
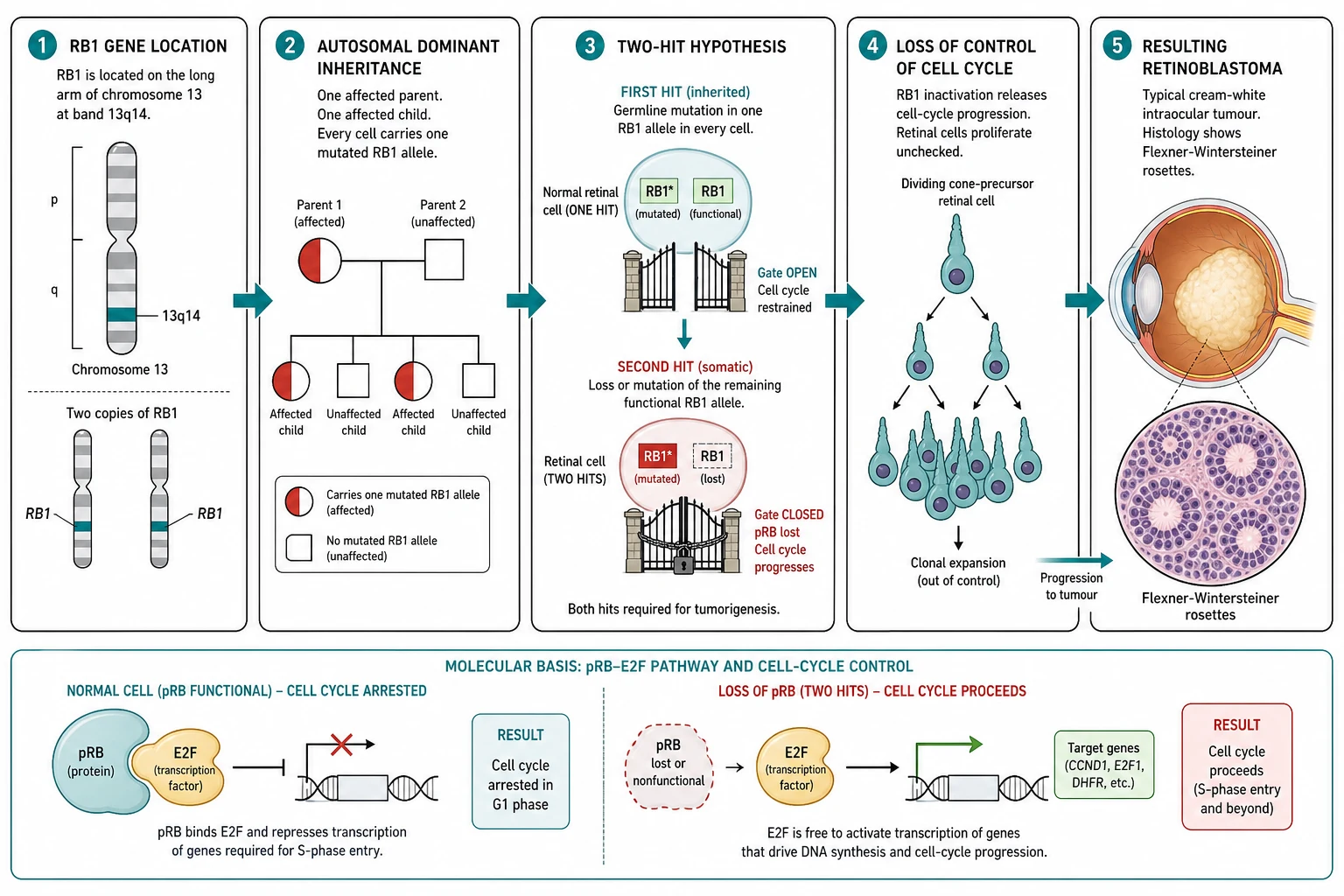
The RB1 gene on the long arm of chromosome thirteen, at the band thirteen-q-fourteen, is the prototypical tumour suppressor gene, and it is the gene that gave the world the concept. The RB1 gene encodes the pRB protein, a nuclear phosphoprotein that regulates the transition from the G1 to the S phase of the cell cycle. The pRB protein binds the E2F family of the transcription factors, and in the hypophosphorylated state it sequesters the E2F and arrests the cell cycle at the G1 restriction point. When the cell is ready to divide, the cyclin-dependent kinases phosphorylate the pRB, which releases the E2F and allows the cell to progress into the S phase. The loss of both copies of the RB1 gene removes the brake on the cell cycle, and the retinal cell proliferates uncontrollably into the retinoblastoma. [4][7]
The Knudson two-hit hypothesis is the model that unified the heritable and the non-heritable forms, and it is one of the most elegant ideas in oncology. Knudson observed that the bilateral retinoblastoma presented earlier and with the multiple tumours, while the unilateral retinoblastoma presented later and with a single tumour, and he reasoned that the difference reflected the number of the mutational events required. In the heritable form, the child inherits one mutated RB1 copy in every cell, and only one further somatic hit in a single retinal cell is needed to remove the remaining functional copy and to trigger the tumour; because there are millions of retinal cells, the probability of the second hit is high, and the disease is bilateral, multifocal and early. In the non-heritable form, both hits must occur as somatic events in a single retinal cell, and the probability is low, so the disease is unilateral, unifocal and later. The hypothesis predicted the existence of the tumour suppressor gene two decades before the RB1 gene was cloned, and it changed the science of cancer. [5][6][7]
The heritable retinoblastoma carries the risk of the second primary malignancy, and it is the risk that defines the survivorship. The child with the germline RB1 mutation carries the mutated RB1 copy in every cell, and the loss of the remaining copy in any tissue removes the tumour suppressor brake. The second primary malignancies include the osteosarcoma, the soft-tissue sarcoma, the melanoma and the brain tumour, and the risk is increased several-fold by the external beam radiotherapy, which is the reason the radiotherapy is now avoided wherever possible. The trilateral retinoblastoma, the combination of the bilateral retinoblastoma with the midline intracranial tumour of the pineal or the suprasellar region, is the most feared of the intracranial second primaries, and it carries a poor prognosis despite the treatment. [8][9]
Clinical Presentation
The child with retinoblastoma presents through several doors, and each presentation carries its own clue and its own examination favourite. The commonest presentation is the leukocoria, the white pupillary reflex, which is detected by the parents in the flash photograph or by the clinician at the red reflex test, and which accounts for roughly sixty percent of the presentations in the high-income setting. The leukocoria of the retinoblastoma is the cream-white mass behind the lens, and it may be the constant reflex or the reflex that appears only in the certain gaze or the certain light. The strabismus is the second commonest presentation, and it accounts for roughly twenty percent of the cases, because the tumour that sits behind the fovea destroys the central vision and the eye drifts; any new strabismus in a child under two years is retinoblastoma until the fundus is examined. [1][2]
The less common presentations are the ones that the boards probe, because they are the points where the diagnosis is missed or the harm is done. The red painful eye, the glaucoma and the pseudo-inflammatory presentation may mimic the orbital cellulitis or the uveitis, and the leukocoria may be hidden behind the inflammation. The proptosis and the orbital mass are the presentations of the advanced extraocular disease, and they are far more common in the low-resource setting where the diagnosis is delayed. The failure to thrive, the anorexia and the neurological signs are the presentations of the trilateral retinoblastoma with the intracranial mass. The family history of retinoblastoma in the infant under surveillance is the presentation that should never reach the leukocoria, because the surveillance detects the tumour before the symptom. [1][3]
