Paeds · paediatric-dermatology
Vascular birthmarks and infantile haemangioma
Also known as Infantile haemangioma · Strawberry naevus · Port-wine stain · Capillary malformation · Salmon patch · PHACE syndrome · Sturge-Weber syndrome · Vascular anomaly
Fellowship topic on vascular birthmarks and infantile haemangioma: the ISSVA split of vascular anomalies into proliferating tumours (infantile haemangioma, congenital haemangiomas, kaposiform haemangioendothelioma) and structural malformations (capillary, venous, lymphatic, arteriovenous and combined); the GLUT1-positive endothelial-proliferation biology of infantile haemangioma and its proliferate-then-involutive natural history; the segmental-haemangioma syndromes PHACE and LUMBAR and their screening; the GNAQ somatic-mosaic biology of port-wine stain and Sturge-Weber syndrome; propranolol 2 to 3 mg per kg per day as first-line therapy for problematic infantile haemangioma with topical timolol for small superficial lesions; pulsed-dye laser for port-wine stain; the Kasabach-Merritt phenomenon of kaposiform haemangioendothelioma; and ANZ, UK, US and Canada guidance.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
The ISSVA fork — tumour versus malformation
Overview & Definition
Picture a six-week-old brought to the clinic because a bright red lump has appeared on the cheek over three weeks, growing faster than anything the parents have seen. They have been told it is a birthmark, but it was not there at birth. This is the everyday face of infantile haemangioma, the commonest tumour of infancy, and the question on every parent's mind — will it go away, and does it need treatment — is exactly the question the clinician must answer with confidence. [2] [11]
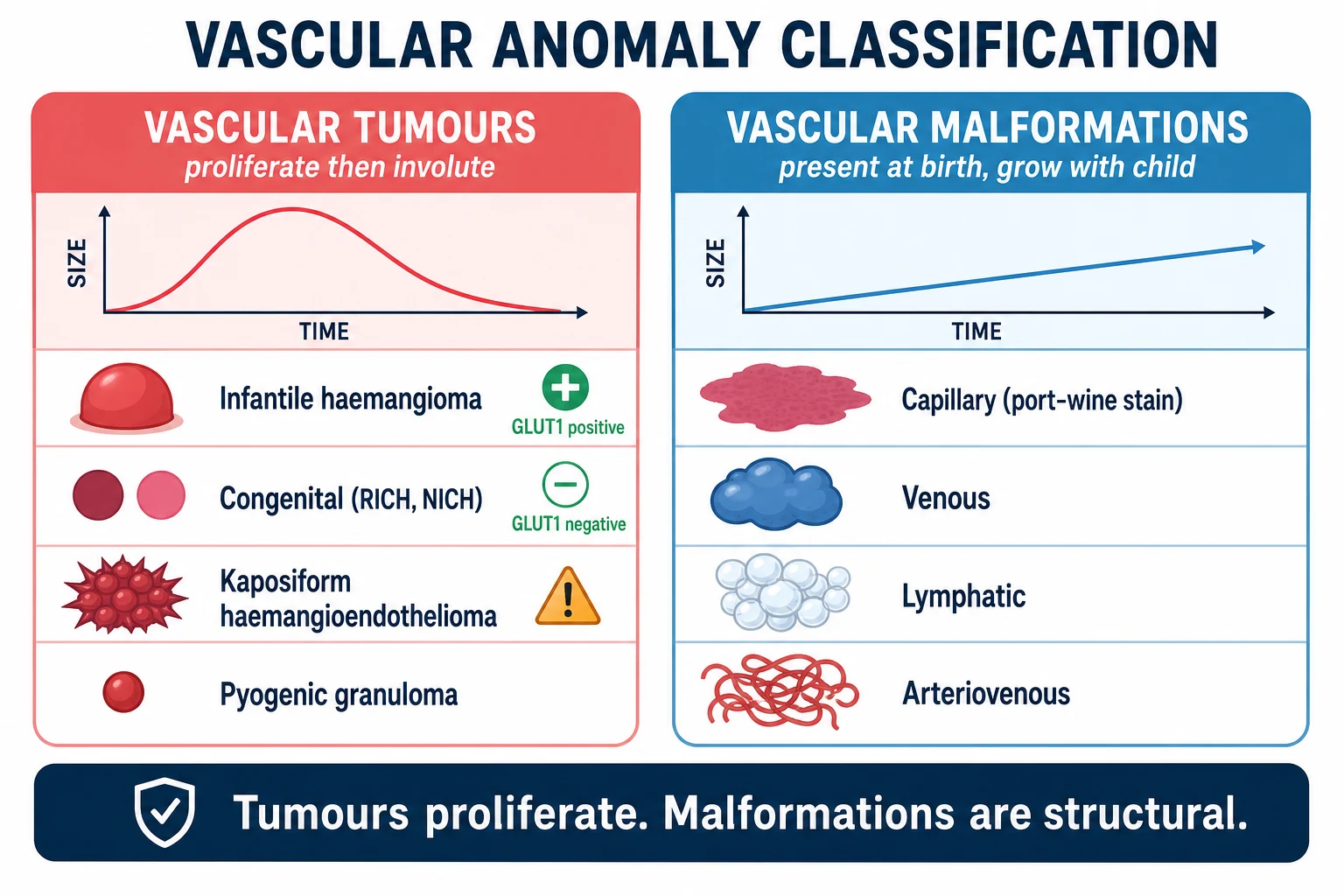
Vascular birthmarks are visible anomalies of blood or lymphatic vessels present in or appearing soon after infancy. They are not one disease. The Mulliken and Glowacki classification, refined into the living International Society for the Study of Vascular Anomalies framework, separates them into two families by cellular behaviour. Vascular tumours arise from endothelial proliferation: the prototype is infantile haemangioma, joined by the congenital haemangiomas and the rarer kaposiform haemangioendothelioma. Vascular malformations are errors of vessel morphogenesis — capillary, venous, lymphatic, arteriovenous, or combinations — present at birth and expanding only in proportion to the child. [5] [6]
The clinician's task has three layers. The first is to name the lesion correctly, because calling a malformation a haemangioma (or vice versa) sends management down the wrong road. The second is to find the lesions that are not harmless — the ones that threaten the airway or the eye, the large and segmental lesions that hide a syndrome, and the multifocal lesions that hide hepatic disease. The third is to treat at the right time: propranolol works in the proliferative phase, so recognising the problematic lesion early is what changes the outcome. [2] [1]
Classification
Classify every vascular birthmark by the two-family fork, because the family decides whether the lesion will shrink on its own, whether propranolol will work, and whether a syndrome is hiding underneath. The fork is the single most useful piece of knowledge in the field, and it is the one examiners test most. [5] [6]
Vascular tumours are proliferative. Infantile haemangioma is the prototype and the commonest: it appears after birth, proliferates rapidly, then involutes. It is GLUT1 positive, which distinguishes it from every look-alike. The congenital haemangiomas — rapidly involuting (RICH), non-involuting (NICH) and partially involuting (PICH) — are fully grown at birth, are GLUT1 negative, and do not proliferate after birth; RICH regresses swiftly within the first year while NICH persists. Kaposiform haemangioendothelioma and tufted angioma are rare, aggressive tumours that cause the Kasabach-Merritt phenomenon and are not infantile haemangiomas. Pyogenic granuloma is a common friable bleeding nodule of older infants and children. [11] [6]
