Paeds · paediatric-dermatology
Genodermatoses and neurocutaneous skin findings
Also known as Neurofibromatosis type 1 · NF1 · Tuberous sclerosis complex · TSC · Sturge-Weber syndrome · Phakomatosis · Neurocutaneous syndrome · Café-au-lait macule · Angiofibroma · Port-wine stain
Fellowship topic on paediatric genodermatoses and neurocutaneous skin findings: the three classic phakomatoses — neurofibromatosis type 1 with its café-au-lait macules, skinfold freckling, neurofibromas and the NIH diagnostic criteria; tuberous sclerosis complex with its hypomelanotic ash-leaf macules, facial angiofibromas, shagreen patch and the major-minor diagnostic criteria; and Sturge-Weber syndrome with its forehead port-wine stain, leptomeningeal angioma and glaucoma. The shared biology is the RAS-MAPK and mTOR signalling pathways — NF1 loss unleashes RAS-MAPK through absent neurofibromin, TSC1 or TSC2 loss drives mTOR overactivation, and a somatic mosaic GNAQ mutation fixes the Sturge-Weber port-wine stain. Management turns on recognising that the skin lesion is the entry point to brain and systemic disease: optic pathway glioma and malignant peripheral nerve sheath tumour surveillance in NF1, vigabatrin for infantile spasms and everolimus for subependymal giant cell astrocytoma in TSC, and seizure, glaucoma and stroke-like-episode care in Sturge-Weber — with ANZ, UK, US and Canada guidance.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
One pathway family, three syndromes — the RAS-MAPK link
Overview & Definition
Picture a four-year-old brought in because the teacher has noticed brown patches on the skin and some difficulty with reading, or a newborn with a dark-red stain covering half the forehead, or an infant suddenly flexing in clusters with a faint white leaf-shape on the back. Each is the everyday face of a neurocutaneous syndrome, and each demands that the clinician see the skin sign as a window onto the brain. [3] [11]
Genodermatoses are inherited skin disorders, and the neurocutaneous syndromes — historically called phakomatoses — are those in which the skin lesion travels with disease of the brain, eye and other organs. The three that dominate paediatric practice are neurofibromatosis type 1, tuberous sclerosis complex and Sturge-Weber syndrome. They are united less by a single lesion than by a lesson: the visible cutaneous marker is often the earliest and most reliable clue to hidden neurological, ophthalmological and systemic disease. [1] [4]
The clinician's task has three layers. The first is to recognise the skin sign and name the syndrome against a set of diagnostic criteria, because the criteria drive the surveillance that follows. The second is to find the hidden complications early — the optic pathway glioma, the epilepsy, the glaucoma, the cardiac or renal tumour, the malignant transformation — before they damage the developing brain. The third is to coordinate the multidisciplinary care that these multisystem diseases demand, because no single organ defines the outcome. [1] [5]
Classification
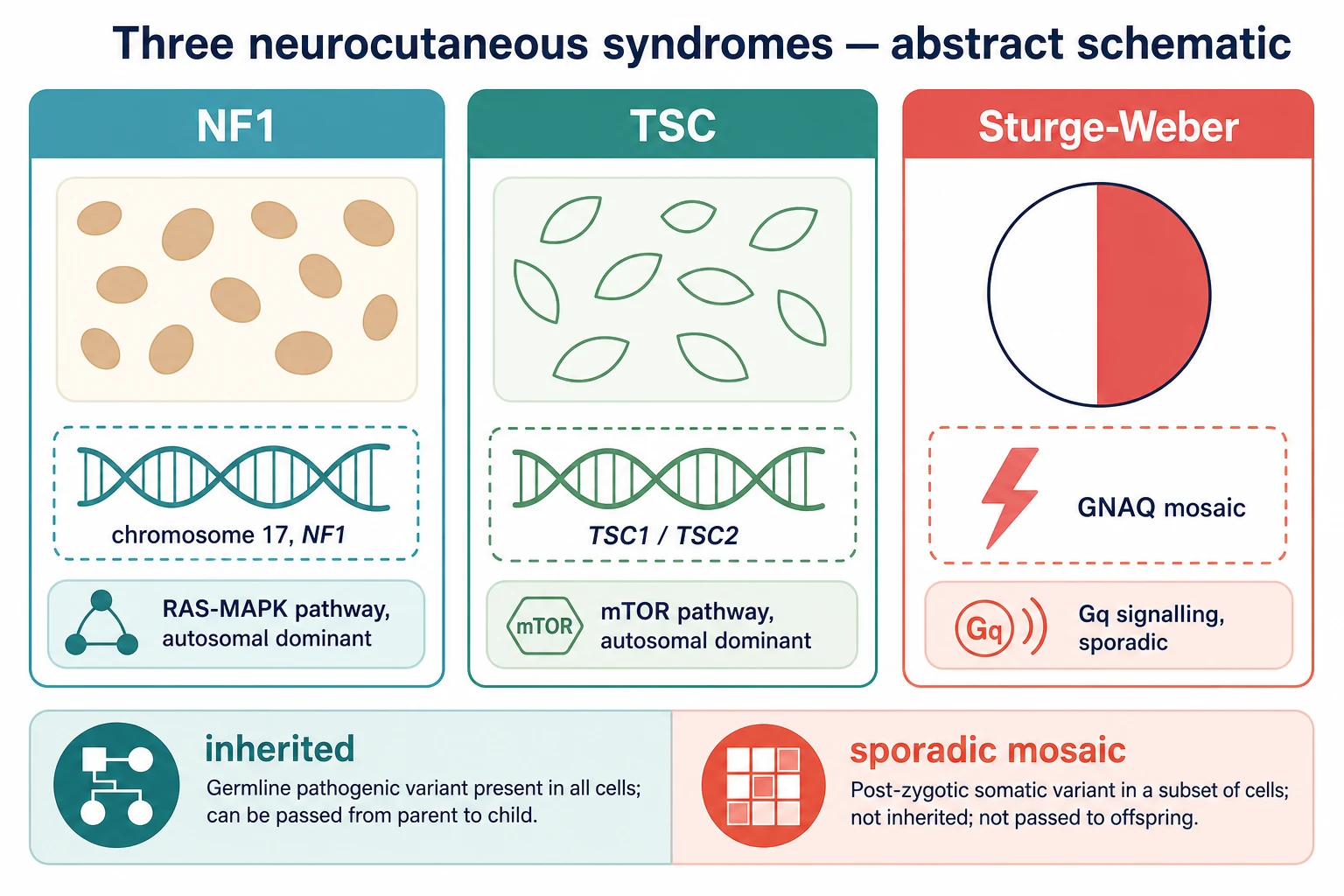
Classify these children by the diagnostic criteria of each syndrome, because the criteria are what turn a skin observation into a surveillance pathway. Each syndrome has its own rulebook, and the examiner tests the criteria relentlessly. [2] [6]
Neurofibromatosis type 1 is diagnosed by the NIH consensus criteria, which require two or more of the following: six or more café-au-lait macules of at least five millimetres before puberty or fifteen millimetres after puberty; axillary or inguinal freckling; two or more neurofibromas of any type or one plexiform neurofibroma; optic pathway glioma; two or more Lisch nodules; a characteristic osseous lesion such as sphenoid wing dysplasia or tibial pseudarthrosis; or a first-degree relative with neurofibromatosis type 1. About half of cases are familial and half arise from a new mutation. [2] [1]
Tuberous sclerosis complex is diagnosed by a pathogenic TSC1 or TSC2 variant, or by clinical criteria — two major features, or one major plus two minor features, make a definite diagnosis. The major features include three or more hypomelanotic macules, three or more angiofibromas or a fibrous cephalic plaque, two or more ungual fibromas, the shagreen patch, cortical dysplasia, subependymal nodules, the subependymal giant cell astrocytoma, cardiac rhabdomyoma and renal angiomyolipoma. The minor features include confetti skin lesions, dental enamel pits and non-renal hamartomas. [6] [5]