The global disparity in the presentation is the fact that defines the prognosis, and the Global Retinoblastoma Study of the JAMA Oncology showed it with the clarity that the boards reward. In the high-income countries, over seventy percent of the children present with the leukocoria or the strabismus, and under five percent present with the proptosis or the extraocular disease. In the low-income countries, the leukocoria is missed, the specialist is far, and the child presents with the proptosis, the orbital cellulitis, the fungating mass or the metastatic disease, and roughly forty percent of the children die. The single greatest determinant of the outcome of retinoblastoma is the national income, because the income determines the speed of the referral and the access to the specialist treatment. [2][3]
Intra-arterial melphalan for the ophthalmic artery chemosurgery
Dose
Approximately three to five milligrams of melphalan per eye per session in the young child, often combined with topotecan, titrated to the age and the eye, with the dose derived from the body surface area to avoid the neutropenia
Differential Diagnosis
The differential diagnosis of leukocoria is the one that the boards probe most directly, and the task is to separate the retinoblastoma from the long list of the conditions that can produce the white pupillary reflex. The congenital cataract is the commonest cause of the unilateral leukocoria in the high-income setting, and it presents as the opacity within the lens itself, visible to the naked eye or with the direct ophthalmoscope, and it is distinguished from the retinoblastoma by the lens location of the opacity and the absence of the retinal mass. The Coats disease is the retinal telangiectasia with the exudative retinal detachment of the young boy, typically between four and ten years, and it produces the yellow-white reflex of the detached retina behind the lens, and it is distinguished by the older age and the telangiectatic vessels on the imaging. [1][3]
The retinopathy of prematurity is the fibrovascular proliferation of the premature infant, and the leukocoria is the late stage with the retinal detachment and the fibrotic mass behind the lens, and it is distinguished by the history of the prematurity, the low birth weight and the oxygen exposure. The persistent fetal vasculature, formerly the persistent hyperplastic primary vitreous, is the remnants of the hyaloid vessel, and it presents with the microphthalmia, the shallow anterior chamber and the retrolental fibrotic mass, and it is distinguished by the microphthalmia that the retinoblastoma does not cause. The toxocariasis is the nematode larval granuloma of the child exposed to the puppy soil, and it presents with the unilateral leukocoria or the endophthalmitis, and it is distinguished by the older age and the positive serology. The retinal coloboma is the sectoral defect of the inferior retina, and the retinal detachment may produce the leukocoria. [1][3]
Retinoblastoma
retinal mass
- Cream-white mass behind the lens
- Infant or toddler under three years
- May be bilateral, strabismus common
- MRI shows the enhancing intraocular mass with calcification
Congenital cataract
lens opacity
- Opacity within the lens itself
- Visible at the naked eye
- May be unilateral or bilateral
- Demands urgent surgery to prevent the amblyopia
Coats disease
telangiectasia
- Older boy four to ten years
- Unilateral exudative retinal detachment
- Telangiectatic retinal vessels
- No calcification on the ultrasound
Retinopathy of prematurity
premature infant
- Premature low-birth-weight infant
- Stage four or five retinal detachment
- History of oxygen exposure
- Bilateral in the typical case
The differential of the strabismus in the young child is the one that connects retinoblastoma to the general paediatric examination, and the principle is simple and absolute. Any new strabismus in a child, particularly under two years and particularly the unilateral, demands the fundus examination to exclude the retinoblastoma, because the tumour that sits behind the fovea destroys the central vision and the eye drifts. The infantile esotropia, the accommodative esotropia and the fourth-nerve palsy are the common causes of the childhood strabismus, but the fundus examination is the non-negotiable step in any new presentation, because the retinoblastoma must not hide behind a benign squint. The candidate who performs the red reflex test on every child with the strabismus is the candidate who does not miss the cancer. [1][2]
Clinical & Bedside Assessment
