Paeds · paediatric-dermatology
Skin manifestations of systemic disease
Also known as Cutaneous signs of systemic disease · Dermatologic markers of internal disease · Skin as a window to systemic disease · Neurocutaneous syndromes · Reactive erythemas · Paraneoplastic dermatoses
Fellowship topic on cutaneous manifestations of systemic disease in children: the skin as a window to internal disease. Covers the reactive erythemas and panniculitides (erythema nodosum signalling streptococcal infection, sarcoidosis and inflammatory bowel disease; erythema multiforme and mycoplasma-induced rash and mucositis after HSV and Mycoplasma; Gianotti-Crosti papular acrodermatitis as a viral exanthem), the neutrophilic dermatoses (Sweet syndrome and pyoderma gangrenosum linked to inflammatory bowel disease and malignancy), the metabolic and endocrine markers (acanthosis nigricans as insulin resistance and obesity and type 2 diabetes; necrobiosis lipoidica and xanthomas), the gastrointestinal and nutritional dermatoses (dermatitis herpetiformis as the cutaneous face of coeliac disease; acrodermatitis enteropathica as zinc deficiency), the haematological and neoplastic markers (petechiae and purpura in leukaemia and septicaemia, the blueberry muffin neonate), and the neurocutaneous phakomatoses (neurofibromatosis type 1 with its revised diagnostic criteria, tuberous sclerosis complex and the port-wine stain of Sturge-Weber syndrome). Built around recognise, investigate the underlying disease, and refer, grounded in the erythema nodosum and erythema multiforme and acanthosis nigricans reviews, the coeliac disease ESsCD guideline, the dermatitis herpetiformis update, the revised NF1 and updated TSC diagnostic criteria, the port-wine birthmark and Sweet syndrome reviews, the acrodermatitis enteropathica paper and the Gianotti-Crosti review.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
The six families of skin-to-systemic signals
Overview & Definition
Picture a ten-year-old sent to the clinic with three weeks of tender red lumps on both shins. She is otherwise well, but the lesions hurt when she walks and her mother is frightened. The lumps are erythema nodosum — a reactive inflammation of the subcutaneous fat — and the first question is not what cream to prescribe but what the skin is reacting to: in her age group, a recent streptococcal sore throat is the commonest answer. The skin has spoken for a systemic event, and the clinician's job is to listen. [1]
Cutaneous manifestations of systemic disease are skin findings that arise from, or signal, a disorder beneath the skin. They span immune-complex reactions such as erythema nodosum, antibody-mediated blistering such as dermatitis herpetiformis, metabolic deposition such as acanthosis nigricans and xanthomas, nutrient depletion such as the zinc deficiency of acrodermatitis enteropathica, malignant infiltration such as leukaemia cutis and the blueberry muffin neonate, and the developmental mosaicism of the neurocutaneous syndromes. What unites them is that the visible lesion is a clue, and the correct response is to chase the internal disease it points to. [3] [8]
The clinician's task has three layers. The first is recognition — naming the skin sign and resisting the urge to treat it as a primary skin disease. The second is the targeted work-up — choosing the investigations that the lesion itself dictates, whether coeliac serology for dermatitis herpetiformis or a full blood count for the purpuric child. The third is the multidisciplinary referral — to dermatology, gastroenterology, endocrinology, haematology or clinical genetics, because these are conditions whose management belongs to a team, not to a single prescription pad. [5] [7]
Classification
Group the cutaneous signals by the mechanism that links them to the internal disease, because each mechanism carries its own differential and its own work-up. The reactive erythemas and panniculitides lead the list: erythema nodosum, the tender nodular panniculitis of the shins, is the archetype of a skin sign that demands a search for streptococcal infection, tuberculosis, sarcoidosis and inflammatory bowel disease. Erythema multiforme, with its target lesions, and mycoplasma-induced rash and mucositis sit alongside it as hypersensitivity reactions to infection and drugs. [1] [2]
The neutrophilic dermatoses form a second family. Sweet syndrome (acute febrile neutrophilic dermatosis) presents with fever and painful erythematous plaques and a raised neutrophil count, and it can be the first sign of acute myeloid leukaemia, inflammatory bowel disease or a drug reaction. Pyoderma gangrenosum begins as a painful papulopustule that ulcerates into a destructive edge, and it too travels with inflammatory bowel disease and arthritis. These conditions share sterile neutrophilic infiltration of the skin and a strong association with systemic inflammation and malignancy. [10]
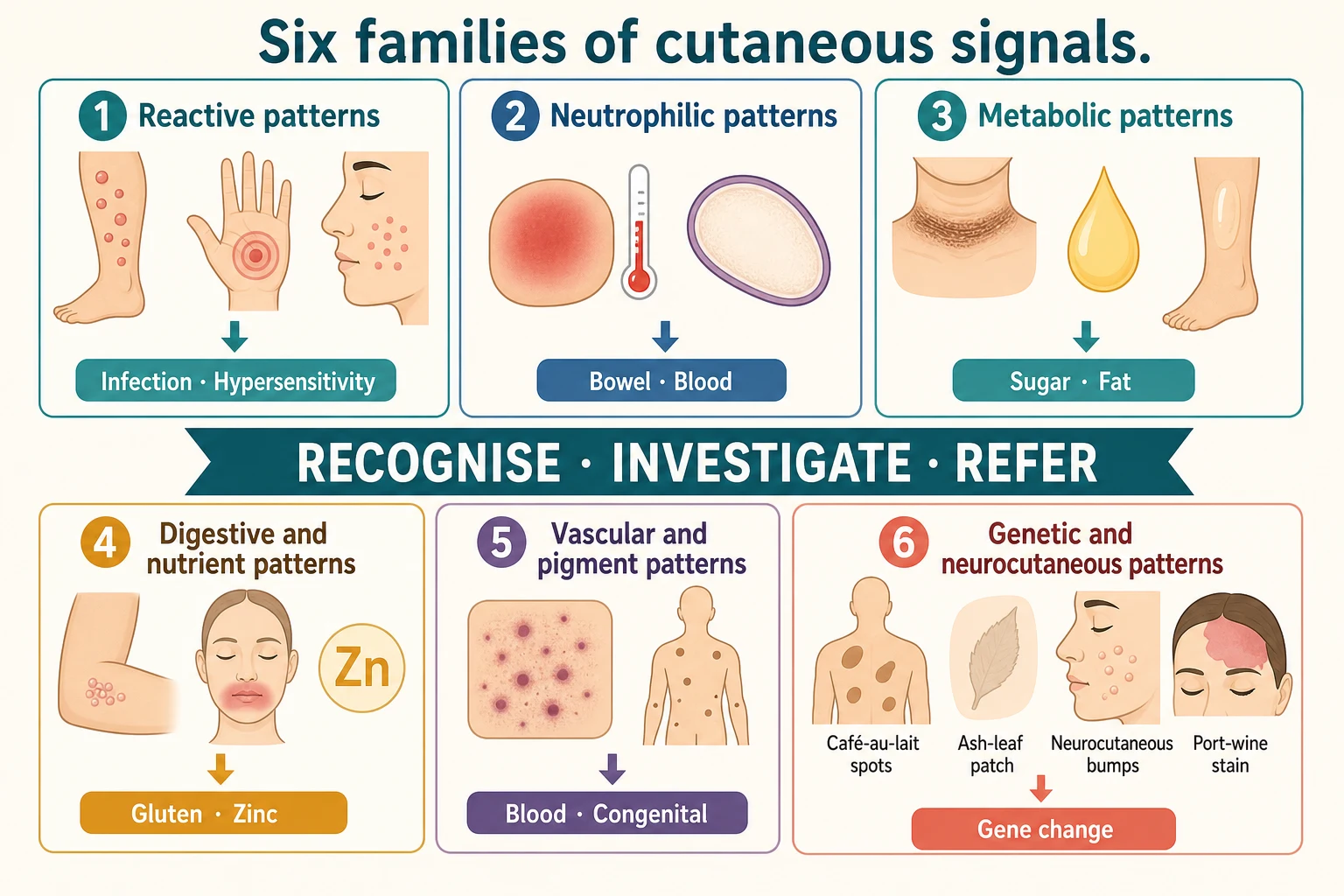
The metabolic and endocrine markers form a third family. Acanthosis nigricans is the velvety, hyperpigmented thickening of the neck, axillae and groin that marks insulin resistance and obesity, and it is increasingly common in children as the prevalence of overweight rises. Necrobiosis lipoidica produces yellow-brown, atrophic plaques on the shins of a child with diabetes, and xanthomas — yellow lipid-laden nodules and tendinous deposits — flag hyperlipidaemia. The skin here mirrors a metabolic state. [3] [4]
The gastrointestinal and nutritional dermatoses form a fourth family. Dermatitis herpetiformis is the intensely pruritic, grouped vesicular eruption on the elbows, knees and buttocks that is the cutaneous face of coeliac disease. Acrodermatitis enteropathica is the periorificial and acral dermatitis, with diarrhoea and alopecia, of zinc deficiency — inherited from SLC39A4 mutations or acquired from inadequate intake. The skin reads the gut and the diet. [6] [11]