Vascular malformations are structural and present at birth. Capillary malformation, the port-wine stain, is a flat pink or deep-red patch that never fades and darkens with age. Venous malformations are soft, blue, compressible masses. Lymphatic malformations are cystic, fluid-filled swellings, common in the neck. Arteriovenous malformations are warm, pulsatile masses with a shunt. Combined malformations produce the syndromes: Klippel-Trenaunay (capillary-lymphaticovenous malformation with limb overgrowth), Sturge-Weber (facial port-wine stain with leptomeningeal angioma), and others. The salmon patch — the fading pink stain of the eyelid, forehead and nape in newborns — is a benign fading capillary stain, not a true port-wine stain, and it resolves in the first years. [6] [5]
Epidemiology & Risk Factors
Infantile haemangioma affects about four to five percent of infants, making it the most common tumour of infancy. The risk factors cluster around prematurity and low birth weight: the more premature and the smaller the infant, the higher the risk, with prevalence rising substantially in very-low-birth-weight infants. Girls are affected two to three times more often than boys, and white infants carry the highest risk. [2] [11]
Vascular malformations are far less common individually but are present at birth, so they are recognised in the neonatal period. Port-wine stain occurs in roughly three per thousand infants. The combined syndromes are rare but disproportionately important because they carry neurological and cardiovascular risk that a skin lesion alone would never suggest. The GNAQ mutation that underlies both isolated port-wine stain and Sturge-Weber syndrome is somatic and mosaic, occurring after fertilisation, which is why the lesion follows a developmental territory and why siblings are not affected. [4] [6]
The lesions that change the epidemiology into a syndrome are the segmental ones. A segmental infantile haemangioma — a large lesion mapping to a developmental facial territory rather than a small focal blob — carries a several-fold higher risk of PHACE syndrome when it is on the face, and of occult spinal dysraphism when it is in the lumbosacral midline. The lesion pattern is the epidemiological signal that triggers investigation. [8] [12]
In Australia and Aotearoa New Zealand, care for complex vascular anomalies is coordinated through specialist multidisciplinary vascular-anomalies services in the major paediatric centres, with outreach and telehealth linking rural and remote families. Timely access to propranolol, ophthalmology and laser services shapes outcome, and equity of access to these services across distance is a recurring theme in regional practice. [2]
Pathophysiology
Why does infantile haemangioma appear after birth, race ahead for months, then melt away — while a port-wine stain sits unchanged from the day of birth? The answer is that one is a proliferating tumour and the other is a fixed structural error, and their biology is entirely different. [5] [11]
Infantile haemangioma arises from a clone of endothelial cells driven into rapid proliferation. The proliferating cells express GLUT1, the glucose transporter, which is the single most useful immunohistochemical marker because it persists in the lesion and is absent from congenital haemangiomas and vascular malformations. The proliferative phase is fuelled by pro-angiogenic signalling including the RAS pathway, and it runs from the first weeks of life to around nine months. Involution then follows as the proliferating endothelium undergoes apoptosis and is replaced by fibrofatty tissue, leaving a smaller, paler residue over years. [11] [5]
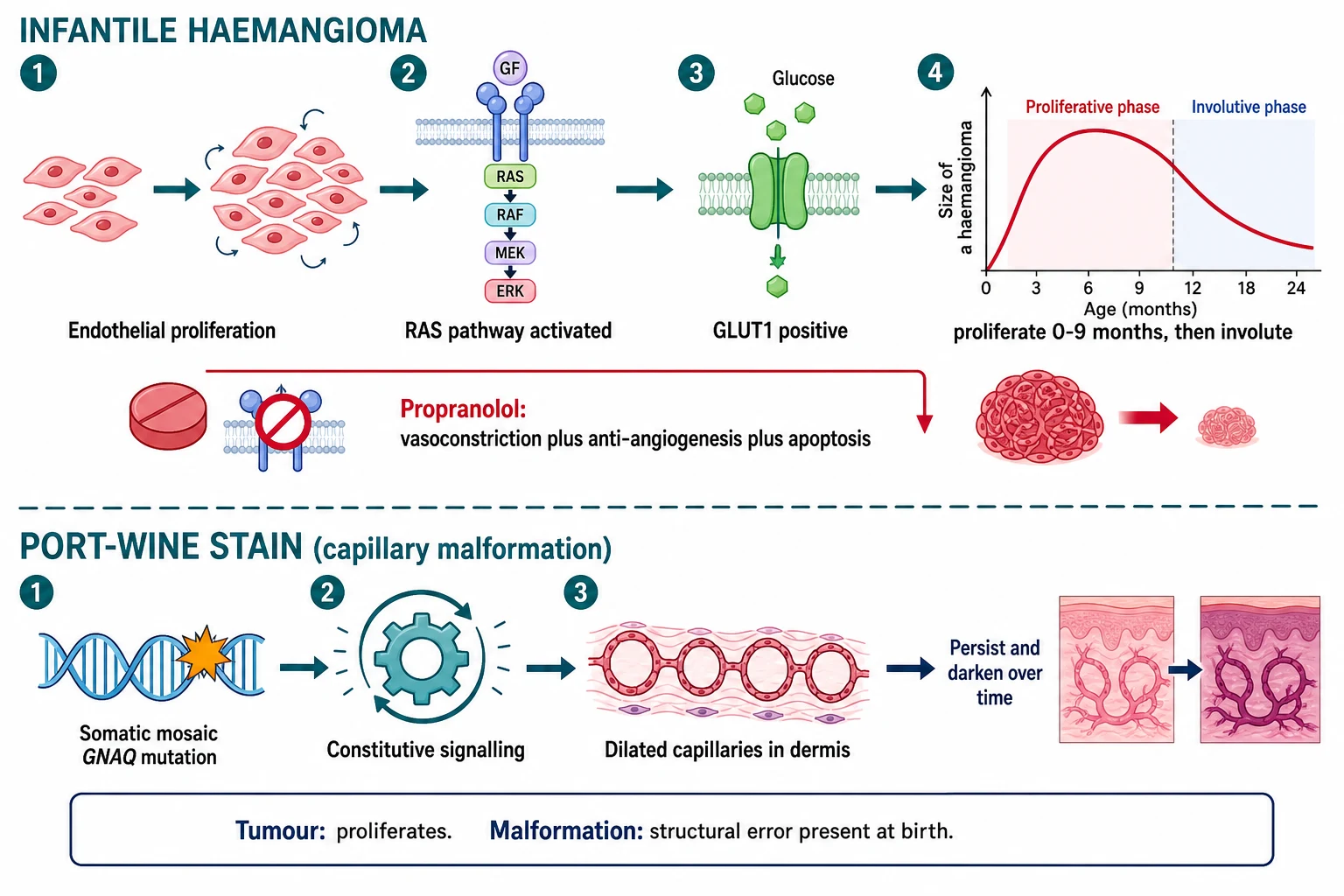
The beta-blocker story transformed the field. Propranolol halts proliferation and accelerates involution through three mechanisms: early vasoconstriction that reduces flow, down-regulation of angiogenic signalling that stops new vessel growth, and induction of endothelial apoptosis that removes the proliferated cells. This is why a tumour that proliferates responds to a beta-blocker, and why a static malformation does not. [1] [7]