The bedside assessment of the child with a suspected leukocoria begins with the red reflex test, and it is the single most important skill in the topic. The test is performed in the dim room with the direct ophthalmoscope held close to the examiner's eye, at a distance of about thirty centimetres from the child, with the ophthalmoscope set to the zero or a low dioptre. The light is shone at both pupils simultaneously, and the examiner compares the colour and the brightness of the reflex in the two eyes. The normal reflex is the warm orange-red, symmetric between the two eyes, and it fills the pupil. The abnormal reflex is the white, the yellow-white, the asymmetric or the absent, and any abnormality demands the urgent ophthalmology referral. The test is performed at the newborn examination, at the six-week check and at every well-child visit, and the parent's observation of the white glow in the photograph is treated with the same urgency. [1][2]
The general examination of the child adds the context that the boards reward. The growth parameters, the dysmorphic features and the neurodevelopment are assessed, because the child with the syndromic context may carry the associated condition. The strabismus is sought with the cover test and the Hirschberg test, and any new strabismus in the young child is examined with the red reflex and the fundus. The skin is examined for the ash-leaf macules and the cafe-au-lait patches that point to the alternative diagnosis, and the abdomen is examined for the hepatosplenomegaly that would point to the metastatic or the alternative disease. The lymph nodes are mapped, and the nervous system is examined for the signs of the raised intracranial pressure or the focal deficit that would point to the trilateral retinoblastoma. [1][3]
The family history is the third pillar of the assessment, and the candidate who takes it carefully is demonstrating the breadth the boards reward. The parents are asked about the childhood eye disease, the enucleation, the blindness and the cancer in the family, because the heritable retinoblastoma is passed with the near-complete penetrance and the family may carry the undiagnosed mutation. The construction of the three-generation pedigree is the step that identifies the at-risk family and that triggers the genetic testing and the surveillance of the at-risk infant. The candidate who links the family history to the genetic counselling and the offspring risk is the candidate who holds the heritable dimension of the topic. [1][4]
Investigations
The investigation of the child with a suspected retinoblastoma is built around the examination under anaesthesia, the imaging and the genetic testing, and the biopsy is avoided because the tumour may seed along the needle track. The examination under anaesthesia is the cornerstone of the ocular assessment, and it combines the dilated fundus examination by the ophthalmologist with the indirect ophthalmoscopy, the fundus photography and the measurement of the tumour. The ophthalmologist maps every tumour, assesses the group and the laterality, and plans the focal therapy. The examination under anaesthesia is repeated at each cycle of the treatment to assess the response and to apply the focal consolidation. [1][3]
The imaging confirms the intraocular mass, assesses the extraocular extension and excludes the trilateral retinoblastoma. The ocular ultrasound is the first-line modality, and it shows the intraocular mass with the characteristic calcification that is the hallmark of the retinoblastoma and that distinguishes it from the Coats disease and the persistent fetal vasculature. The contrast-enhanced magnetic resonance imaging of the orbits and the brain is the standard for the staging, and it defines the intraocular tumour, the optic nerve invasion, the extraocular extension and the pineal or the suprasellar mass of the trilateral retinoblastoma. The computed tomography is avoided because of the radiation and the increased risk of the second malignancy in the heritable disease. The bone scan and the bone marrow are reserved for the suspected metastatic or the extraocular disease, which is rare in the high-income presentation. [1][8]