The haematological and neoplastic markers form a fifth family, and they are often the most urgent. Non-blanching petechiae and purpura in a febrile or unwell child raise meningococcal septicaemia, disseminated intravascular coagulation and acute leukaemia. The blueberry muffin baby — a neonate with widespread dark blue-red papules of dermal extramedullary haematopoiesis — signals congenital infection (cytomegalovirus, rubella, toxoplasmosis), haemolytic disease or congenital leukaemia or neuroblastoma. These are not outpatient problems. [1] [9]
The neurocutaneous syndromes — the phakomatoses — form the sixth family, and they carry their own diagnostic criteria. Neurofibromatosis type 1 shows café-au-lait macules, skinfold freckling, neurofibromas and Lisch nodules. Tuberous sclerosis complex shows hypomelanotic ash-leaf macules, angiofibromas, the shagreen patch and periungual fibromas. Sturge-Weber syndrome shows the facial port-wine stain of a congenital capillary malformation in the forehead or upper eyelid, paired with leptomeningeal angiomatosis and glaucoma. Each has a gene, a skin sign and a surveillance schedule. [7] [8] [9]
Epidemiology & Risk Factors
The epidemiology of cutaneous manifestations of systemic disease is the epidemiology of the diseases themselves, because the skin sign follows the underlying condition. Erythema nodosum is the commonest panniculitis of children, and in paediatric series group A streptococcal pharyngitis is the leading identified cause, followed by tuberculosis and sarcoidosis in endemic regions, and inflammatory bowel disease in older children and adolescents. A tender shin nodule in the weeks after a sore throat is the classic presentation. [1]
Acanthosis nigricans has become common as childhood overweight and obesity have risen, and it tracks closely with insulin resistance, a family history of type 2 diabetes, and non-white ethnicity, in whom the threshold for screening is applied more liberally. An adolescent with a velvety neck and a body mass index above the ninety-fifth centile is the typical presentation, and the skin sign is an opportunity to intervene on a modifiable metabolic trajectory before glucose intolerance develops. [3] [4]
The neurocutaneous syndromes are individually rare but collectively a fixture of paediatric examination. Neurofibromatosis type 1 affects roughly one in three thousand people and is the commonest autosomal dominant single-gene disorder that a general paediatrician will meet, with half of cases familial and half new mutations. Tuberous sclerosis complex affects roughly one in six thousand to ten thousand, and Sturge-Weber syndrome affects roughly one in twenty to fifty thousand. Knowing the skin signs is how these are first recognised — often by the generalist, not the geneticist. [7] [8] [9]
In Australia and Aotearoa New Zealand, several regional factors shape the epidemiology and the work-up. Tuberculosis remains a consideration for erythema nodosum in migrant, refugee and Indigenous communities, and in communities with higher rates of latent infection, so a Mantoux or interferon-gamma release assay enters the differential of a tender shin nodule in the right child. Specialist paediatric dermatology and clinical genetics concentrate in major centres, so a child with a suspected phakomatosis in a rural or remote community may depend on outreach and telehealth to reach the multidisciplinary clinic that confirms the diagnosis and sets up surveillance. Equity of access to those services is part of the management. [1] [8]
Pathophysiology
The mechanisms that link a skin sign to an internal disease are as varied as the diseases, and naming the mechanism narrows the differential. In the reactive erythemas, immune complex deposition and delayed hypersensitivity drive the response. Erythema nodosum is a type IV hypersensitivity reaction in the subcutaneous fat — immune complexes and T-cell activity produce a septal panniculitis in which neutrophils and then lymphocytes and giant cells surround the fibrous septa, and the overlying skin reddens from the inflammation beneath it. [1]
Erythema multiforme is the classic immune-mediated target lesion. It is most often triggered by herpes simplex virus, and by Mycoplasma pneumoniae, and the term mycoplasma-induced rash and mucositis now names the distinct mucosa-dominant syndrome that mycoplasma produces, separated from Stevens-Johnson syndrome and from classical erythema multiforme because its skin involvement is mild and its mucositis severe. The mechanism is a cell-mediated immune response to microbial antigens deposited in the skin and mucosa. [2]
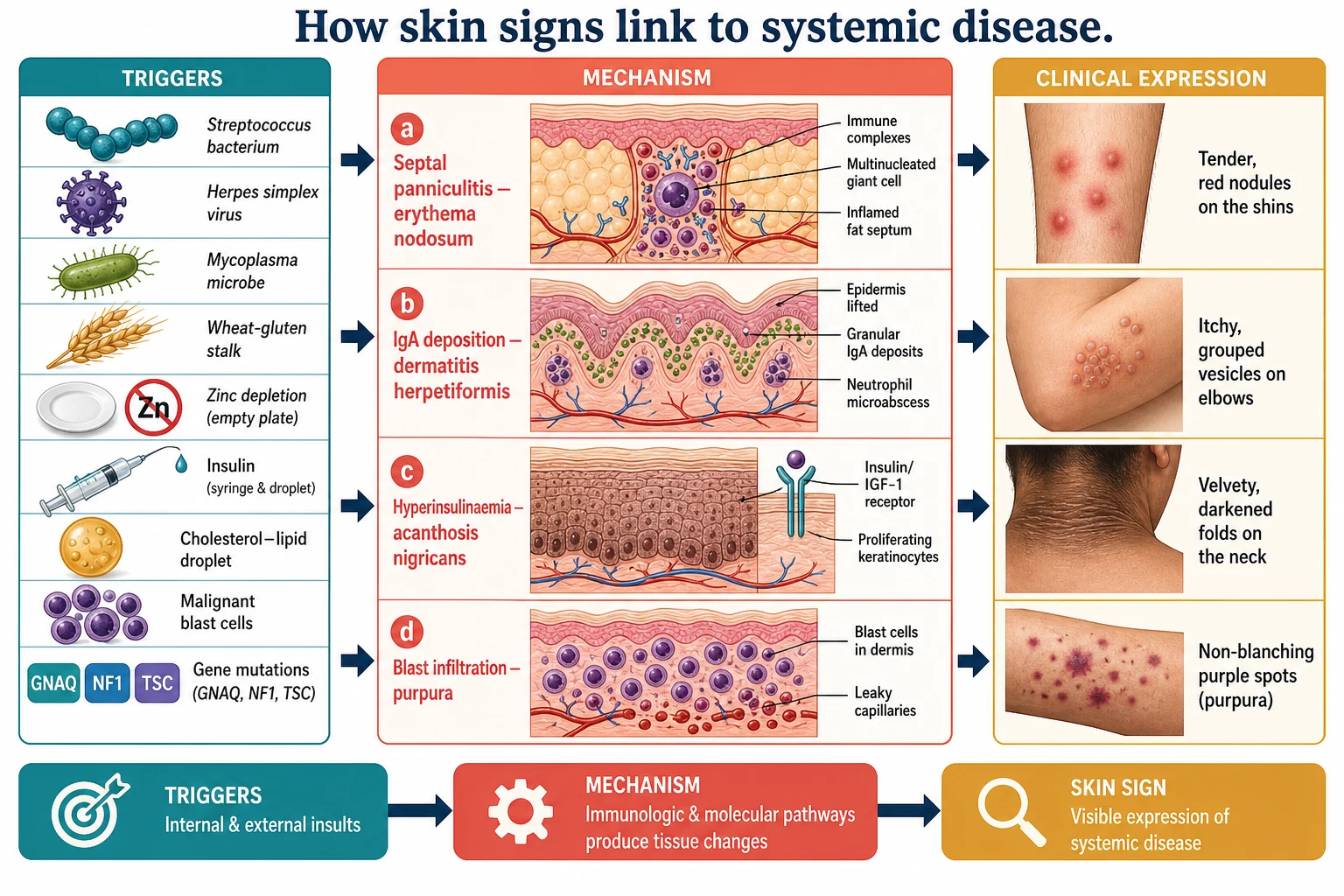
In the antibody-mediated dermatoses, the skin is the target of an immune response to an internal antigen. Dermatitis herpetiformis is the cutaneous face of coeliac disease: IgA autoantibodies against epidermal transglutaminase deposit in the dermal papillae, complement activates, neutrophils collect, and the resultant microabscesses lift the epidermis into the intensely itchy grouped vesicles that define it. The same gluten-driven immune process that flattens the small-intestinal villi blisters the elbows and knees. [5] [6]
In the metabolic markers, a circulating substrate or hormone reshapes the skin. In acanthosis nigricans, hyperinsulinaemia from insulin resistance drives epidermal keratinocyte and fibroblast proliferation through insulin-like growth factor receptors, producing the velvety thickening and hyperpigmentation of the flexures. In necrobiosis lipoidica, microangiopathy and collagen degeneration in the dermis produce the yellow, atrophic shin plaques of diabetes; in xanthomas, lipid-laden macrophages collect in the skin and tendons of the child with a familial hyperlipidaemia. [3] [4]
In the nutritional dermatoses, depletion of a single nutrient dismantles the epidermis. Zinc is a cofactor for hundreds of enzymes of replication and keratinisation, so its deficiency — from the inherited SLC39A4 transporter defect of acrodermatitis enteropathica or from inadequate intake — produces a well-defined periorificial and acral dermatitis with diarrhoea, alopecia and irritability that resolves within days of zinc replacement. The skin is exquisitely sensitive to a supply it cannot store long. [11]