Port-wine stain runs on different biology. It is caused by a somatic mosaic activating mutation in GNAQ, the same mutation found in Sturge-Weber syndrome. The mutation drives constitutive signalling in the affected capillaries, leaving a network of dilated, thin-walled vessels in the superficial dermis that are present from birth. Because the vessels are structurally fixed, the lesion does not regress; instead it darkens from pink to deep purple and thickens into cobblestoned nodules over decades. The same mutation in leptomeningeal vessels produces the angioma of Sturge-Weber syndrome and its seizure risk. [4] [6]
Clinical Presentation
The classic infantile haemangioma appears in the first two to eight weeks of life as a faint red macule or a pale precursor patch, then grows rapidly into its characteristic form. A superficial haemangioma is bright red, raised, lobulated and strawberry-like with a sharply demarcated border. A deep haemangioma shows only a bluish swelling beneath normal skin. A mixed lesion has both components. Growth is fastest in the first few months and is largely complete by nine to twelve months, after which the lesion stabilises and begins the slow, pale involution that may continue for years. [11] [2]
The natural history is the key counselling point. Most of the involution happens in the first few years, but the residue — a paler, flatter, sometimes telangiectatic or fibrofatty patch — often persists, and lesions on the face can leave a permanent cosmetic mark. The decision to treat turns on whether the lesion threatens function, risks a permanent scar, or signals a syndrome, not merely on its existence. [2] [11]
[5] [6]Complications are part of the presentation because they often bring the child to attention. Ulceration is the commonest, a painful, bleeding breakdown usually over the lip, neck fold or perineum, prone to infection and scarring. A periocular haemangioma threatens vision through astigmatism, amblyopia or optic nerve compression. A subglottic or airway haemangioma presents with biphasic stridor worsening over weeks, worse on crying, and is an emergency. Large or hepatic haemangiomas cause high-output cardiac failure, and multifocal skin lesions raise the question of liver involvement. [2] [11]
Differential Diagnosis
For the bright-red growing nodule, the differential turns on whether it is a tumour, a malformation, or something else entirely. The most common error in the field is calling a malformation a haemangioma or vice versa, and the two-family fork resolves most cases: if it appeared after birth and is proliferating, it is infantile haemangioma; if it was present at birth and is static, it is a malformation. [5] [6]
Pyogenic granuloma is a friable, rapidly growing, easily bleeding nodule that develops over weeks and bleeds on minor trauma — it is not a haemangioma and is treated by excision or cautery. Congenital haemangiomas (RICH and NICH) are fully formed at birth, which alone separates them from infantile haemangioma. A Spitz naevus is a pink or brown flat nodule in older infants that can mimic a haemangioma. The salmon patch of the newborn eyelid, glabella and nape fades in the first years and must not be mistaken for a port-wine stain. [11] [6]
For the flat pink patch present at birth, the differential is between the fading salmon patch, the permanent port-wine stain, and an early vascular malformation of another type. A port-wine stain on the forehead or upper eyelid (the ophthalmic or first-trigeminal territory) raises Sturge-Weber syndrome, while a patch on the limb raises Klippel-Trenaunay syndrome when overgrowth and venous or lymphatic disease accompany it. [4] [6]
Clinical & Bedside Assessment
The assessment begins with two questions at the bedside: was the lesion present at birth, and how has it changed since? A lesion that appeared after birth and is proliferating is a tumour; one present at birth and growing with the child is a malformation. The history of onset and growth is the most powerful single diagnostic tool. Ask about the rate of growth, any ulceration or bleeding, and any functional impact — feeding difficulty, noisy breathing, or a change in the appearance of the eye. [11] [2]