The genetic testing is the investigation that defines the heritable form and the offspring risk, and it is now the standard for every child with the retinoblastoma. The RB1 gene is sequenced from the blood and, where possible, from the tumour tissue of the enucleated eye, and the identification of the germline mutation confirms the heritable form and the fifty-percent offspring risk. The child with the bilateral disease has the heritable form by the definition, and the genetic testing is directed at the specific mutation for the family counselling and the prenatal or the preimplantation testing. The child with the unilateral disease may carry the germline mutation in roughly fifteen percent, and the genetic testing is performed to identify these children and to guide the surveillance and the offspring counselling. [4][7]
Management — Resuscitation
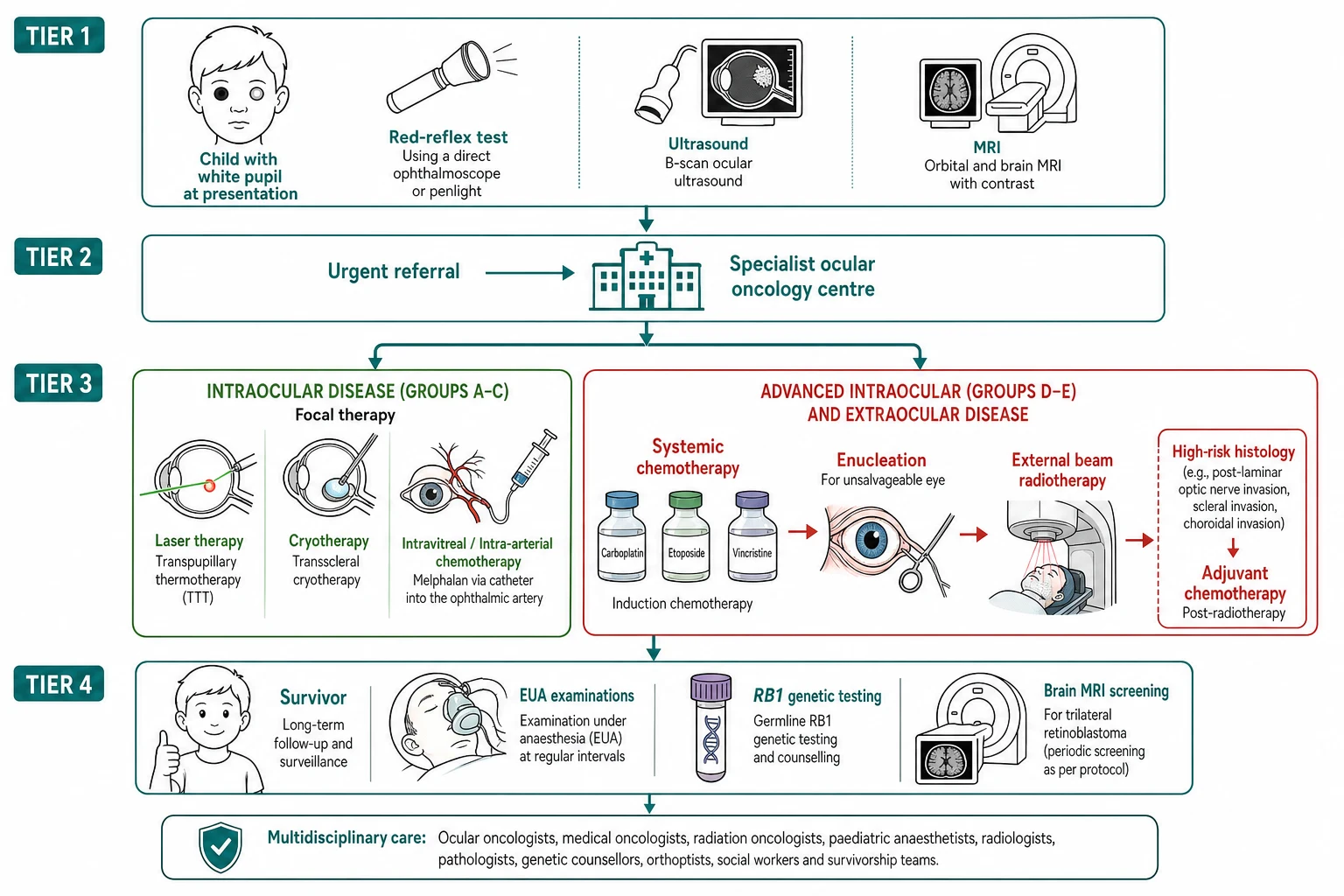
The resuscitation of the child with a newly suspected retinoblastoma is governed by the principle that the dangerous element is the delay, and the management begins with the urgent referral to the ophthalmology service. The child with the leukocoria or the abnormal red reflex is referred the same day, because the retinoblastoma grows fast and the delay of the weeks can convert a curable intraocular tumour into the lethal extraocular disease. The child with the proptosis, the orbital cellulitis or the signs of the raised intracranial pressure is managed as the emergency, with the imaging, the intravenous antibiotics if the infection is suspected and the urgent transfer to the specialist centre. [1][2]
The child with the advanced disease may present with the acute complications that demand the immediate management. The neovascular glaucoma causes the severe pain and the corneal oedema, and it is managed with the analgesia and the glaucoma control before the definitive treatment. The retinal detachment and the vitreous haemorrhage are managed by the specialist, and the enucleation may be the only option for the painful blind eye. The child with the trilateral retinoblastoma and the raised intracranial pressure is managed with the dexamethasone, the urgent imaging and the neurosurgical and the oncological pathway. The child with the metastatic disease in the bone or the marrow, which is the presentation of the delayed low-resource disease, is managed with the systemic chemotherapy before the definitive ocular treatment. [1][9]
The urgent pathway for the child with the leukocoria
Perform the red reflex test in the dim room and document the finding
Any white, yellow-white, asymmetric or absent reflex is abnormal
Refer the child the same day to the ophthalmology service
Do not wait, do not review in the weeks, because the delay costs the eye and the life
The ophthalmology service performs the examination under anaesthesia and the imaging
The ocular ultrasound and the magnetic resonance imaging confirm the retinoblastoma and stage the disease
Confirm the diagnosis and assign the International Intraocular Retinoblastoma Classification group
The group A through E drives the risk-adapted treatment
Begin the genetic testing for the RB1 gene
The heritable form is defined and the family counselling is begun
Management — Definitive & Stepwise
[1] [3]The definitive treatment of retinoblastoma is risk-adapted, and the goal is the cure of the life first, the salvage of the eye second and the preservation of the vision third, in that order. The treatment is delivered by the multidisciplinary ocular oncology team, and the plan is built around the laterality, the group and the staging. The focal therapy is the first line for the small tumour of the group A and the consolidation after the chemotherapy. The laser photocoagulation and the transpupillary thermotherapy are applied to the small tumour away from the macula, and the cryotherapy is applied to the anterior tumour. The focal therapy alone may cure the group A, and it is the consolidation that destroys the residual tumour after the chemotherapy has reduced the bulk. [1][3]
The ophthalmic artery chemosurgery is the modern advance that has transformed the treatment of the group D and the recurrent disease, and it is the treatment that the boards reward the candidate who names. The technique delivers the chemotherapy directly into the ophthalmic artery through the femoral catheter advanced under the fluoroscopic guidance by the interventional neuroradiologist, and it achieves the high intraocular concentration of the melphalan and the topotecan with the low systemic exposure. The melphalan is given at approximately three to five milligrams per eye per session, often combined with the topotecan, and the cycles are repeated every three to four weeks for two to six cycles. The chemosurgery salvages the eyes that would previously have been enucleated, and it has reduced the reliance on the external beam radiotherapy and its second-malignancy risk. [10]