In the haematological and neoplastic markers, the skin is infiltrated by cells it should not contain. Meningococci and other bacteria seed the vessel walls in septicaemia to produce purpura; leukaemic blasts infiltrate the dermis as leukaemia cutis or thrombocytopenia produces petechiae; and in the blueberry muffin neonate, persistent dermal extramedullary haematopoiesis — a fetal response to congenital infection, severe haemolysis or congenital leukaemia or neuroblastoma — leaves nests of haematopoietic cells in the dermis as dark blue-red papules. [9]
In the neurocutaneous syndromes, an inherited or somatic gene mutation dysregulates cell growth in both skin and nervous system. Neurofibromatosis type 1 arises from NF1 loss and the loss of neurofibromin's brake on Ras signalling; tuberous sclerosis complex arises from TSC1 or TSC2 loss and disinhibited mTOR signalling, producing both the skin hamartomas and the brain tubers; Sturge-Weber syndrome arises from a somatic activating GNAQ mutation in the vascular endothelium, producing the port-wine stain and the leptomeningeal angiomatosis in the same embryonic distribution. The skin sign and the brain lesion share a single developmental event. [7] [8] [9]
Clinical Presentation
The reactive erythemas announce themselves acutely. Erythema nodosum presents with bilateral, symmetric, tender, warm, erythematous nodules over the anterior shins — the child may have a fever, joint aches and malaise, and the lesions evolve from red to purple to bruise-yellow over three to six weeks without ulcerating. Erythema multiforme presents with target lesions — three zones of concentric colour change — on the palms, soles and extensor surfaces, often with mild mucosal involvement, and it recurs with reactivation of herpes simplex. [1] [2]
Mycoplasma-induced rash and mucositis deserves its own recognition. After a respiratory illness, the child develops prominent mucositis of at least two sites — lips, oral mucosa, conjunctivae, urethra — with relative sparing of the skin, unlike Stevens-Johnson syndrome where skin detachment is the rule. Naming it correctly changes the work-up and the prognosis, because the target is mycoplasma and the outcome is usually favourable. [2]
Gianotti-Crosti syndrome (papular acrodermatitis of childhood) presents in a preschool child with a characteristic eruption of monomorphic, flat-topped, pink-to-flesh-coloured papules over the cheeks, buttocks and the extensor surfaces of the limbs, strikingly sparing the trunk. It follows a viral infection — historically hepatitis B, now most often Epstein-Barr virus, enteroviruses, respiratory viruses and many others — and it resolves over weeks without treatment. The clue to the systemic link is the distribution, not the morphology. [12]
The metabolic and endocrine markers are chronic and often found incidentally. Acanthosis nigricans shows a velvety, brown thickening in the neck folds, axillae, groin and sometimes the knuckles, and the affected adolescent is usually overweight. Necrobiosis lipoidica shows sharply demarcated, yellow-brown, shiny, atrophic plaques with telangiectasia over the shins, sometimes preceding or accompanying diabetes. Xanthomas show yellow-orange papules, nodules or tendinous thickenings in a child with a markedly abnormal lipid profile. [3] [4]
The neutrophilic dermatoses are dramatic and unwell. Sweet syndrome presents with fever, malaise and painful, erythematous, oedematous plaques that can look pseudovesicular, distributed over the face, trunk and limbs, with a raised neutrophil count and sometimes arthralgia. Pyoderma gangrenosum begins as a painful papulopustule that rapidly breaks down into a deep ulcer with a violaceous, undermined, boggy edge, often on the legs, and classically pathergic — worsening with debridement. Both demand a search for inflammatory bowel disease, arthritis and malignancy. [10]
The haematological and neoplastic markers are emergencies. A febrile child with non-blanching petechiae or purpura is meningococcal septicaemia or leukaemia until proven otherwise. A neonate with blueberry muffin lesions — firm, dark blue-red papules and plaques over the trunk, limbs and sometimes the scalp — has dermal extramedullary haematopoiesis from congenital infection, haemolysis or malignancy. The skin sign is a warning siren, not a curiosity. [9]
The neurocutaneous syndromes are read off the skin in childhood. Neurofibromatosis type 1 shows six or more café-au-lait macules, axillary and inguinal freckling, cutaneous and subcutaneous neurofibromas, and — on slit-lamp — Lisch nodules of the iris. Tuberous sclerosis complex shows hypomelanotic ash-leaf macules (best seen under Wood's lamp), facial angiofibromas over the nasolabial folds, the shagreen patch in the lumbosacral area, and periungual fibromas. Sturge-Weber syndrome shows a port-wine stain (capillary malformation) in the forehead and upper eyelid distribution, and the child is at risk of seizures, stroke-like episodes, developmental delay and glaucoma. [7] [8] [9]
Differential Diagnosis
The differential for each cutaneous signal is the differential for the underlying disease it suggests, so the task is to keep the systemic question open. For erythema nodosum, the differential is the cause list: in children, group A streptococcal pharyngitis leads, then tuberculosis and sarcoidosis in the right population, inflammatory bowel disease in the older child, and drugs such as the oral contraceptive pill, sulfonamides and penicillins. The streptococcal cause is confirmed by throat swab and a rising antistreptolysin-O titre. [1]
For an acral target lesion, the differential is erythema multiforme against urticaria, fixed drug eruption and, when mucosal involvement is severe, Stevens-Johnson syndrome. Mycoplasma-induced rash and mucositis is separated from Stevens-Johnson syndrome by its prominent mucositis and minimal skin detachment, and identifying it correctly avoids the wrong work-up. A drug history, a herpes history and a mycoplasma respiratory history close the loop. [2]