Sturge-Weber syndrome has no diagnostic criteria list in the same sense; it is a clinical diagnosis defined classically by the triad of a facial port-wine stain in the forehead or upper-eyelid territory, an ipsilateral leptomeningeal angioma and glaucoma. The port-wine stain distribution matters most: involvement of the forehead and upper eyelid carries the highest risk of brain and eye involvement, and an isolated port-wine stain elsewhere is far less likely to be syndromic. [9] [11]
Epidemiology & Risk Factors
Neurofibromatosis type 1 is the commonest of the three, with a prevalence of about one in three thousand and no ethnic predilection. It is autosomal dominant with complete penetrance but highly variable expression, so two children with the same mutation can look entirely different. About half of cases are familial and half are new mutations, so a negative family history never excludes it. [4] [3]
Tuberous sclerosis complex affects about one in six thousand to one in ten thousand live births. Like neurofibromatosis type 1 it is autosomal dominant with variable expression, and two-thirds of cases arise from new mutations. TSC2 mutations tend to produce a more severe phenotype than TSC1, and the contiguous-gene deletion of TSC2 and PKD1 causes severe polycystic kidney disease in infancy. [5] [6]
Sturge-Weber syndrome is rare, affecting roughly one in twenty to one in fifty thousand, and is entirely sporadic. The somatic mosaic GNAQ mutation arises after fertilisation, so the lesion is confined to the developmental territory it arises in, siblings are not at risk, and the parents can be reassured that recurrence is close to zero. The same GNAQ mutation, in a different territory and burden, produces an isolated port-wine stain without brain involvement — which is why most port-wine stains are not Sturge-Weber. [9] [12]
The epidemiological signal that changes practice is the distribution of the skin lesion. A port-wine stain on the forehead or upper eyelid carries a materially higher risk of leptomeningeal angioma and glaucoma than one on the cheek or limb. A segmental or large facial lesion is the trigger to image the brain and review the eye, while a lesion in a non-forehead territory can usually be managed as an isolated birthmark. [10] [11]
In Australia and Aotearoa New Zealand, the neurocutaneous syndromes are managed through specialist genetics, neurology and ophthalmology services in the major paediatric centres, with outreach clinics and telehealth linking rural and remote families to multidisciplinary surveillance. Equity of access to magnetic resonance imaging, electroencephalography and ophthalmology shapes the developmental outcome, particularly for Aboriginal and Torres Strait Islander and Maori and Pasifika children in remote settings. [5]
Pathophysiology
Why do café-au-lait macules, ash-leaf macules and a port-wine stain each travel with brain disease? The answer is that all three are disorders of cell-growth signalling, and the skin, brain and eye share the affected pathway. [4] [5]
Neurofibromatosis type 1 is caused by loss-of-function of the NF1 gene on chromosome 17, whose product neurofibromin is a brake on the RAS proto-oncogene. Without neurofibromin, RAS stays active and the RAS-MAPK and mTOR cascades overfire, driving the proliferation of melanocytes that produces café-au-lait macules and of Schwann cells that produces neurofibromas. The same pathway dysregulation underlies the optic pathway glioma, the learning and behavioural phenotype, and the small lifetime risk of malignant peripheral nerve sheath tumour. [4] [1]
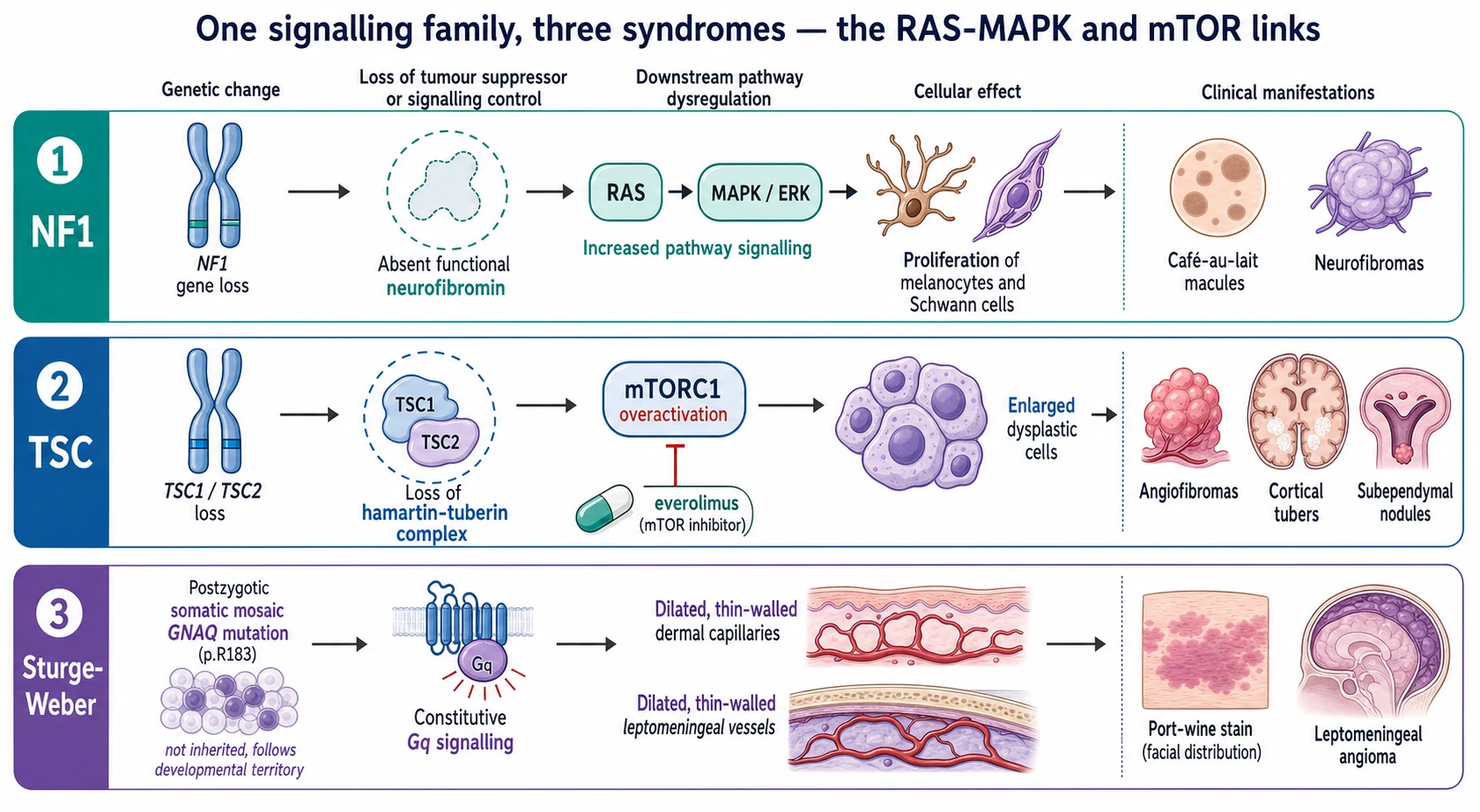
Tuberous sclerosis complex is caused by loss-of-function of TSC1 (encoding hamartin) or TSC2 (encoding tuberin). The hamartin-tuberin complex is the brake on mTORC1, so its loss drives unchecked mTORC1 signalling and the growth of dysplastic hamartomatous cells in the skin, brain, kidney, heart and lung. This produces the cortical tubers, subependymal nodules and subependymal giant cell astrocytomas in the brain, the angiofibromas and shagreen patch in the skin, and the cardiac rhabdomyoma and renal angiomyolipoma. It is why an mTOR inhibitor can shrink the astrocytoma and the angiomyolipoma. [5] [8]