The systemic chemotherapy is the treatment for the advanced intraocular disease, the extraocular disease and the high-risk histology, and it uses the combination of the carboplatin, the etoposide and the vincristine delivered over the several cycles. The systemic chemotherapy reduces the tumour bulk to allow the focal consolidation, and it treats the optic nerve invasion and the high-risk histology that carries the risk of the metastasis. The intravitreal melphalan is the salvage for the refractory vitreous seeds, delivered by the injection into the vitreous under the controlled conditions to avoid the tumour seeding. The external beam radiotherapy is reserved for the refractory disease, because the radiation increases the risk of the second malignancy and the facial growth disturbance in the heritable disease. [1][10]
The treatment journey of the intraocular retinoblastoma
The enucleation is the treatment of the unsalvageable eye, and it is the decision that the boards probe for the reasoning. The enucleation is performed for the group E eye with no hope of the useful vision, the neovascular glaucoma, the anterior chamber invasion or the optic nerve invasion, and it removes the source of the disease and allows the histology that defines the high-risk features. The enucleated eye is sent for the histology, and the optic nerve margin, the choroidal invasion and the anterior segment invasion are assessed, because the high-risk histology guides the adjuvant chemotherapy. The orbital implant is placed at the time of the enucleation to support the prosthesis and the facial growth, and the cosmetic and the psychological support is part of the management. [1][3]
Specific Subtypes & Scenarios
The trilateral retinoblastoma is the scenario that links the ocular disease to the intracranial danger, and it is the one the boards probe most directly. The trilateral retinoblastoma is the combination of the bilateral heritable retinoblastoma with the primitive neuroectodermal tumour of the pineal or the suprasellar region, and it arises from the cells that share the retinal lineage. The presentation is the raised intracranial pressure, the headache, the vomiting and the focal deficit, and it may appear months to years after the ocular diagnosis. The prognosis is poor, with the survival under twelve months despite the treatment, and the routine brain magnetic resonance imaging at the diagnosis and through the surveillance is the standard for the heritable disease. The Rodjan group showed that the routine brain screening on the admission detects the asymptomatic trilateral retinoblastoma, and it is the practice that the boards reward. [8][9]
The heritable retinoblastoma is the scenario that demands the genetic counselling and the lifelong surveillance, and it is the one that captures the depth of the topic. The child with the bilateral or the multifocal disease has the germline RB1 mutation, and the mutation is passed with the near-complete penetrance and the fifty-percent offspring risk. The genetic counselling addresses the diagnosis, the offspring risk, the prenatal and the preimplantation testing, and the lifelong surveillance for the second malignancy. The surveillance includes the regular skin examination, the avoidance of the radiation, the prompt assessment of any new mass or pain, and the vigilance for the bone tumour, the soft-tissue sarcoma and the melanoma. The candidate who links the genetics to the counselling and the surveillance is demonstrating the reasoning the boards reward. [4][7]
The five International Intraocular Retinoblastoma Classification groups
The unilateral retinoblastoma is the scenario that carries the surprise, and it is the one that the candidate must not dismiss. The unilateral disease is the non-heritable somatic form in the majority, but roughly fifteen percent of the children with the unilateral retinoblastoma carry the germline RB1 mutation, and these children carry the risk of the second tumour in the fellow eye and the second primary malignancy. The genetic testing is therefore performed on every child with the retinoblastoma, including the unilateral disease, because the identification of the germline mutation changes the surveillance and the counselling. The candidate who tests the genetics on every child is the candidate who does not miss the heritable form. [1][4]
Complications & Pitfalls