For acanthosis nigricans, the differential of the metabolic cause is insulin resistance, obesity and type 2 diabetes, and the rare endocrine tumour, but in a child the assumption is insulin resistance until screening proves otherwise. For dermatitis herpetiformis, the differential of an itchy vesicular elbow eruption includes scabies, atopic dermatitis and linear IgA bullous dermatosis of childhood, and the distinction is coeliac serology, the granular IgA on direct immunofluorescence of perilesional skin, and the response to a gluten-free diet. [3] [6]
For a port-wine stain, the differential is a common naevus flammeus against the risk of Sturge-Weber syndrome when the stain lies in the forehead or upper eyelid distribution, and the question is whether the leptomeninges and the eye are involved — answered by neuroimaging and tonometry. For the blueberry muffin neonate, the differential is the congenital infection panel (cytomegalovirus, rubella, toxoplasmosis, syphilis), haemolytic disease of the newborn, and congenital leukaemia or neuroblastoma — a broad and serious list that demands urgent investigation. [9]
Clinical & Bedside Assessment
Begin with a focused history anchored to the skin sign. For a tender shin nodule, ask about a sore throat, fever, cough and abdominal symptoms in the preceding weeks, and about travel, tuberculosis contact and bowel symptoms. For an itchy elbow eruption, ask about gluten sensitivity, gut symptoms, iron deficiency and a family history of coeliac disease or autoimmune disease. For a purpuric child, ask about fever, unwellness, bruising, bleeding and recent infection. For a suspected phakomatosis, take a three-generation family history and ask about seizures, learning, development, vision and hearing. [1] [7]
Examine the whole skin in good light, and examine it systematically. Describe the primary lesion — a nodule, a target, a vesicle, a plaque, a macule, a port-wine stain — and its distribution. For a suspected neurocutaneous syndrome, count the café-au-lait macules and measure them, look for axillary and ingueral freckling, examine the Wood's lamp for ash-leaf macules, look at the face for angiofibromas and the nails for periungual fibromas, and examine the spine and skin for the shagreen patch. A Wood's lamp and a tape measure belong in the assessment of a child with unusual pigmentation. [7] [8]
Then look beyond the skin to the system the sign implicates. For erythema nodosum, examine the throat and listen to the chest, and feel for lymphadenopathy and hepatosplenomegaly. For acanthosis nigricans, measure height, weight, body mass index and blood pressure, and look for the acanthosis behind the neck and in the axillae. For dermatitis herpetiformis, look for the finger-prick marks of scratching, assess growth, and check for dental enamel defects and iron deficiency. For a port-wine stain in the V1 distribution, examine the eyes for glaucoma and assess the neurology. [1] [9]
Measure severity and impact where a validated tool exists, but more often the bedside assessment is the decision about how quickly to investigate and whom to refer to. A febrile purpuric child is a resuscitation, not a clinic booking. A port-wine stain over the forehead is an urgent neuroimaging and ophthalmology referral. A velvety neck in an overweight adolescent is a fasting glucose and lipid panel and a conversation about weight. Match the urgency to the sign. [3] [9]
Investigations
The investigations are dictated by the lesion itself, because each cutaneous signal names its own work-up. For erythema nodosum, arrange a throat swab and an antistreptolysin-O titre, a chest radiograph, a tuberculin skin test or interferon-gamma release assay, and — in the right child — inflammatory markers and a stool study. The streptococcal cause is the commonest finding in children, and confirming it closes the diagnosis. [1]
For dermatitis herpetiformis, arrange coeliac serology — immunoglobulin A anti-tissue transglutaminase and anti-endomysial antibodies — and confirm coeliac disease with a duodenal biopsy while the child is on a gluten-containing diet. Direct immunofluorescence of perilesional skin shows granular IgA deposition in the dermal papillae, and a skin biopsy for routine histology shows subepidermal blistering with neutrophil microabscesses. The skin biopsy and the gut biopsy together secure the diagnosis. [5] [6]
For acanthosis nigricans, arrange a fasting glucose or an oral glucose tolerance test, a fasting lipid panel, and a glycosylated haemoglobin in the right child, and screen for the metabolic syndrome. The skin sign is the opportunity to find insulin resistance early and to act on it with lifestyle change, which is the intervention most likely to change the trajectory. [3] [4]
For the neutrophilic dermatoses, arrange a full blood count and film, inflammatory markers, and — guided by the clinical picture — a search for inflammatory bowel disease and malignancy, because Sweet syndrome may be the first presentation of acute myeloid leukaemia. A skin biopsy shows the dense dermal neutrophilic infiltrate that defines Sweet syndrome and excludes infection. [10]
For the neurocutaneous syndromes, the diagnosis is clinical and criteria-based. For neurofibromatosis type 1, apply the revised diagnostic criteria — two or more of café-au-lait macules, skinfold freckling, neurofibromas, optic pathway glioma, Lisch nodules, osseous dysplasia and a first-degree relative. For tuberous sclerosis complex, apply the major and minor gene criteria of the updated international consensus, with genetic testing for a TSC1 or TSC2 pathogenic variant now sufficient for the diagnosis. For a port-wine stain in the V1 distribution, arrange neuroimaging to define the leptomeningeal angiomatosis and urgent ophthalmology for glaucoma. [7] [8] [9]