Sturge-Weber syndrome runs on different biology entirely. A somatic activating mutation in GNAQ, classically the p.R183 residue, arises in a cell during early embryonic development and is passed only to the descendants of that cell, confining the disease to one developmental territory. The mutation drives constitutive Gq signalling, which leaves a network of dilated thin-walled capillaries and veins in the dermis — the port-wine stain — and in the leptomeninges, where the impaired venous drainage underlies seizures, stroke-like episodes and the progressive neurological decline. [9] [10]
Clinical Presentation
The café-au-lait macule is the gateway sign of neurofibromatosis type 1. It is a flat, uniformly tan or light-brown patch with a smooth border, present from the first year of life and increasing in number and size through early childhood. Skinfold freckling — small freckles in the axillae, groin and base of the neck — appears later, around three to five years. Cutaneous neurofibromas, soft flesh-coloured nodules that buttonhole through the skin, emerge in late childhood and adolescence, while the larger plexiform neurofibroma is a congenital diffuse soft-tissue swelling that may be present from birth. [3] [4]
The non-cutaneous face of neurofibromatosis type 1 is what does the harm. The optic pathway glioma presents with visual loss, proptosis or precocious puberty, though most are asymptomatic and found on surveillance. Learning difficulty, attention-deficit and behavioural problems are the commonest neurodevelopmental feature and often the presenting concern. Hypertension may reflect renal artery stenosis or a phaeochromocytoma, scoliosis and long-bone bowelling reflect the bony dysplasia, and a plexiform neurofibroma that grows rapidly, becomes painful or develops a new neurological deficit raises the spectre of malignant peripheral nerve sheath tumour. [1] [4]
The ash-leaf macule is the earliest cutaneous sign of tuberous sclerosis, often visible at birth as a white, leaf-shaped hypopigmented patch best seen under ultraviolet light. The facial angiofibromas — pink-red papules over the nose and cheeks once called adenoma sebaceum — appear in mid-childhood, as does the shagreen patch, a flesh-coloured cobblestoned plaque in the lumbosacral area, and the ungual fibromas around the nails. The cardiac rhabdomyoma may cause a murmur or arrhythmia in the newborn, while renal angiomyolipoma and epilepsy declare themselves across childhood. [5] [7]
[6] [2]For Sturge-Weber syndrome the port-wine stain is present at birth as a flat, pink to dark-red patch in the forehead and upper-eyelid territory, growing only with the child and never fading. The neurological presentation is seizures — often onset in the first two years, frequently prolonged or in clusters, sometimes infantile spasms — with hemiparesis, developmental delay and visual field defects on the side opposite the stain. Glaucoma may present with a cloudy cornea, tearing or buphthalmos in infancy, and stroke-like episodes with transient weakness or visual loss often accompany intercurrent illness. [10] [12]
Differential Diagnosis
A child with multiple café-au-lait macules raises the question of neurofibromatosis type 1, but café-au-lait macules occur in other RASopathies too. Legius syndrome, caused by SPRED1 loss, produces multiple café-au-lait macules and skinfold freckling but not the neurofibromas, Lisch nodules or tumours of neurofibromatosis type 1, and it is the key differential when the skin signs are present but the full criteria are not. The other RASopathies — Noonan, Noonan-with-lentigines, Costello and cardio-facio-cutaneous syndromes — add short stature, characteristic facies and cardiac disease. [4] [3]
A few café-au-lait macules in an otherwise well child are common and benign. The diagnostic threshold of six or more, in the required size for age, exists precisely to separate the common isolated macule from neurofibromatosis type 1, so a single or small number of macules in a child with no family history and no other features needs only reassurance and watchful surveillance. [2] [4]
A facial port-wine stain must be distinguished from the fading salmon patch of the newborn eyelid and nape, which resolves in the first years and never signifies Sturge-Weber. A port-wine stain is present at birth, is darker, does not fade and grows with the child. The syndromic concern rises with the territory: a stain in the forehead or upper eyelid carries the risk of leptomeningeal angioma and glaucoma, while a stain confined to the lower face or limb rarely does. [9] [11]