Examine the lesion's morphology and distribution. Describe whether it is superficial, deep or mixed, its colour and border, and its size. Crucially, determine whether it is focal or segmental — a large lesion mapping to a facial developmental territory, rather than a discrete blob, is the trigger to screen for PHACE syndrome. Examine the face fully for midline and segmental patterns, look in the mouth for airway involvement, and assess the eye for any periocular lesion that threatens vision. [2] [8]
Examine the whole skin, not just the obvious lesion, because the presence of five or more cutaneous haemangiomas signals a risk of hepatic haemangiomas that need ultrasound screening. For a lumbosacral or perineal lesion, examine the overlying skin for other markers of spinal dysraphism — a dimple, a tuft of hair, a tail, or a deviated gluteal cleft — and palpate the limbs for the overgrowth of Klippel-Trenaunay syndrome. [12] [2]
Assess the functional and systemic red flags directly. Stridor in an infant with a cutaneous haemangioma, particularly a beard-distribution or segmental facial lesion, demands airway assessment. Tachycardia, a gallop rhythm or hepatomegaly in an infant with a large or multifocal lesion signals high-output cardiac failure and the need for echocardiography. [2] [11]
Investigations
Most infantile haemangiomas are diagnosed clinically, and no test is needed for the typical focal lesion in a well infant. The history of postnatal appearance and proliferation, the strawberry morphology, and the distribution make the diagnosis. The investigation effort goes into the at-risk lesion, not the routine one. [2] [11]
The single most important investigation is the syndrome screen triggered by the segmental or large lesion. A large or segmental facial haemangioma warrants brain and neck MRI with MR angiography to look for the structural and arterial anomalies of PHACE syndrome, echocardiography for aortic arch and cardiac anomalies, and ophthalmology review for the eye anomalies. A midline lumbosacral or perineal haemangioma warrants spinal ultrasound in the young infant or spinal MRI for occult spinal dysraphism. These are the investigations that change management, and they are driven by the lesion pattern. [8] [12]
For five or more cutaneous haemangiomas, screen the liver with ultrasound for hepatic haemangiomas, because liver involvement can cause high-output cardiac failure and, through type 3 iodothyronine deiodinase expression in the tumour, hypothyroidism. Check thyroid function in any infant with a large or multifocal haemangioma. [2] [11]
Tissue diagnosis with biopsy and GLUT1 staining is reserved for the genuinely ambiguous lesion, the lesion that does not behave as expected, or the suspicion of a malignancy or kaposiform haemangioendothelioma. A lesion that is fully grown at birth, behaves aggressively, or is associated with thrombocytopenia should prompt specialist referral and biopsy rather than empirical propranolol, because the diagnosis may not be infantile haemangioma. [11] [6]
Management — Resuscitation
Resuscitation belongs to the few haemangiomas that threaten life or function at presentation. The emergencies are the compromised airway, the failing circulation, and the lesion hiding a consumptive coagulopathy. Each needs urgent recognition, not a dermatology appointment. [2] [11]
An infant with a cutaneous haemangioma in a beard or segmental facial distribution who develops worsening stridor has a presumptive subglottic haemangioma until proven otherwise. Assess and secure the airway with paediatric ENT and anaesthetic support, confirm with airway endoscopy, and start systemic propranolol promptly — propranolol is both diagnostic support and treatment for airway disease. Bilateral or large segmental facial lesions carry the highest airway risk. [2] [11]