The complications of retinoblastoma divide into the disease-related and the treatment-related, and the candidate who holds both together is the one who manages the survivor as well as the acute patient. The disease-related complications are the loss of the vision, the neovascular glaucoma, the retinal detachment, the vitreous haemorrhage, the optic nerve invasion, the extraocular extension and the metastasis. The metastatic disease seeds to the bone, the bone marrow and the brain, and it is the presentation of the delayed low-resource disease, with the survival under twelve months despite the intensive treatment. The trilateral retinoblastoma is the intracranial complication of the heritable disease. [1][9]
The treatment-related complications are the immediate toxicities and the late effects, and the late effects dominate the survivorship. The systemic chemotherapy causes the myelosuppression, the febrile neutropenia, the hearing loss from the carboplatin and the secondary leukaemia from the etoposide. The ophthalmic artery chemosurgery causes the transient orbital oedema, the eyelid erythema, the neutropenia and the rare retinal vascular occlusion or the brain ischaemic event. The external beam radiotherapy causes the facial growth disturbance, the cataract, the dry eye and, most importantly, the increased risk of the second malignancy in the heritable disease. The enucleation removes the eye and demands the orbital implant and the psychological support. [1][10]
The classic diagnostic pitfalls are the ones the examiner probes, because they are the points where the diagnosis is missed or the harm is done. The leukocoria attributed to the photographic artefact, the strabismus attributed to the benign squint without the fundus examination, the red eye attributed to the conjunctivitis or the cellulitis without the dilation, and the proptosis attributed to the trauma are the errors that delay the diagnosis. The failure to perform the red reflex test at the newborn and the well-child examinations, the failure to refer the leukocoria the same day, and the failure to image and to test the genetics on the unilateral disease are the mistakes that change the outcome. [1][2]
Prognosis & Disposition
The prognosis of retinoblastoma is the most resource-dependent of any childhood cancer, and the survival figures span the range from the near-universal cure of the high-income presentation to the fatal outcome of the low-resource presentation. The high-income presentation, with the leukocoria and the strabismus, the intraocular disease and the access to the specialist treatment, carries a survival above ninety-five percent. The low-income presentation, with the proptosis and the extraocular disease and the delayed access, carries a survival below forty percent. The Global Retinoblastoma Study showed that the survival of retinoblastoma is the proxy for the national income, and it is the fact that the boards reward the candidate who can name with the clarity and the moral weight. [2][3]
The prognostic factors are the stage at the presentation, the laterality, the histology and the access to the treatment, and the candidate who holds them together is the one who reasons the prognosis. The intraocular disease carries an excellent prognosis, while the extraocular and the metastatic disease carry a poor prognosis. The unilateral disease carries the excellent prognosis of the localised tumour, while the bilateral disease carries the heritable form with its second-malignancy risk. The high-risk histology, with the optic nerve invasion beyond the lamina cribrosa, the massive choroidal invasion or the anterior segment invasion, guides the adjuvant chemotherapy. The heritable disease carries the lifelong risk of the second primary malignancy, which is the leading cause of the death in the adult survivor of the heritable retinoblastoma. [1][4]
The disposition of the child is the specialist ocular oncology centre, because the diagnosis, the staging, the genetic testing and the risk-adapted treatment demand the multidisciplinary team and the protocol-based care. The child with the heritable disease is managed in the centre that delivers the ophthalmic artery chemosurgery and the genetic counselling, and the child with the extraocular disease is managed jointly with the paediatric oncology. The survivor returns to the primary care and the long-term follow-up clinic, where the second malignancy is watched, the vision is supported, and the transition to the adult care and the genetic counselling for the family planning is planned. [1][4]
Special Populations