Clinical & Bedside Assessment
Assessment begins with the skin under good light and a Wood or ultraviolet lamp. Count and measure the café-au-lait macules, mapping each against the age-based NIH threshold, and look specifically in the axillae and groin for freckling. Examine for cutaneous and plexiform neurofibromas, assess the face for angiofibromas and the lumbosacral area for a shagreen patch, and examine the nails for ungual fibromas. Under ultraviolet light, look for the hypopigmented ash-leaf and confetti macules of tuberous sclerosis. [2] [6]
Examine the eyes in every child with a suspected neurocutaneous syndrome. Inspect for the iris hamartomas — the Lisch nodules — of neurofibromatosis type 1, assess visual acuity and eye movements for an optic pathway glioma, and measure for glaucoma and check the red reflex in the infant with a port-wine stain. The ophthalmological finding often confirms the diagnosis before any imaging. [1] [10]
Measure the blood pressure in every child with neurofibromatosis type 1, because hypertension from renal artery stenosis or phaeochromocytoma is a treatable and easily missed complication. Examine the spine for scoliosis, the long bones for bowing or a pseudarthrosis, and any plexiform lesion for change in size, texture or tenderness. For tuberous sclerosis, listen to the heart for a rhabdomyoma murmur and examine the abdomen for a renal mass. [4] [7]
Take a careful three-generation family history, because neurofibromatosis type 1 and tuberous sclerosis are autosomal dominant and a parent may be mildly affected. Examine the parents where possible — their skin and eyes may reveal an unsuspected diagnosis that refines the child's risk and the family's counselling. A negative family history is expected in Sturge-Weber, where the reassurance that the condition is sporadic and non-recurrent is itself part of the assessment. [3] [9]
Investigations
Most neurocutaneous syndromes are diagnosed clinically against criteria, and the investigation effort goes into the surveillance of the hidden complications rather than into confirming what the skin already shows. Genetic testing confirms and refines, but it rarely makes the diagnosis alone when the clinical criteria are met. [1] [5]
For neurofibromatosis type 1, genetic testing of the NF1 gene is increasingly available and useful when the clinical criteria are incomplete, in young children, or for counselling and trial eligibility. Surveillance imaging is selective: routine magnetic resonance imaging of the brain in an asymptomatic child is not required, but a child with visual symptoms or precocious puberty needs dedicated optic pathway imaging, and a changing plexiform lesion needs imaging and consideration of biopsy. Annual blood pressure, growth, and developmental and learning assessment are the core surveillance. [4] [1]
For tuberous sclerosis, the brain is imaged at diagnosis with magnetic resonance imaging to map the cortical tubers, subependymal nodules and any subependymal giant cell astrocytoma, and at intervals thereafter because the astrocytoma can grow and obstruct the foramen of Monro. An electroencephalogram is obtained for any seizure suspicion, and an echocardiogram and renal ultrasound screen for the cardiac rhabdomyoma and renal angiomyolipoma. Genetic testing of TSC1 and TSC2 confirms the diagnosis in the majority. [5] [7]
For Sturge-Weber syndrome, a contrast-enhanced brain magnetic resonance image with gadolinium defines the leptomeningeal angioma, the cortical calcification and any asymmetry, and is the cornerstone of confirming the brain involvement that the port-wine stain threatens. An electroencephalogram characterises the seizures, and ophthalmology with tonometry assesses for glaucoma. Genetic testing for the GNAQ mutation is available but the diagnosis remains clinical in most cases. [10] [12]
Management — Resuscitation
Resuscitation belongs to the acute neurological and ophthalmological presentations, not to the stable child with a skin sign. The emergencies are status epilepticus, raised intracranial pressure from a growing astrocytoma, and acute visual loss or refractory glaucoma. [7] [10]