An infant with a large or hepatic haemangioma and high-output cardiac failure — tachycardia, a gallop, hepatomegaly, poor feeding — needs urgent echocardiography, cardiac support and specialist treatment of the underlying lesion. A rapidly enlarging, firm, purpuric vascular tumour with bruising and a falling platelet count is kaposiform haemangioendothelioma with the Kasabach-Merritt phenomenon: check the full blood count and coagulation, and refer urgently to a vascular-anomalies or haematology-oncology specialist for sirolimus or vincristine, not propranolol. [11] [2]
Keep resuscitation separate from routine care in your mind: the well infant with a focal haemangioma needs observation or propranolol in clinic, while the infant with stridor, cardiac failure or a consumptive coagulopathy needs the hospital, the airway team and the specialist. The timing of propranolol in the proliferative phase is what matters for the common lesion; the recognition of the emergency is what matters for the rare one. [2] [1]
Management — Definitive & Stepwise
Definitive management is a stepwise pathway matched to risk, because most haemangiomas need nothing and a few need propranolol now. The decision turns on whether the lesion is harmless, threatens function or appearance, or signals a syndrome. The art is treating the lesions that need it, early, and reassuring the family about the rest. [2] [3]
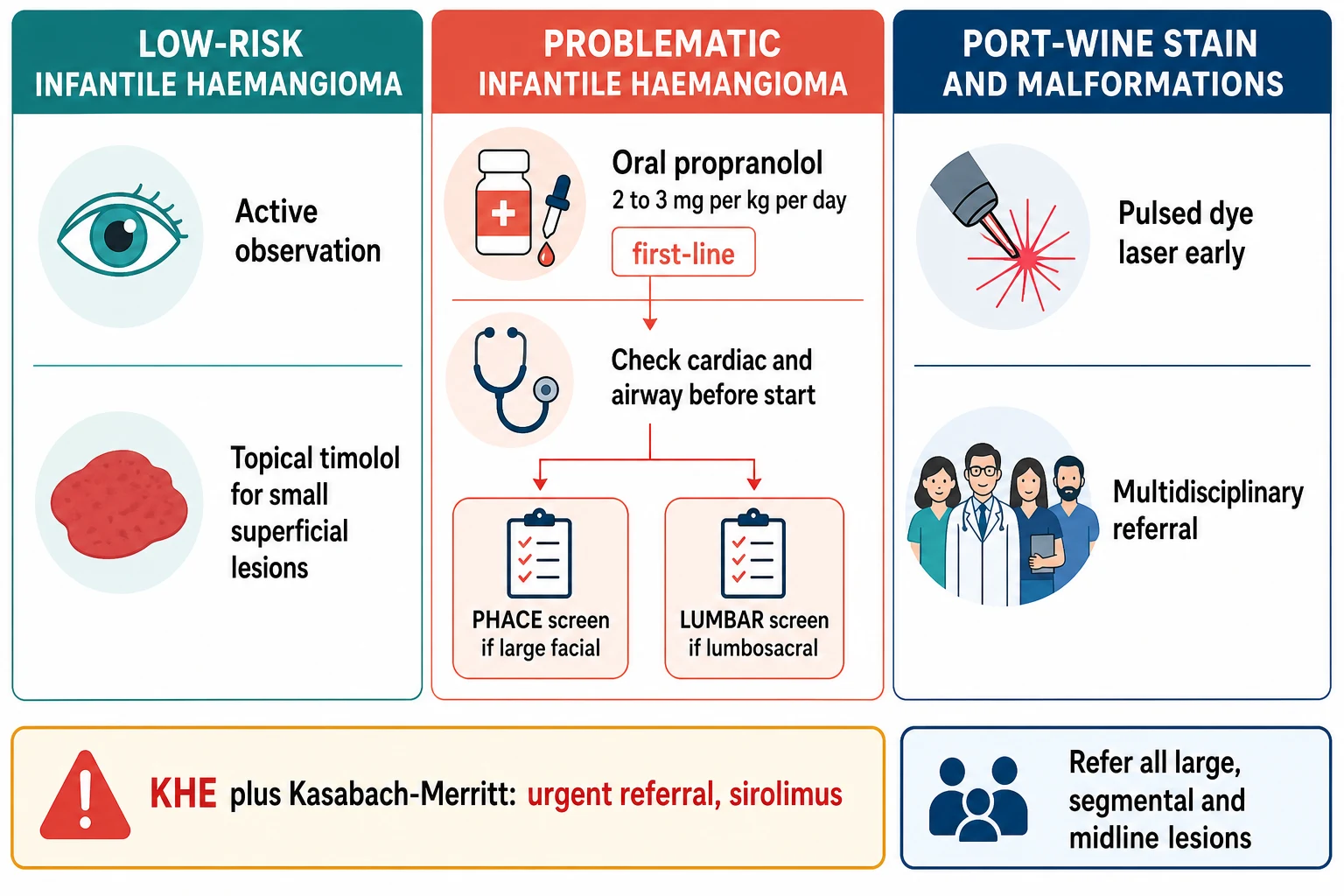
For the low-risk focal haemangioma in a non-critical site — a small lesion on the trunk or limb, not ulcerated, not near the eye or airway — the management is active observation and parent counselling on the natural history. Reassure the family that the lesion will grow for a few months and then involute over years, and review at intervals. No treatment is needed for the majority of infantile haemangiomas. [2] [3]
For the small superficial haemangioma where treatment is desired, topical timolol 0.5 percent gel-forming solution applied to the lesion surface is effective for thin superficial lesions, supported by the randomised trial evidence. It is the first choice for small, superficial, non-ulcerated lesions where a topical agent is enough, avoiding the systemic exposure of oral therapy. [10] [3]
For problematic infantile haemangioma — periocular, airway, large, segmental, ulcerated, or risking permanent disfigurement — oral propranolol is first-line. The standard dose is 2 to 3 mg per kg per day in two or three divided doses (the licensed oral solution is approved at 3 mg per kg per day in two divided doses), typically initiated at around 1 mg per kg per day and titrated upward. Treatment is started in the proliferative phase and continued to around twelve months of age, when the proliferative drive has passed. A basic cardiac check before starting excludes significant bradycardia, heart block or aortic obstruction, which are cautions rather than absolute contraindications. [1] [7]
Infantile haemangioma treatment ladder (AAP / consensus — confirm locally)
In Australia and New Zealand the Royal Children's Hospital Melbourne guideline and the major paediatric vascular-anomalies services align with the AAP clinical practice guideline: propranolol for problematic infantile haemangioma at 2 to 3 mg per kg per day, topical timolol for small superficial lesions, and multidisciplinary referral for large, segmental and syndromic lesions. Pulses of care are coordinated so that rural and remote families can initiate and monitor propranolol with outreach and telehealth support. [2]
The important cautions with propranolol are hypoglycaemia, particularly with intercurrent illness and poor feeding, bradycardia and hypotension, and reactive airway disease. Families are counselled to maintain feeding during illness, to recognise the limp or pale infant as possible hypoglycaemia, and to avoid propranolol during severe respiratory illness. Initiation for an infant with suspected PHACE syndrome and cerebral arterial anomalies needs specialist supervision because of the rare risk of stroke from haemodynamic change. [7] [8]
The stepwise vascular-birthmark pathway
Classify the lesion by the tumour-versus-malformation fork: did it appear after birth and proliferate, or was it present at birth and grow with the child?