In Australia and New Zealand, the access pathway for genetic testing and specialist surveillance of the neurocutaneous syndromes runs through state-funded clinical genetics and paediatric multidisciplinary clinics, but these concentrate in major centres. A child in a rural or remote community may reach the clinic only through outreach and telehealth, so the generalist's role in recognition, early work-up and referral is the gatekeeping step. Document the skin findings clearly and photograph them, because the criteria are visual and the clinic may be a long way away. [8] [9]
Management — Resuscitation
Resuscitation belongs to the dangerous end of the spectrum, where the skin sign warns of a life-threatening systemic disease. A febrile child with rapidly evolving non-blanching purpura is meningococcal septicaemia until proven otherwise — give parenteral antibiotics within the first hour, support the airway and circulation, and transfer to a paediatric intensive care setting. Do not wait for the rash to confirm the diagnosis; the rash is a late sign and the window is narrow. [9]
The blueberry muffin neonate is a neonatal emergency. The dark blue-red papules signal congenital infection, severe haemolysis or congenital leukaemia or neuroblastoma, so isolate and investigate urgently — a congenital infection screen, a full blood count and film, and specialist paediatric and haematology input. The skin sign here is the first evidence of a condition that may threaten the neonate's hearing, vision, neurodevelopment or life. [9]
The port-wine stain over the forehead or upper eyelid is an urgent neurology and ophthalmology referral, because the Sturge-Weber syndrome it may signal carries a risk of glaucoma in infancy and of seizures, stroke-like episodes and developmental impairment. Laser treatment of the stain itself is part of the longer-term plan, but the immediate priority is to define the brain and eye involvement and to set up surveillance. [9]
Keep resuscitation separate from the routine clinic work-up in your mind. The vast majority of cutaneous manifestations of systemic disease are not emergencies — the tender shin nodule, the velvety neck, the itchy elbow are clinic problems that demand a work-up and a referral, not a resuscitation bay. But the purpuric child, the blueberry muffin neonate and the V1 port-wine stain are the exceptions, and recognising them is the single most important judgment the skin examination asks of you. [1] [9]
Management — Definitive & Stepwise
Definitive management follows the principle that the skin sign is a door to the systemic disease, so the treatment is the treatment of the disease beneath it. The stepwise pathway for each family begins with recognition and a targeted work-up, moves to confirm the underlying diagnosis, and then treats the cause — with the skin often improving as the systemic disease comes under control. [1] [5]
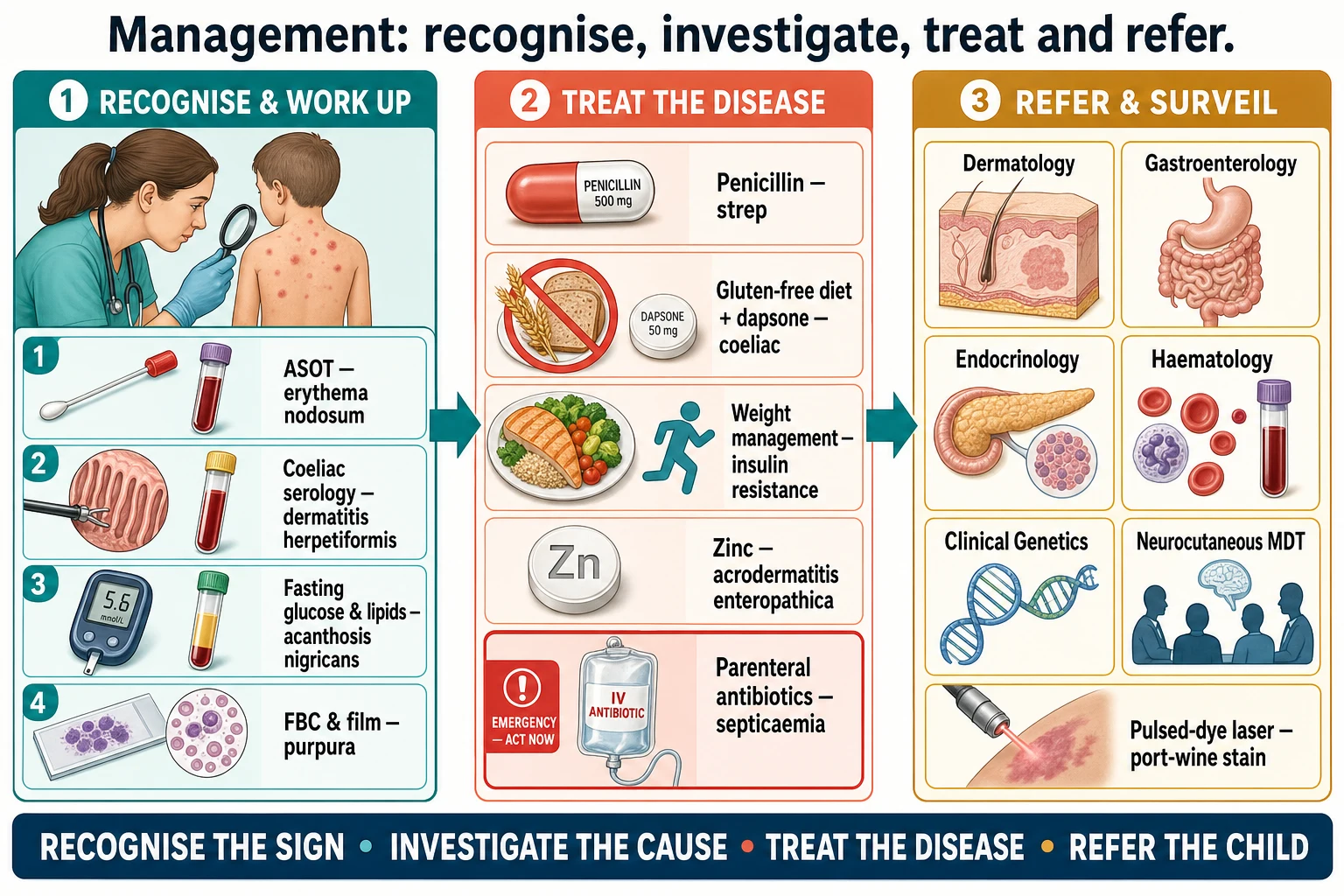
For erythema nodosum, treat the trigger. Confirm and treat the streptococcal pharyngitis, investigate and manage tuberculosis or sarcoidosis where found, and address inflammatory bowel disease with the gastroenterology team. The nodules themselves are treated with rest, elevation and non-steroidal anti-inflammatory drugs, and they resolve over weeks as the underlying cause settles. Potassium iodide is occasionally used for prolonged disease, but the principle is to treat the cause, not the nodule. [1]
For dermatitis herpetiformis, the definitive treatment is a strict, lifelong gluten-free diet for the underlying coeliac disease, guided by the coeliac guideline, plus dapsone for rapid control of the itch while the diet takes effect over months. Dapsone requires screening for glucose-6-phosphate dehydrogenase deficiency and monitoring for haemolysis and methaemoglobinaemia, and the skin lesions clear on a gluten-free diet, so the long-term management is dietary, not pharmacological. [5] [6]
Targeted therapy for selected cutaneous-systemic diseases (confirm local formularies)
For acanthosis nigricans, the treatment is the management of insulin resistance — family-based behavioural change, nutrition support, physical activity and weight management — and the velvety thickening improves as the metabolic state improves. Screen for and manage type 2 diabetes and dyslipidaemia where present. There is no effective topical treatment for the skin sign itself, because it mirrors the metabolic state. [3] [4]
For acrodermatitis enteropathica, oral zinc replacement is specific and dramatic, with the dermatitis resolving within days to weeks of starting supplementation. The inherited form requires lifelong zinc, and the acquired form resolves once intake is restored. Confirm the deficiency with a serum zinc level, but do not delay treatment in the classic clinical picture. [11]
For the neurocutaneous syndromes, management is surveillance and multidisciplinary care, not cure. Neurofibromatosis type 1 requires annual review of growth, blood pressure, neurodevelopment and the skin, with imaging for optic pathway glioma and assessment of learning and behaviour. Tuberous sclerosis complex requires surveillance for brain tubers and subependymal giant cell astrocytoma, renal angiomyolipomas, cardiac rhabdomyomas and epilepsy, with mTOR inhibitors such as everolimus for progressive lesions. Sturge-Weber syndrome requires seizure management, glaucoma treatment and low-dose aspirin for stroke-like episodes, and the port-wine stain is treated with pulsed-dye laser. [7] [8] [9]
The recognise, investigate, treat and refer pathway
Recognise the skin sign and resist treating it as a primary skin disease; name the lesion and the family it belongs to.
Build the differential for the underlying disease the sign suggests — infection, metabolic, gastrointestinal, haematological or genetic.
Order the targeted work-up the lesion dictates: throat swab and ASOT for erythema nodosum, coeliac serology and biopsy for dermatitis herpetiformis, fasting glucose and lipids for acanthosis nigricans, full blood count for purpura.
Resuscitate the emergencies first: parenteral antibiotics for meningococcal purpura, urgent investigation for the blueberry muffin neonate, neuroimaging and ophthalmology for the V1 port-wine stain.