An infant with tuberous sclerosis presenting with infantile spasms needs an electroencephalogram and prompt treatment, because early control of the spasms and the hypsarrhythmia protects the developing brain. Vigabatrin is the first-line agent for infantile spasms in tuberous sclerosis, started without delay once the diagnosis is confirmed, with ophthalmological awareness of the irreversible visual-field toxicity that accompanies its use. A child who fails vigabatrin needs paediatric neurology and consideration of adrenocorticotropic hormone or other therapies. [7] [5]
A child with a subependymal giant cell astrocytoma that is enlarging and obstructing the foramen of Monro presents with signs of raised intracranial pressure — headache, vomiting, deteriorating vision or declining function. This is a neurosurgical and oncological emergency: urgent imaging, neurosurgical decompression where there is hydrocephalus, and the mTOR inhibitor everolimus for the growing lesion. [8] [7]
For Sturge-Weber, the emergency is the prolonged or clustering seizure, the stroke-like episode with acute hemiparesis, and the infant with acute glaucoma. Treat the seizure actively along the standard status epilepticus pathway, assess for raised intraocular pressure in any irritable infant with a port-wine stain, and involve neurology and ophthalmology early. Keep resuscitation separate from routine surveillance: the stable child needs multidisciplinary clinic review, while the acutely presenting child needs the hospital and the specialist team. [10] [12]
Management — Definitive & Stepwise
Definitive management is a lifelong, syndrome-specific surveillance pathway matched to risk, because these are chronic multisystem diseases whose complications declare themselves across childhood. The unifying principle is that the skin sign identifies the child, and the multidisciplinary team surveils the brain, eye and organs. [1] [5]
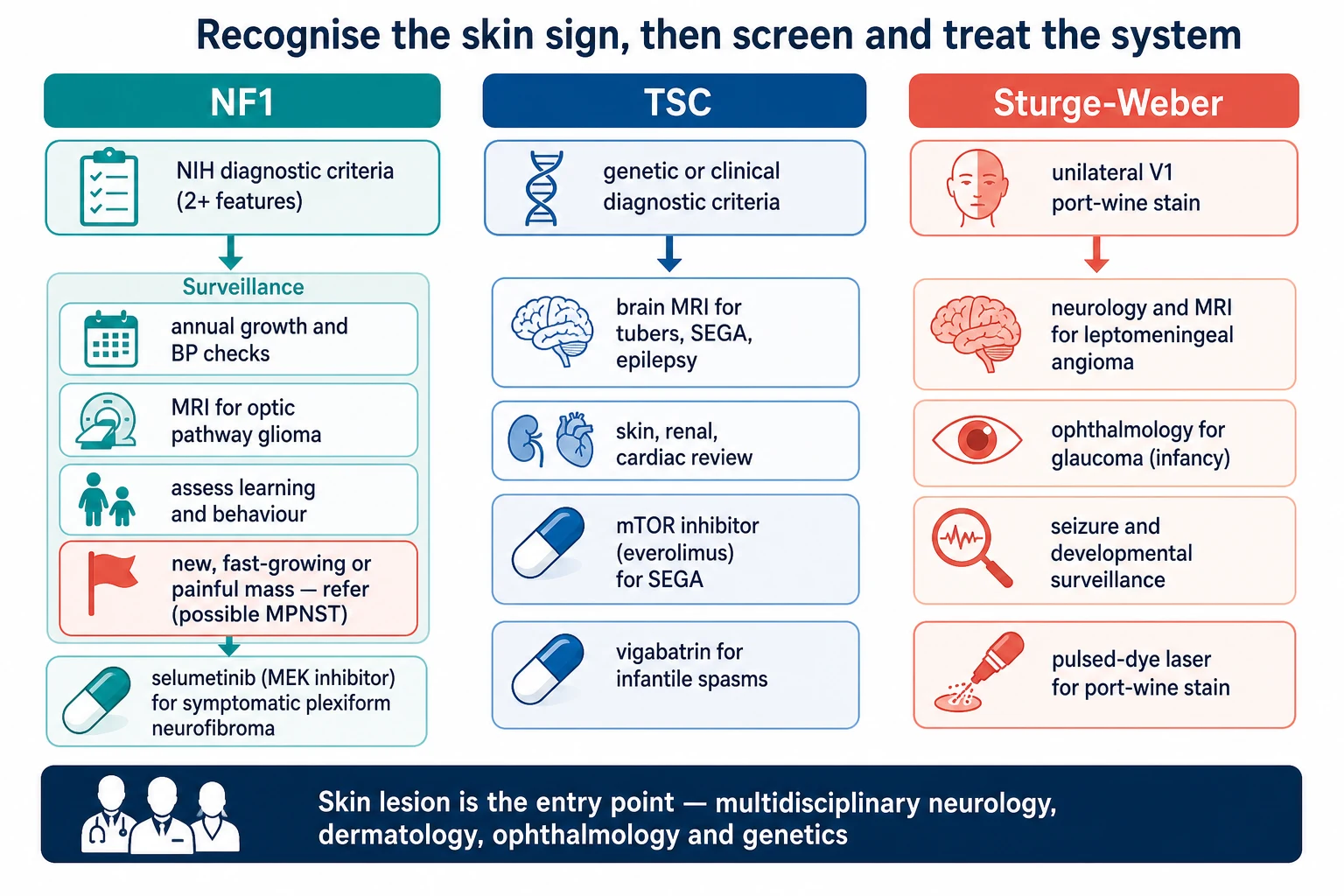
For neurofibromatosis type 1, the core of management is annual review in a medical home: growth and puberty, blood pressure, skin and plexiform lesion assessment, and developmental, learning and behavioural surveillance. Vision screening for the optic pathway glioma is undertaken at least annually through the early school years, with dedicated imaging for any visual change or precocious puberty. A changing, painful or rapidly enlarging plexiform lesion is referred urgently for imaging and biopsy to exclude a malignant peripheral nerve sheath tumour, and a symptomatic, inoperable plexiform neurofibroma may be treated with a MEK inhibitor such as selumetinib under specialist care. [4] [1]
For tuberous sclerosis complex, management follows the consensus surveillance schedule. The brain is imaged at diagnosis and at intervals for the subependymal giant cell astrocytoma, epilepsy is treated early and aggressively — vigabatrin first for infantile spasms, then standard antiseizure medicines, with epilepsy surgery considered early for drug-resistant focal epilepsy — and the growing astrocytoma is treated with everolimus, the mTOR inhibitor that reduced lesion volume in the phase-three EXIST-1 trial. The skin, kidney and heart are screened and managed across the multidisciplinary clinic, with everolimus also used for growing renal angiomyolipoma. [7] [8]
Neurocutaneous syndrome therapy ladder (consensus — confirm locally)
For Sturge-Weber syndrome, management is shared between neurology, ophthalmology and dermatology. Seizures are treated with standard antiseizure medicines, with low-dose aspirin used in many centres to reduce the stroke-like episodes that drive neurological decline, and developmental surveillance runs throughout childhood. Glaucoma is managed by ophthalmology with topical intraocular-pressure-lowering agents and surgery, often beginning in infancy. The port-wine stain is treated early with pulsed-dye laser, which lightens the lesion and reduces later thickening. [10] [12]
In Australia and New Zealand the consensus surveillance schedules of the International Tuberous Sclerosis Complex Consensus Conference and the major paediatric neurology and genetics services guide care: annual multidisciplinary review, magnetic resonance imaging of the brain at intervals for the astrocytoma, vigabatrin first for infantile spasms, and everolimus for the growing lesion. Outreach and telehealth link rural and remote families to these services so that surveillance is not delayed by distance. [7]
The neurocutaneous syndrome pathway
Recognise the skin sign and name the syndrome against the NIH criteria for NF1, the major-minor criteria for TSC, or the forehead port-wine stain of Sturge-Weber.