Decide risk: is it focal and non-critical, or problematic (periocular, airway, large, segmental, ulcerated, multifocal)?
Low-risk focal lesion: active observation and parent counselling on the natural history; review through the proliferative phase.
Small superficial lesion where treatment is desired: topical timolol 0.5 percent gel-forming solution.
Problematic infantile haemangioma: oral propranolol 2 to 3 mg per kg per day, cardiac check first, treat in the proliferative phase to around twelve months.
Large or segmental facial lesion: screen for PHACE syndrome with brain and neck MRI and MRA, echocardiography and ophthalmology.
Midline lumbosacral or perineal lesion: screen for occult spinal dysraphism with spinal ultrasound or MRI.
Five or more cutaneous lesions: screen the liver with ultrasound and check thyroid function.
Port-wine stain: refer early for pulsed-dye laser; if on the forehead or upper eyelid, screen for Sturge-Weber syndrome.
Rapidly enlarging purpuric tumour with thrombocytopenia: suspect kaposiform haemangioendothelioma and Kasabach-Merritt; refer urgently for sirolimus or vincristine.
For the port-wine stain, management is early referral for pulsed-dye laser, because results are best when treatment begins in infancy when the skin is thin and the vessels are small. Laser does not cure the lesion but lightens it and reduces later thickening. A forehead or upper-eyelid stain triggers evaluation for Sturge-Weber syndrome with neurological assessment and ophthalmology for glaucoma surveillance. The malformations — venous, lymphatic and arteriovenous — are managed in a specialist multidisciplinary vascular-anomalies service, not with propranolol. [4] [6]
Specific Subtypes & Scenarios
A focal haemangioma on the trunk or limb. This is the bread-and-butter case. Reassure the family about the proliferate-then-involutive natural history, confirm the lesion is not ulcerated and not near a critical site, and review at intervals. Most need no treatment. [2] [11]
A periocular haemangioma. This threatens vision through astigmatism, amblyopia or optic nerve compression and needs urgent ophthalmology and systemic propranolol. The risk is permanent visual loss from amblyopia if the lesion is not treated early, so a periocular lesion is treated, not observed. [2] [11]
A large or segmental facial haemangioma. This is the lesion that mandates a syndrome screen. Perform brain and neck MRI with MR angiography, echocardiography and ophthalmology review for PHACE syndrome. Treatment is propranolol under specialist supervision, because the cerebral arterial anomalies of PHACE carry a small stroke risk during propranolol initiation that requires monitoring. [8] [9]
A lumbosacral or perineal haemangioma. This raises LUMBAR syndrome and the risk of occult spinal dysraphism, tethered cord and genitourinary and anorectal anomalies. Image the spine with ultrasound in the young infant or MRI, and refer to the specialist team. The skin lesion is the visible marker of a hidden spinal problem. [12]
An infant with stridor and a beard-distribution or segmental facial haemangioma. Presume a subglottic airway haemangioma. Assess and secure the airway with ENT and anaesthetic support, confirm with endoscopy, and start systemic propranolol. Airway haemangiomas can compromise the airway rapidly, and the cutaneous distribution is the warning sign. [2] [11]
Five or more cutaneous haemangiomas. Screen the liver with ultrasound for hepatic haemangiomas and check thyroid function for the hypothyroidism of large haemangiomatosis. Hepatic involvement can cause high-output cardiac failure, so the infant with multiple skin lesions is a cardiac and hepatic patient until screened. [2] [11]
A port-wine stain on the forehead or upper eyelid. Evaluate for Sturge-Weber syndrome with neurological assessment and ophthalmology for glaucoma, because the leptomeningeal angioma underlies seizures and developmental risk and glaucoma can develop early. Refer early for pulsed-dye laser for the skin lesion. [4] [6]
A rapidly enlarging purpuric tumour with bruising and thrombocytopenia. This is kaposiform haemangioendothelioma with the Kasabach-Merritt phenomenon, not infantile haemangioma. Refer urgently to a specialist for sirolimus or vincristine; propranolol is not effective and is not the treatment here. [11] [6]
Complications & Pitfalls
The complications of infantile haemangioma are ulceration, functional obstruction, cardiac failure, permanent disfigurement and the syndromes. Ulceration is the commonest and is painful, bleeds, and scars; it occurs most over the lip, neck fold and perineum, and is treated with wound care, pain relief and propranolol. Functional obstruction — the airway, the eye — threatens life or vision and is the indication for urgent propranolol. [2] [11]
The cardinal pitfall is misclassification. Calling a malformation a haemangioma leads to ineffective propranolol and missed laser; calling a haemangioma a malformation leads to missed propranolol in the window where it works. The two-family fork, applied with the history of onset and growth, resolves most cases and is the single most important skill in the field. [5] [6]
A second pitfall is treating the trivial and missing the dangerous. A small focal haemangioma needs no treatment, while a periocular, airway or segmental lesion needs propranolol now and a syndrome screen. Failing to recognise the segmental pattern, the periocular site or the airway warning sign is how visual loss, airway compromise and missed PHACE happen. [2] [8]