Confirm the underlying diagnosis and treat the cause — penicillin for strep, gluten-free diet and dapsone for coeliac, weight management for insulin resistance, zinc for acrodermatitis enteropathica.
Refer to the right specialist and set up surveillance — dermatology, gastroenterology, endocrinology, haematology or clinical genetics and the neurocutaneous multidisciplinary clinic.
Review the skin sign as a marker of disease control, because the skin often improves as the systemic disease comes under control.
Specific Subtypes & Scenarios
A school-age child with tender red nodules on the shins. This is the bread-and-butter reactive erythema. Confirm the diagnosis of erythema nodosum clinically, then chase the cause — a throat swab and antistreptolysin-O titre, a chest radiograph and a tuberculosis screen. Streptococcal pharyngitis is the commonest finding in children, and treating it settles the nodules over weeks with rest and non-steroidal anti-inflammatory drugs. Reassure the family that the nodules do not ulcerate or scar. [1]
An overweight adolescent with a velvety dark neck. Recognise acanthosis nigricans as the cutaneous marker of insulin resistance, measure body mass index and blood pressure, and arrange a fasting glucose and lipid panel. The intervention is family-based weight management — nutrition, activity and behavioural change — and the skin improves as the metabolic state improves. Screen the family for type 2 diabetes, because the sign often runs with a family history. [3] [4]
A child with intensely itchy blisters on the elbows and buttocks. Recognise dermatitis herpetiformis, confirm coeliac disease with immunoglobulin A anti-tissue transglutaminase serology and a duodenal biopsy, and treat with a strict lifelong gluten-free diet and dapsone for rapid itch control. Screen for iron deficiency and assess growth, because the gut may be silent while the skin is not. The skin biopsy shows granular IgA in the dermal papillae on immunofluorescence. [5] [6]
An infant with a facial port-wine stain over the forehead. Recognise the Sturge-Weber risk, arrange urgent neuroimaging for the leptomeningeal angiomatosis and urgent ophthalmology for glaucoma, and set up surveillance for seizures and developmental delay. The port-wine stain itself is treated with pulsed-dye laser, often beginning in infancy, but the brain and eye involvement is the priority. Counsel the family about the somatic GNAQ origin and the absence of inheritance. [9]
A neonate with widespread dark blue-red papules. This is the blueberry muffin baby — a neonatal emergency. Isolate and investigate urgently with a congenital infection screen (cytomegalovirus, rubella, toxoplasmosis, syphilis), a full blood count and film, and specialist paediatric and haematology input. The differential includes haemolytic disease and congenital leukaemia or neuroblastoma, and the outcome depends on the cause. [9]
A child with café-au-lait macules and axillary freckling. Apply the revised neurofibromatosis type 1 diagnostic criteria, examine for neurofibromas, Lisch nodules and optic pathway glioma, and set up annual surveillance of growth, blood pressure, neurodevelopment and the skin. Refer to clinical genetics, because half of cases are familial, and counsel the family about the autosomal dominant inheritance. The skin sign is the entry point to a lifelong surveillance programme. [7]
A child with seizures and ash-leaf macules. Recognise tuberous sclerosis complex, apply the updated international criteria, arrange genetic testing for a TSC1 or TSC2 pathogenic variant, and set up multidisciplinary surveillance for epilepsy, subependymal giant cell astrocytoma, renal angiomyolipoma and cardiac rhabdomyoma. The skin signs — ash-leaf macules, angiofibromas, shagreen patch, periungual fibromas — are the most accessible criteria, often present long before the seizures. [8]
Complications & Pitfalls
The principal complication of a cutaneous manifestation of systemic disease is the complication of the underlying disease itself, which is why the recognition matters so much. Untreated coeliac disease brings growth failure, iron deficiency, osteoporosis and an increased risk of lymphoma. Unrecognised insulin resistance progresses to type 2 diabetes and cardiovascular disease. The neurocutaneous syndromes carry seizures, glaucoma, renal failure, disfigurement and learning difficulty. The skin sign ignored is the systemic disease untreated. [5] [8]
The first pitfall is treating the skin sign as a primary skin disease. Prescribing a topical steroid for dermatitis herpetiformis, a moisturiser for acanthosis nigricans or an antibiotic for erythema nodosum without chasing the cause misses the diagnosis and delays the real treatment. The reflex to treat the visible lesion must yield to the discipline of investigating the disease beneath it. [1] [6]
The second pitfall is underestimating the purpuric child or the blueberry muffin neonate. Non-blanching purpura in a febrile child is a time-critical emergency, and waiting for the diagnosis to declare itself costs lives. The blueberry muffin neonate is not a benign birthmark, and a delay in the congenital infection screen and the blood count delays the diagnosis of congenital leukaemia, neuroblastoma or a treatable infection. Treat both as emergencies. [9]
The third pitfall is missing the V1 port-wine stain's Sturge-Weber risk. A facial port-wine stain in the forehead or upper eyelid distribution carries a substantial risk of leptomeningeal angiomatosis and glaucoma, and the absence of seizures in infancy does not exclude the syndrome. Arrange neuroimaging and ophthalmology early, because the glaucoma may be present from infancy and the seizures may follow. [9]
The fourth pitfall is confusing mycoplasma-induced rash and mucositis with Stevens-Johnson syndrome. The two look alike — mucositis is prominent in both — but the target is mycoplasma in one and a drug in the other, the skin detachment is minimal in the former and extensive in the latter, and the management differs. A careful drug history, a mycoplasma respiratory history, and attention to the pattern of skin detachment separate them. [2]