Examine the eyes (Lisch nodules, optic pathway, glaucoma, red reflex), measure the blood pressure, and take a three-generation family history.
Order syndrome-specific imaging: dedicated optic pathway views or surveillance for NF1, brain MRI and echocardiogram and renal ultrasound for TSC, and contrast brain MRI for Sturge-Weber.
NF1: annual medical-home review of growth, blood pressure, skin, vision and learning; urgent referral for any changing, painful or rapidly enlarging plexiform lesion.
TSC: treat infantile spasms with vigabatrin, image for the subependymal giant cell astrocytoma, and start everolimus for the growing lesion.
Sturge-Weber: treat seizures actively, review the eye for glaucoma from infancy, and consider pulsed-dye laser and low-dose aspirin.
Coordinate multidisciplinary care across genetics, neurology, ophthalmology, dermatology and development throughout childhood.
Counsel the family on inheritance, surveillance and the specific red flags that need urgent review.
Specific Subtypes & Scenarios
A child with six or more café-au-lait macules. Apply the NIH criteria in full, examining for skinfold freckling, neurofibromas, Lisch nodules and a family history, and consider Legius syndrome if only the macules and freckling are present. If criteria are not yet met, especially in a young child whose freckling and neurofibromas have not emerged, arrange surveillance and review as the phenotype evolves. [2] [4]
An infant with tuberous sclerosis and infantile spasms. Confirm with an electroencephalogram showing hypsarrhythmia, start vigabatrin promptly, and image the brain for the tubers and any astrocytoma. Early control of the spasms protects development, so vigabatrin is started as soon as the diagnosis is clear rather than after exhaustive investigation. [7] [5]
An infant with a forehead port-wine stain. Evaluate for Sturge-Weber syndrome with a neurological assessment, a contrast brain magnetic resonance image for the leptomeningeal angioma, and ophthalmology for glaucoma, because the forehead and upper-eyelid territory carries the highest risk of brain and eye involvement. Refer early for pulsed-dye laser for the stain itself. [10] [9]
A child with NF1 and a changing plexiform lesion. A plexiform neurofibroma that grows rapidly, becomes painful or firm, or produces a new neurological deficit raises the concern of a malignant peripheral nerve sheath tumour. Refer urgently for imaging, positron-emission tomography where appropriate, and specialist review for biopsy, because early recognition of this aggressive tumour changes the outcome. [4] [1]
A child with TSC and a growing lesion near the foramen of Monro. A subependymal giant cell astrocytoma that enlarges threatens obstructive hydrocephalus. Image urgently, involve neurosurgery for any hydrocephalus, and start everolimus, which reduced lesion volume in the EXIST-1 trial and is now standard for the growing astrocytoma. [8] [7]
A child with Sturge-Weber and stroke-like episodes. Transient weakness, visual loss or deterioration with intercurrent illness reflects the impaired venous drainage of the angioma. Treat the seizures actively, consider low-dose aspirin as used in many specialist centres, and rehabilitate the residual deficit, recognising that the episodes drive the long-term cognitive outcome. [10] [11]
Complications & Pitfalls
The complications of these syndromes are neurological, ophthalmological and neoplastic. In neurofibromatosis type 1 they are the optic pathway glioma, learning and behavioural difficulty, scoliosis and bony dysplasia, hypertension, and the malignant peripheral nerve sheath tumour. In tuberous sclerosis they are the epilepsy and developmental delay, the subependymal giant cell astrocytoma, the cardiac rhabdomyoma, the renal angiomyolipoma and, in later life, lymphangioleiomyomatosis. In Sturge-Weber they are the drug-resistant epilepsy, the progressive hemiparesis and cognitive decline, the glaucoma and the stroke-like episodes. [4] [10]
The cardinal pitfall is treating the skin and missing the brain and eye. A child assessed for café-au-lait macules without a blood pressure, a vision check or a learning assessment has had an incomplete workup. The skin sign is the entry point, and the surveillance it triggers is the management. [1] [4]
A second pitfall is delaying vigabatrin for TSC infantile spasms. The spasms and the hypsarrhythmia damage the developing brain, and vigabatrin is first-line and time-critical in tuberous sclerosis. Waiting for exhaustive imaging or a second opinion before treating squanders the window in which control best protects cognition. [7] [5]
A third pitfall is missing the malignant peripheral nerve sheath tumour. In a known neurofibromatosis type 1 lesion, persistent pain, rapid growth, a change in texture or a new neurological deficit is malignant transformation until proven otherwise, and reassurance without imaging is how the diagnosis is delayed. [4] [1]
A fourth pitfall is dismissing the forehead port-wine stain. A stain in the forehead or upper-eyelid territory is the one that harbours leptomeningeal involvement and glaucoma, and reassurance without a brain scan and an eye review misses the Sturge-Weber diagnosis at the moment when early intervention most helps. [9] [11]