A third pitfall is confusing Kasabach-Merritt with infantile haemangioma. The consumptive thrombocytopenia belongs to kaposiform haemangioendothelioma and tufted angioma, and propranolol does not treat it. A rapidly enlarging purpuric tumour with a falling platelet count is a different disease needing urgent specialist care. [11] [6]
A fourth pitfall is failing to screen for PHACE and LUMBAR. A large facial segmental haemangioma without a brain and vascular imaging study, or a lumbosacral haemangioma without spinal imaging, is an incomplete assessment. The skin lesion is the marker of a hidden structural anomaly, and the syndrome screen is part of the management of the lesion. [8] [12]
A final pitfall is counselling falsely. Telling a family a haemangioma will vanish completely ignores the fibrofatty residue that often persists, especially on the face. Honest counselling about residual change — and timely propranolol or laser for the lesions that will scar — builds the trust that lets the family navigate the long involution. [11] [2]
Prognosis & Disposition
Most infantile haemangiomas follow a benign course: rapid proliferation to around nine to twelve months, then slow involution over several years, with the bulk of resolution in early childhood. The focal, non-critical lesion leaves little or no residue. The prognosis is shaped by site and size — facial, segmental and ulcerated lesions carry the highest risk of permanent cosmetic residue, which is why early propranolol for these lesions matters. [2] [11]
Disposition follows risk. Treat with propranolol in the proliferative phase the periocular, airway, large, segmental and ulcerated lesions, and screen the segmental and multifocal lesions for their syndromes. Observe the focal non-critical lesion with counselling and review. Admit the infant with airway compromise, high-output cardiac failure or the Kasabach-Merritt phenomenon. The threshold for specialist referral of the segmental, midline and multifocal lesion is deliberately low. [2] [1]
At the end of propranolol therapy, review the residue. Telangiectasia, fibrofatty tissue and scarring may remain, and pulsed-dye laser or surgery can address the cosmetic residue after involution. For the port-wine stain, laser lightens and reduces thickening but the lesion persists for life, so ongoing laser review and Sturge-Weber surveillance where relevant are part of the long-term plan. [6] [4]
Recurrence of a fully involuted haemangioma is uncommon, but the family needs to know that minor re-expansion can occur with intercurrent illness in some children still in the involution phase. The long-term outcome is shaped by how early the problematic lesion was treated, which is why recognition in the proliferative phase is the whole skill. [11] [2]
Special Populations
Premature and low-birth-weight infants carry the highest risk of infantile haemangioma, and their lesions can be larger and more numerous. The threshold to assess and screen is lower, because multifocal and hepatic disease cluster here, and thyroid function needs checking in the infant with large or multifocal lesions. [2] [11]
Infants with large or segmental facial haemangiomas are the PHACE-syndrome population. Their care is multidisciplinary — dermatology, neurology, cardiology, ophthalmology and neuroradiology — because the structural brain, arterial, cardiac and ocular anomalies demand coordinated surveillance. Propranolol initiation under specialist supervision addresses the rare stroke risk of cerebral arterial anomalies. [8] [9]
Infants with lumbosacral or perineal haemangiomas are the LUMBAR-syndrome population. Occult spinal dysraphism, tethered cord, and genitourinary and anorectal anomalies may accompany the skin lesion, and spinal imaging and urology or neurosurgery referral are part of the assessment. [12]
Aboriginal and Torres Strait Islander, Maori and Pasifika children, and families in rural and remote areas need equitable access to propranolol initiation, laser services and specialist multidisciplinary care. Distance should not delay treatment in the proliferative phase, so outreach, telehealth and clear referral pathways are part of delivering the standard of care to every infant regardless of location. [2]
Children with Sturge-Weber syndrome and Klippel-Trenaunay syndrome need lifelong multidisciplinary care — neurology, ophthalmology and developmental surveillance for Sturge-Weber, and vascular, orthopaedic and lymphology services for Klippel-Trenaunay. The skin lesion is the entry point to a multisystem disease, and the general paediatrician often coordinates the team. [4] [6]
Evidence, Guidelines & Regional Differences
The treatment evidence for infantile haemangioma was transformed by the 2008 observation that propranolol halted proliferation and accelerated involution, an accidental finding that became the standard of care within a few years. The Cochrane review of interventions for infantile haemangioma synthesises the trial evidence for propranolol, timolol and other therapies, and the consensus conferences established the dosing and initiation protocols in widespread use. [1] [3]
Topical timolol for early superficial infantile haemangioma
Population: Infants with small superficial infantile haemangiomas in the early proliferative stage, randomised to topical timolol versus vehicle in a controlled trial.
Key finding
Topical timolol significantly reduced lesion volume and colour compared with vehicle in the early proliferative stage, with a favourable safety profile, supporting its use for small superficial lesions.
Practice change
For thin, superficial, non-ulcerated haemangiomas where a topical agent is enough, timolol avoids the systemic exposure of oral propranolol while delivering meaningful reduction.