The fifth pitfall is failing to apply the diagnostic criteria for the neurocutaneous syndromes. The revised neurofibromatosis type 1 criteria and the updated tuberous sclerosis complex criteria are clinical tools that a general paediatrician can apply at the bedside, and they distinguish the true syndrome from isolated café-au-lait macules or a single ash-leaf patch. A Wood's lamp, a tape measure and the criteria card are the tools of recognition. [7] [8]
Prognosis & Disposition
The prognosis is the prognosis of the underlying disease, and the disposition is shaped by the urgency of the sign. Most cutaneous manifestations of systemic disease are managed in the outpatient clinic, with a targeted work-up and a referral to the appropriate specialist. Erythema nodosum resolves over weeks as the trigger settles. Dermatitis herpetiformis clears on a gluten-free diet. Acanthosis nigricans improves with weight loss. Gianotti-Crosti syndrome resolves spontaneously over weeks without treatment. [1] [12]
The emergencies alter the disposition. The febrile purpuric child is admitted for resuscitation and parenteral antibiotics. The blueberry muffin neonate is admitted to a neonatal unit for urgent investigation. The V1 port-wine stain is referred urgently to neurology and ophthalmology. The suspected phakomatosis is referred to clinical genetics and the relevant multidisciplinary clinic for surveillance. Match the disposition to the urgency of the sign. [8] [9]
At each review, the skin sign is a marker of disease control, because the skin often improves as the systemic disease comes under control. The velvety neck that thins as the weight comes off, the dermatitis herpetiformis that clears as the gluten leaves the diet, and the erythema nodosum that fades as the strep is treated — each is a visible report card on the management of the disease beneath it. Use the skin to read the system. [3] [6]
Special Populations
Neonates present the most time-critical cutaneous signs of systemic disease. The blueberry muffin baby, the vesicular eruption of congenital infection, the vesicopustules of neonatal disease and the port-wine stain of Sturge-Weber all demand urgent investigation, because the neonatal skin is often the first and only sign of a condition that threatens the brain, the eye, the hearing and the blood. The threshold to investigate is low, and the threshold to involve neonatology and specialist services is lower still. [9]
Overweight and obese adolescents carry the rising tide of acanthosis nigricans and the insulin resistance behind it. The skin sign is an opportunity to intervene on a modifiable metabolic trajectory before glucose intolerance develops, and the family-based approach — nutrition, activity and behavioural change — is the intervention most likely to change both the skin and the metabolic risk. Treat the weight at the same consultation as the skin. [3] [4]
Children with a family history of a neurocutaneous syndrome or coeliac disease need proactive surveillance, because both conditions are autosomal dominant or familial and the skin sign may be the first evidence in an at-risk child. Apply the diagnostic criteria at the annual review, and refer to clinical genetics where the criteria are met, because early surveillance detects the optic pathway glioma, the renal angiomyolipoma and the coeliac enteropathy before they harm. [5] [7]
Children in rural, remote and disadvantaged communities, and Indigenous families face real inequity of access to specialist dermatology, clinical genetics and the multidisciplinary neurocutaneous clinic, all of which concentrate in major centres. Deliver care through outreach and telehealth, document and photograph the skin findings clearly because the criteria are visual, and advocate for the service pathways that make surveillance achievable where the family lives. Distance should never be the reason a port-wine stain is not assessed for Sturge-Weber risk. [8] [9]
Immunocompromised children with cutaneous signs of systemic disease need coordinated care between dermatology and the treating immunology, oncology or transplant team, because the differential of a skin sign in an immunocompromised child is broader — infection, drug eruption, graft-versus-host disease and malignancy all enter — and the threshold to biopsy and investigate is lower. Involve the team early. [10]
Evidence, Guidelines & Regional Differences
The evidence base for cutaneous manifestations of systemic disease is built from disease-specific reviews and consensus criteria rather than single trials, because the field is a constellation of conditions rather than one disease. The erythema nodosum review consolidates the cause list and the work-up that anchors the reactive erythemas, and the acanthosis nigricans updated review frames the insulin-resistance link and the metabolic work-up. These are the teaching references for the metabolic and reactive families. [1] [3]
Revised diagnostic criteria for neurofibromatosis type 1 and Legius syndrome
Population: An international consensus panel, convened to revise the National Institutes of Health diagnostic criteria for neurofibromatosis type 1 and to distinguish it from Legius syndrome, the related Rasopathy caused by SPRED1 loss.
Key finding
The revised criteria retain the clinical hallmarks — six or more café-au-lait macules, skinfold freckling, neurofibromas, optic pathway glioma, Lisch nodules, osseous dysplasia and a first-degree relative — and add the option of a pathogenic NF1 variant, while clarifying the Legius syndrome differential that shares the café-au-lait macules and freckling.
Practice change
A general paediatrician can apply the revised criteria at the bedside to confirm neurofibromatosis type 1 on the skin signs alone, while recognising that Legius syndrome is the differential when café-au-lait macules and freckling appear without neurofibromas.