A final pitfall is counselling falsely on inheritance. Reassuring a family that an autosomal dominant syndrome cannot recur, or alarming them that a sporadic mosaic Sturge-Weber stain will recur in a sibling, both spring from not knowing the genetics. Neurofibromatosis type 1 and tuberous sclerosis carry a one-in-two recurrence risk in offspring; Sturge-Weber is sporadic and close to non-recurrent. [3] [9]
Prognosis & Disposition
The prognosis of the neurocutaneous syndromes is set by how early the hidden complications are found and controlled. Neurofibromatosis type 1 has a variable but often mild course in childhood, with the optic pathway glioma usually low-grade and indolent and life expectancy near-normal — the dominant burdens are learning, cosmetic and the lifelong small risk of malignancy. Tuberous sclerosis prognosis hinges on the epilepsy and the developmental outcome, which are improved by early seizure control and by timely treatment of the astrocytoma and renal lesions. Sturge-Weber prognosis is driven by the seizure burden and the frequency of stroke-like episodes, which together shape the cognitive trajectory. [4] [10]
Disposition follows the syndrome and the acute presentation. The stable child is managed in a multidisciplinary outpatient clinic with surveillance at intervals; the infant with spasms, the child with a growing astrocytoma or raised intracranial pressure, and the child with status epilepticus or acute glaucoma need the hospital and the specialist. The threshold for involving genetics, neurology and ophthalmology is deliberately low. [5] [12]
Because these are lifelong diseases, the disposition includes planned transition to adult care. The child with neurofibromatosis type 1 needs ongoing blood pressure and malignancy surveillance; the adolescent with tuberous sclerosis needs renal and pulmonary surveillance and reproductive counselling; the young person with Sturge-Weber needs continued seizure and glaucoma management. Building the transition early preserves the continuity that shapes the long-term outcome. [1] [11]
Special Populations
Children with neurocutaneous syndromes and disability or neurodiversity are the core of the practice. Learning difficulty, attention-deficit, autism-spectrum features and behavioural problems are common in neurofibromatosis type 1 and tuberous sclerosis, and developmental surveillance with educational support is part of the management, not an afterthought. Recognising and supporting the neurocognitive phenotype changes the child's trajectory as much as treating the seizures. [4] [5]
Complex, chronic and technology-dependent children — those with drug-resistant epilepsy, a shunted astrocytoma, renal failure or chronic respiratory disease — need coordinated medical-home care with clear escalation plans. The general paediatrician often holds the overview while the subspecialty teams deliver the organ-specific care, and a written emergency plan prevents the crises that distance and complexity otherwise create. [1] [7]
Aboriginal and Torres Strait Islander, Maori and Pasifika children, and families in rural and remote areas need equitable access to the multidisciplinary surveillance that defines good care in these diseases. Distance should not delay the brain scan, the electroencephalogram or the ophthalmology review, so outreach clinics, telehealth and clear referral pathways are part of delivering the standard of care to every child regardless of location. [5]
Families facing a new genetic diagnosis need clear counselling on inheritance, recurrence and the surveillance plan, and access to genetic services for testing and family cascade screening. The sporadic, non-recurrent nature of Sturge-Weber is itself a powerful reassurance, while the one-in-two recurrence risk of neurofibromatosis type 1 and tuberous sclerosis shapes reproductive decisions for the affected family. [3] [9]
Evidence, Guidelines & Regional Differences
The diagnostic and surveillance framework for neurofibromatosis type 1 rests on the 1988 National Institutes of Health consensus criteria, which remain in everyday use, and on modern reviews that have refined the multidisciplinary management. The criteria are deliberately clinical so that the diagnosis can be made at the bedside, and the surveillance schedule — vision, blood pressure, growth, development and skin — is built around the complications that the criteria predict. [2] [4]
For tuberous sclerosis, the evidence base is the 2012 International Tuberous Sclerosis Complex Consensus Conference, whose diagnostic-criteria and surveillance-and-management papers set the global standard, updated in 2021 to reflect molecular diagnosis and refined surveillance. The transformative therapeutic evidence is the EXIST-1 phase-three trial, which showed that everolimus reduced the volume of the subependymal giant cell astrocytoma and established mTOR inhibition as standard of care for the growing lesion. [6] [8]
Everolimus for subependymal giant cell astrocytoma in TSC (EXIST-1)
Population: Patients with tuberous sclerosis complex and subependymal giant cell astrocytomas, randomised to the mTOR inhibitor everolimus versus placebo in a multicentre phase-three trial.
Key finding
Everolimus significantly reduced the volume of the subependymal giant cell astrocytoma compared with placebo, with a manageable adverse-event profile.
Practice change
mTOR inhibition is now standard of care for the growing astrocytoma, transforming a neurosurgical disease into a medical one for many children and informing the broader use of everolimus for TSC-related renal and other lesions.