The classification and syndrome evidence is anchored by the Mulliken and Glowacki classification, refined into the International Society for the Study of Vascular Anomalies framework that organises the field into tumours and malformations. The discovery of the somatic mosaic GNAQ mutation in Sturge-Weber syndrome and port-wine stain united the biology of isolated and syndromic capillary malformation and explained why the lesion follows a developmental territory. [5] [4]
The PHACE-syndrome consensus statements established the diagnostic criteria and the care recommendations — including the brain and neck vascular imaging, echocardiography and ophthalmology that make up the screen for a large facial segmental haemangioma. The neuroradiological review of lumbosacral infantile haemangioma and spinal dysraphism anchors the LUMBAR-syndrome screen. [9] [12]
The regional policy structure is consistent in principle. The AAP clinical practice guideline for the management of infantile haemangiomas sets the propranolol-first approach with syndrome screening. In Australia and New Zealand, the Royal Children's Hospital Melbourne guideline and the major paediatric vascular-anomalies services align with this, with outreach and telehealth for rural families. In the UK, the Royal College of Paediatrics and specialist dermatology services follow the same propranolol and laser pathway. In the US and Canada, the AAP guideline and the specialist vascular-anomalies centres set the standard. In every region the principle is the same: classify by the fork, treat the problematic lesion early with propranolol, screen the segmental and midline lesion, and refer the malformations to a multidisciplinary service. [2] [6]
The controversies are real: the optimal age to stop propranolol, the role of nadolol versus propranolol, the long-term developmental outcome of PHACE syndrome, the best laser regimen for port-wine stain, and equitable access to specialist and laser services across distance. The defence against each is the same: early classification, propranolol in the proliferative phase for the lesions that need it, syndrome screening for the segmental and midline lesion, and multidisciplinary referral for the malformations. [11] [3]
Exam Pearls
- Vascular birthmarks divide into proliferating vascular tumours and structural vascular malformations; the history of onset and growth decides which family — the Mulliken and ISSVA fork. [5]
- Infantile haemangioma is absent at birth, appears in the first weeks, proliferates to around nine months then involutes, and is GLUT1 positive — the marker that separates it from congenital haemangiomas and malformations. [11]
- Risk factors are female sex, prematurity and low birth weight; prevalence is about four to five percent of infants. [2]
- Propranolol is first-line for problematic infantile haemangioma at 2 to 3 mg per kg per day in divided doses, started in the proliferative phase and continued to around twelve months. [1] [7]
- Topical timolol 0.5 percent gel-forming solution is for small, superficial, non-ulcerated lesions. [10]
- A large or segmental facial haemangioma mandates a PHACE syndrome screen (brain and neck MRI and MRA, echocardiography, ophthalmology). [8]
- A midline lumbosacral or perineal haemangioma mandates a screen for occult spinal dysraphism (LUMBAR syndrome). [12]
- Five or more cutaneous haemangiomas warrant liver ultrasound for hepatic haemangiomas (cardiac failure) and a thyroid check (hypothyroidism). [2]
- Port-wine stain is a capillary malformation present at birth, caused by somatic mosaic GNAQ mutation, treated with pulsed-dye laser best begun in infancy. [4]
- A forehead or upper-eyelid port-wine stain raises Sturge-Weber syndrome — leptomeningeal angioma, seizures and glaucoma. [4]
- Kasabach-Merritt phenomenon (consumptive thrombocytopenia) is caused by kaposiform haemangioendothelioma, not infantile haemangioma; treat with sirolimus or vincristine, not propranolol. [11]
- The salmon patch of the newborn eyelid and nape fades in the first years — do not mistake it for a port-wine stain. [6]
References
- [1]Léauté-Labrèze C; Dumas de la Roque E; Hubiche T; Boralevi F; et al Propranolol for severe hemangiomas of infancy. N Engl J Med, 2008.PMID 18550886
- [2]Krowchuk DP; Frieden IJ; Mancini AJ; Darrow DH; et al Clinical Practice Guideline for the Management of Infantile Hemangiomas. Pediatrics, 2019.PMID 30584062
- [3]Novoa M; Baselga E; Beltran S; Giraldo L; et al Interventions for infantile haemangiomas of the skin. Cochrane Database Syst Rev, 2018.PMID 29667726
- [4]Shirley MD; Tang H; Gallione CJ; Baugher JD; et al Sturge-Weber syndrome and port-wine stains caused by somatic mutation in GNAQ. N Engl J Med, 2013.PMID 23656586
- [5]Mulliken JB; Glowacki J Hemangiomas and vascular malformations in infants and children: a classification based on endothelial characteristics. Plast Reconstr Surg, 1982.PMID 7063565
- [6]Wassef M; Blei F; Adams D; Alomari A; et al Vascular Anomalies Classification: Recommendations From the International Society for the Study of Vascular Anomalies. Pediatrics, 2015.PMID 26055853
- [7]Drolet BA; Frommelt PC; Chamlin SL; Haggstrom A; et al Initiation and use of propranolol for infantile hemangioma: report of a consensus conference. Pediatrics, 2013.PMID 23266923
- [8]Garzon MC; Epstein LG; Heyer GL; Frommelt PC; et al PHACE Syndrome: Consensus-Derived Diagnosis and Care Recommendations. J Pediatr, 2016.PMID 27659028
- [9]Metry D; Heyer G; Hess C; Garzon M; et al Consensus Statement on Diagnostic Criteria for PHACE Syndrome. Pediatrics, 2009.PMID 19858157
- [10]Muñoz-Garza FZ; Ríos M; Roé-Crespo E; Bernabeu-Wittel J; et al Efficacy and Safety of Topical Timolol for the Treatment of Infantile Hemangioma in the Early Proliferative Stage: A Randomized Clinical Trial. JAMA Dermatol, 2021.PMID 33825828
- [11]Sebaratnam DF; Rodríguez Bandera AL; Wong LF; Wargon O Infantile hemangioma. Part 2: Management. J Am Acad Dermatol, 2021.PMID 34419523
- [12]Schumacher WE; Drolet BA; Maheshwari M; Horii KA; et al Spinal dysraphism associated with the cutaneous lumbosacral infantile hemangioma: a neuroradiological review. Pediatr Radiol, 2012.PMID 22138893