The coeliac disease ESsCD guideline is the official European guidance that anchors the dermatitis herpetiformis link, setting out the serology, the duodenal biopsy and the lifelong gluten-free diet that define the management. The dermatitis herpetiformis update consolidates the diagnosis, the immunofluorescence findings and the dapsone-plus-diet approach. Together they frame the gastrointestinal family. [5] [6]
The neurocutaneous evidence is built on the consensus criteria. The updated international tuberous sclerosis complex criteria set the major and minor gene and clinical criteria, with a pathogenic TSC1 or TSC2 variant now sufficient for the diagnosis, and the surveillance schedule for brain, kidney, heart and skin. The port-wine birthmark and Sturge-Weber review frames the risk assessment and the early laser and the neurology and ophthalmology referral. These are the references the multidisciplinary clinic uses. [8] [9]
The neutrophilic dermatoses are framed by the Sweet syndrome review, which consolidates the malignancy, drug and infection associations and the diagnostic triad of fever, painful plaques and a dermal neutrophilic infiltrate. The mycoplasma-induced rash and mucositis systematic review separates that syndrome from Stevens-Johnson syndrome and erythema multiforme, and the Gianotti-Crosti review consolidates the viral triggers and the characteristic distribution. The acrodermatitis enteropathica paper frames the zinc-deficiency mechanism and the dramatic response to replacement. [10] [2] [12] [11]
The regional policy structure is consistent in principle and varies in access. In the United Kingdom, the NICE and specialist guidance frame the investigation of cutaneous markers of systemic disease. In the United States and Canada, the American and Canadian specialist and genetic guidance frame the work-up and the neurocutaneous surveillance. In Australia and New Zealand, the Therapeutic Guidelines and the state-funded clinical genetics services frame the same pathway, with a particular emphasis on tuberculosis in the erythema nodosum differential in migrant and Indigenous communities, and on equitable access to specialist and genetic services for rural and remote families. The principle is the same everywhere: the skin sign is a door, and the work-up is the room behind it. [1] [8]
The controversies are real: the optimal threshold for metabolic screening in acanthosis nigricans; the role of aspirin and the timing of laser in Sturge-Weber syndrome; the long-term surveillance burden of the neurocutaneous syndromes; and the inequities in access to specialist and genetic care. The defence against each is the same: recognise the sign, investigate the cause, treat the disease, and refer to the team that owns it. [3] [9]
Exam Pearls
- The skin is a window to systemic disease; the framework is recognise, investigate the underlying disease, and refer — the skin sign is a door, not a destination. [1]
- Group the signs by mechanism: reactive erythemas (erythema nodosum, erythema multiforme, Gianotti-Crosti), neutrophilic dermatoses (Sweet, pyoderma gangrenosum), metabolic (acanthosis nigricans, necrobiosis lipoidica, xanthomas), gastrointestinal and nutritional (dermatitis herpetiformis, acrodermatitis enteropathica), haematological and neoplastic (petechiae, blueberry muffin baby) and neurocutaneous (NF1, TSC, Sturge-Weber). [8]
- Erythema nodosum in children is most often group A streptococcal pharyngitis — confirm with a throat swab and antistreptolysin-O titre; also consider tuberculosis, sarcoidosis and inflammatory bowel disease. [1]
- Acanthosis nigricans is the cutaneous marker of insulin resistance — screen with fasting glucose and a lipid panel and manage the weight. [3]
- Dermatitis herpetiformis is the cutaneous face of coeliac disease — confirm with IgA anti-tissue transglutaminase serology and a duodenal biopsy, and treat with a strict lifelong gluten-free diet and dapsone for rapid itch control (screen for G6PD deficiency). [5] [6]
- Mycoplasma-induced rash and mucositis has prominent mucositis and minimal skin detachment and is distinct from Stevens-Johnson syndrome — name it correctly. [2]
- Acrodermatitis enteropathica is zinc deficiency (SLC39A4) — periorificial and acral dermatitis with diarrhoea and alopecia, resolving within days of zinc replacement. [11]
- Neurofibromatosis type 1 revised criteria: six or more café-au-lait macules (≥5 mm prepubertal, ≥15 mm postpubertal), skinfold freckling, neurofibromas, optic pathway glioma, Lisch nodules, osseous dysplasia, first-degree relative. [7]
- Tuberous sclerosis complex: ash-leaf hypomelanotic macules, angiofibromas, shagreen patch, periungual fibromas — a pathogenic TSC1 or TSC2 variant is now diagnostic. [8]
- The emergencies are the febrile purpuric child (meningococcal septicaemia or leukaemia), the blueberry muffin neonate (congenital infection or leukaemia or neuroblastoma) and the V1 port-wine stain (Sturge-Weber — urgent neuroimaging and ophthalmology for glaucoma). [9]
References
- [1]Leung AKC; Leong KF; Lam JM Erythema nodosum. World J Pediatr, 2018.PMID 30269303
- [2]Canavan TN; Mathes EF; Frieden I; Shinkai K Mycoplasma pneumoniae-induced rash and mucositis as a syndrome distinct from Stevens-Johnson syndrome and erythema multiforme: a systematic review. J Am Acad Dermatol, 2015.PMID 25592340
- [3]Leung AKC; Lam JM; Barankin B; Leong KF; et al Acanthosis Nigricans: An Updated Review. Curr Pediatr Rev, 2022.PMID 36698243
- [4]Baselga Torres E; Torres-Pradilla M Cutaneous manifestations in children with diabetes mellitus and obesity. Actas Dermosifiliogr, 2014.PMID 24698434
- [5]Al-Toma A; Volta U; Auricchio R; Castillejo G; et al European Society for the Study of Coeliac Disease (ESsCD) guideline for coeliac disease and other gluten-related disorders. United European Gastroenterol J, 2019.PMID 31210940
- [6]Nguyen CN; Kim SJ Dermatitis Herpetiformis: An Update on Diagnosis, Disease Monitoring, and Management. Medicina (Kaunas), 2021.PMID 34441049
- [7]Legius E; Messiaen L; Wolkenstein P; Pancza P; et al Revised diagnostic criteria for neurofibromatosis type 1 and Legius syndrome: an international consensus recommendation. Genet Med, 2021.PMID 34012067
- [8]Northrup H; Aronow ME; Bebin EM; Bissler J; et al Updated International Tuberous Sclerosis Complex Diagnostic Criteria and Surveillance and Management Recommendations. Pediatr Neurol, 2021.PMID 34399110
- [9]Poliner A; Fernandez Faith E; Blieden L; Kelly KM; et al Port-wine Birthmarks: Update on Diagnosis, Risk Assessment for Sturge-Weber Syndrome, and Management. Pediatr Rev, 2022.PMID 36045161
- [10]Villarreal-Villarreal CD; Ocampo-Candiani J; Villarreal-Martinez A Sweet Syndrome: A Review and Update. Actas Dermosifiliogr, 2016.PMID 26826881
- [11]Sivakumar A; Vageshappa RK; Kumari R Acrodermatitis Enteropathica. JAMA Dermatol, 2024.PMID 37938848
- [12]Leung AKC; Sergi CM; Lam JM; Leong KF Gianotti-Crosti syndrome (papular acrodermatitis of childhood) in the era of a viral recrudescence and vaccine opposition. World J Pediatr, 2019.PMID 31134587