For Sturge-Weber syndrome, the discovery of the somatic mosaic GNAQ mutation united the biology of the port-wine stain and the leptomeningeal angioma and explained why the disease follows a developmental territory and never recurs. Comprehensive reviews guide the multidisciplinary care of seizures, glaucoma and the stroke-like episodes, with low-dose aspirin used in many specialist centres though the evidence remains observational. [9] [10]
The regional policy structure is consistent in principle. In Australia and New Zealand, the major paediatric genetics, neurology and ophthalmology services follow the consensus surveillance schedules with outreach and telehealth for rural families. In the United Kingdom, the Royal College of Paediatrics and specialist neurology services follow the same criteria-based surveillance. In the United States and Canada, the consensus criteria and the GeneReviews surveillance guidance set the standard. In every region the principle is the same: recognise the skin sign, apply the criteria, surveil the brain and eye, treat the complications early, and coordinate the multidisciplinary team. [5] [1]
The controversies are real: the role of routine brain imaging in asymptomatic neurofibromatosis type 1, the optimal duration of everolimus for the astrocytoma, the place of low-dose aspirin in Sturge-Weber, the long-term safety of MEK inhibition, and equitable access to specialist and surgical services across distance. The defence against each is the same — early recognition, criteria-based diagnosis, proactive surveillance and multidisciplinary care. [4] [12]
Exam Pearls
- The three phakomatoses announce themselves in the skin before the brain: café-au-lait macules in NF1, ash-leaf macules in TSC, and the forehead port-wine stain in Sturge-Weber. [1] [9]
- Neurofibromatosis type 1 is diagnosed by the NIH criteria — two or more of six or more café-au-lait macules (at least five millimetres before puberty or fifteen after), skinfold freckling, two or more neurofibromas or a plexiform neurofibroma, optic pathway glioma, two or more Lisch nodules, a characteristic bony lesion, or an affected first-degree relative. [2] [3]
- NF1 is caused by loss of the NF1 gene product neurofibromin, releasing the RAS-MAPK pathway; it is autosomal dominant with prevalence about one in three thousand and about half of cases new mutations. [4]
- NF1 surveillance covers the optic pathway glioma, learning and behaviour, blood pressure (renal artery stenosis, phaeochromocytoma), and the malignant peripheral nerve sheath tumour risk in a changing plexiform lesion. [1] [4]
- Tuberous sclerosis complex is caused by TSC1 or TSC2 loss driving mTOR overactivation; a definite clinical diagnosis needs two major or one major plus two minor criteria, or a pathogenic variant. [6] [5]
- The TSC major features include three or more hypomelanotic macules, three or more angiofibromas or a fibrous plaque, the shagreen patch, cortical tubers, subependymal nodules, the subependymal giant cell astrocytoma, cardiac rhabdomyoma and renal angiomyolipoma. [6]
- Vigabatrin is first-line for infantile spasms in TSC; the growing subependymal giant cell astrocytoma is treated with the mTOR inhibitor everolimus (EXIST-1). [7] [8]
- Sturge-Weber syndrome is caused by a somatic mosaic GNAQ mutation; it is sporadic, non-recurrent, and classically a unilateral forehead or upper-eyelid port-wine stain with ipsilateral leptomeningeal angioma, seizures and glaucoma. [9] [11]
- A port-wine stain on the forehead or upper eyelid carries the highest risk of brain and eye involvement and mandates a contrast brain magnetic resonance image and ophthalmology for glaucoma. [10] [9]
- Legius syndrome (SPRED1) mimics NF1 with café-au-lait macules and freckling but lacks neurofibromas, Lisch nodules and tumours. [4]
- Inheritance matters for counselling: NF1 and TSC are autosomal dominant with a one-in-two recurrence risk, while Sturge-Weber is sporadic and close to non-recurrent in siblings. [3] [9]
References
- [1]Gutmann DH; Aylsworth A; Carey JC; et al The diagnostic evaluation and multidisciplinary management of neurofibromatosis 1 and neurofibromatosis 2. JAMA, 1997.PMID 9207339
- [2]National Institutes of Health Consensus Development Panel Neurofibromatosis. Conference statement. National Institutes of Health Consensus Development Conference. Arch Neurol, 1988.PMID 3128965
- [3]Williams VC; Lucas J; Babcock MA; et al Neurofibromatosis type 1 revisited. Pediatrics, 2009.PMID 19117870
- [4]Gutmann DH; Ferner RE; Listernick RH; et al Neurofibromatosis type 1. Nat Rev Dis Primers, 2017.PMID 28230061
- [5]Northrup H; Aronow ME; Bebin EM; et al Updated International Tuberous Sclerosis Complex Diagnostic Criteria and Surveillance and Management Recommendations. Pediatr Neurol, 2021.PMID 34399110
- [6]Northrup H; Krueger DA; International Tuberous Sclerosis Complex Consensus Group Tuberous sclerosis complex diagnostic criteria update: recommendations of the 2012 International Tuberous Sclerosis Complex Consensus Conference. Pediatr Neurol, 2013.PMID 24053982
- [7]Krueger DA; Northrup H; International Tuberous Sclerosis Complex Consensus Group Tuberous sclerosis complex surveillance and management: recommendations of the 2012 International Tuberous Sclerosis Complex Consensus Conference. Pediatr Neurol, 2013.PMID 24053983
- [8]Franz DN; Belousova E; Sparagana S; et al Efficacy and safety of everolimus for subependymal giant cell astrocytomas associated with tuberous sclerosis complex (EXIST-1): a multicentre, randomised, placebo-controlled phase 3 trial. Lancet, 2013.PMID 23158522
- [9]Shirley MD; Tang H; Gallione CJ; et al Sturge-Weber syndrome and port-wine stains caused by somatic mutation in GNAQ. N Engl J Med, 2013.PMID 23656586
- [10]Lo W; Marchuk DA; Ball KL; et al Updates and future horizons on the understanding, diagnosis, and treatment of Sturge-Weber syndrome brain involvement. Dev Med Child Neurol, 2012.PMID 22191476
- [11]Comi AM Update on Sturge-Weber syndrome: diagnosis, treatment, quantitative measures, and controversies. Lymphat Res Biol, 2007.PMID 18370916
- [12]Yeom S; Comi AM Updates on Sturge-Weber Syndrome. Stroke, 2022.PMID 36263782