The infant with the heritable retinoblastoma is the patient in whom the surveillance transforms the outcome, and the approach is shaped by the early detection. The infant with the affected parent or the known family mutation is enrolled in the surveillance protocol from the birth, with the examinations under anaesthesia every few weeks in the first months of life, and the tumour is detected before the leukocoria and treated with the focal therapy that salvages the vision. The genetic testing is performed on the cord blood or the neonatal sample, and the infant who carries the mutation enters the surveillance, while the infant who does not is spared the examinations. The early detection is the reason the hereditary retinoblastoma in the high-resource setting now presents at the smaller stage and with the better outcome. [1][4]
The child with the heritable disease who reaches the survivorship is the patient in whom the second malignancy is the primary concern, because the heritable form carries the lifelong risk. The survivor is counselled about the second-malignancy risk, the avoidance of the radiation, the sun protection and the prompt assessment of any new mass, bone pain or persistent symptom. The surveillance includes the regular skin examination, the vigilance for the bone and the soft-tissue tumour, and the brain imaging for the trilateral retinoblastoma through the early years. The genetic counselling for the family planning, with the prenatal and the preimplantation testing, is offered to the adult survivor who wishes to have the children, and the candidate who links the survivorship to the counselling is demonstrating the breadth the boards reward. [4][7]
The socioeconomic disadvantage, the remoteness and the migrant or the refugee status affect the access to the red reflex screening, the ophthalmology referral and the specialist treatment, and the indigenous and the remote populations carry the later presentation and the worse outcome in the regions where the access is limited. The global disparity in the survival of retinoblastoma is the equity issue that the candidate who acknowledges the social determinants demonstrates the depth the boards reward. The family from the remote area may face the prolonged separation during the months of the treatment, and the social work, the educational liaison and the telehealth support are part of the multidisciplinary care. [2][3]
Evidence, Guidelines & Regional Differences
The landmark evidence for retinoblastoma is the body of the genetic, the clinical and the global studies that have refined the understanding and the treatment. The Knudson two-hit hypothesis, published in nineteen-seventy-one, unified the heritable and the non-heritable forms and predicted the tumour suppressor gene, and it is the idea that changed the science of cancer. The cloning of the RB1 gene in nineteen-eighty-six confirmed the prediction, and the molecular biology has since defined the RB1 gene on chromosome thirteen, the pRB protein and the cell-cycle regulation. The genomic landscape review of Therault and colleagues summarised the molecular biology, and it established the RB1 gene as the gateway lesion with the additional driver mutations that include the MYCN amplification in the rare RB1-negative retinoblastoma. [4][5][6]
The Global Retinoblastoma Study of the JAMA Oncology, published in twenty-twenty, analysed over four thousand children across the world and showed that the national income is the single greatest determinant of the presentation and the outcome. The high-income countries present with the leukocoria and the strabismus, the intraocular disease and the survival above ninety-five percent, while the low-income countries present with the proptosis and the extraocular disease and the survival below forty percent. The study is the call to the action for the global health, and it is the evidence that the boards reward the candidate who can cite. [2][3]
The standard of care across the high-resource regions, including Australasia, the United Kingdom, the United States and Canada, is the risk-adapted treatment delivered by the multidisciplinary ocular oncology team, with the focal therapy, the ophthalmic artery chemosurgery, the systemic chemotherapy and the enucleation. The ophthalmic artery chemosurgery has reduced the reliance on the external beam radiotherapy and its second-malignancy risk, and it is the standard for the group D and the recurrent disease. The genetic testing of the RB1 gene is the standard for every child, and the surveillance of the at-risk infant with the known family mutation is the standard from the birth. The regional differences are concentrated in the access and the timing, with the rural and the remote populations and the low-resource settings carrying the later presentation and the worse outcome.
[1][2]The controversies are the ones that the candidate can discuss without pretending to resolve them. The optimal combination of the chemosurgery and the systemic chemotherapy, the role of the intravitreal melphalan and the risk of the tumour seeding, the role of the external beam radiotherapy in the heritable disease, and the role of the adjuvant chemotherapy for the high-risk histology are the open questions. The evidence is weakest in the rare RB1-negative retinoblastoma and in the resource-limited setting, where the access to the chemosurgery, the genetic testing and the surveillance is limited. The precision medicine approaches, including the gene therapy and the targeted therapy, are the emerging treatments that the candidate may name as the future direction. [1][10]
Exam Pearls
The high-yield facts for the examination are the ones that the examiner probes and that the candidate must carry without hesitation. The leukocoria as the red-flag sign, the RB1 tumour suppressor gene on chromosome thirteen, the Knudson two-hit hypothesis, the International Intraocular Retinoblastoma Classification of groups A through E, the urgent same-day referral, and the ophthalmic artery chemosurgery are the core of the topic. The heritable bilateral disease with its second-malignancy risk, the trilateral retinoblastoma, and the global disparity in the survival complete the picture. The candidate who carries the red reflex test, the urgent referral and the risk-adapted treatment holds the whole topic. [1][2]
The red reflex test is the single most testable skill in the topic, and the principle is simple and absolute. The red reflex is checked in the dim room with the direct ophthalmoscope at thirty centimetres from the child, comparing the colour and the brightness of the two eyes, and any white, yellow-white, asymmetric or absent reflex is abnormal. The test is performed at the newborn examination, at the six-week check and at every well-child visit, and the parent's observation of the white glow in the photograph is treated with the same urgency. The abnormal red reflex is retinoblastoma until proven otherwise, and it is referred the same day to the ophthalmology service, because the delay of the weeks can convert the curable intraocular tumour into the lethal extraocular disease. [1][2]
References
- [1]Dimaras H, Corson TW, Coburn B, et al Retinoblastoma. Nat Rev Dis Primers, 2015.PMID 27189421
- [2]Global Retinoblastoma Study Group Global Retinoblastoma Presentation and Analysis by National Income Level. JAMA Oncol, 2020.PMID 32105305
- [3]Dimaras H, Dimarts A, Berman H, et al Retinoblastoma, the visible CNS tumor: A review. J Neurosci Res, 2019.PMID 29314142
- [4]Therault BL, Dimaras H, Gallie BL, Corson TW The genomic landscape of retinoblastoma: a review. Clin Exp Ophthalmol, 2014.PMID 24433356
- [5]Chernoff J The two-hit theory hits 50. Mol Biol Cell, 2021.PMID 34735271
- [6]Mastrangelo D, De Francesco S, Hadjistilianou T, Lore C, Gallie B, Megiorni F, Marsilio N, Cevenini G, Pizz L, Moscardini S, Loré C The retinoblastoma paradigm revisited. Med Sci Monit, 2008.PMID 19043380
- [7]Sabado Alvarez C, Rodriguez de la Rua E, Sanchez Sanchez R, et al Molecular biology of retinoblastoma. Clin Transl Oncol, 2008.PMID 18628066
- [8]Rodjan F, de Graaf P, van der Valk P, et al Trilateral retinoblastoma: neuroimaging characteristics and value of routine brain screening on admission. J Neurooncol, 2012.PMID 22802019
- [9]Mouratova T Trilateral retinoblastoma: a literature review, 1971-2004. Bull Soc Belge Ophtalmol, 2005.PMID 16281731
- [10]Taich P, Ceciliano O, Buitrago E, et al Topotecan Delivery to the Optic Nerve after Ophthalmic Artery Chemosurgery. PLoS One, 2016.PMID 26959658