Paeds · preventive-and-community-paediatrics
Newborn bloodspot screening for inherited metabolic disease and follow-up
Also known as Heel-prick test · Dried blood spot screening · Newborn metabolic screening · Guthrie test · NBS bloodspot · Neonatal bloodspot programme
Fellowship guide to dried bloodspot newborn screening: valid collection, result states, critical positives in well neonates, CH PKU MCADD galactosaemia CAH CF SCID pathways, incomplete cards, family counselling and regional panel differences.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
Overview & Definition
Newborn bloodspot screening is a population programme that tests dried blood from a heel prick to find serious treatable conditions before the baby looks unwell. It sits beside hearing screening, critical congenital heart disease pulse oximetry and the newborn examination. Those other checks do not replace the card. [2] [17]
Screening is not the same as diagnosis. A presumptive positive means the baby needs a defined confirmatory pathway. A clear result lowers risk for the conditions on that panel. It does not prove the child will never develop any metabolic or endocrine disease. Unsatisfactory means the sample failed quality rules. Incomplete means the test was never done, never arrived, or never had an owner after discharge. [1] [14]
Wilson and Jungner principles still frame the programme. There must be an important health problem, a latent stage, a suitable test, an accepted treatment, and a system that can deliver confirmation and care. Modern reviews keep those ideas alive as panels expand and genomic options appear. A laboratory printout without follow-through is not screening. [1] [2]
Closed loop from heel to treatment
Collect a valid card
Correct timing window, warm heel, filled circles, air dry, accurate identifiers, special-situation notes for preterm, TPN or transfusion.
Protect transport and analysis
Prompt despatch. Laboratory methods include MS/MS and targeted assays. Second-tier tests may refine some markers.
Classify the result state
Clear, presumptive positive, borderline, unsatisfactory, incomplete. Each state has a different action and urgency.
Contact and confirm
Critical positives need same-day family contact. Confirmatory bloods and specialist pathways start without waiting for the baby to look sick.
Treat and hand over
Start protocolised treatment when indicated. Educate for illness plans. Document community follow-up and residual risk.
Classification
Think first in result states, then in disease names. [14] [17]
Clear or negative means continue routine care and still listen if the infant later looks metabolic or endocrine unwell. Presumptive positive or screen positive means a defined confirmatory pathway. Some programmes mark a subset as critical, needing same-day phone contact. Borderline means timed repeat or second-tier testing per protocol. Unsatisfactory means the card failed quality and is not a free pass. Incomplete or not collected means system failure until a valid sample is documented. Carrier-informative results, where reported, need careful genetic counselling language rather than disease labelling. [1] [14] [17]
Condition groups on many panels include endocrine disease such as congenital hypothyroidism (CH) and congenital adrenal hyperplasia (CAH); amino acidopathies such as phenylketonuria (PKU); fatty-acid oxidation disorders such as medium-chain acyl-CoA dehydrogenase deficiency (MCADD); organic acidurias and urea-cycle disorders where included; classic galactosaemia; biotinidase deficiency; cystic fibrosis (CF); haemoglobinopathies; and T-cell receptor excision circle (TREC) screening for severe combined immunodeficiency (SCID) where the programme includes it. Exact lists differ by country and by Australian state or territory. Never recite one jurisdiction’s list as universal. [2] [18]
Tandem mass spectrometry (MS/MS) expanded panels by measuring many amino acids and acylcarnitines from one spot. Targeted assays still underpin TSH, 17-hydroxyprogesterone, immunoreactive trypsinogen (IRT), galactose metabolites, TREC and other markers. Second-tier testing uses a more specific assay on the same card to cut false positives for selected conditions without abandoning the primary screen. [13] [14]
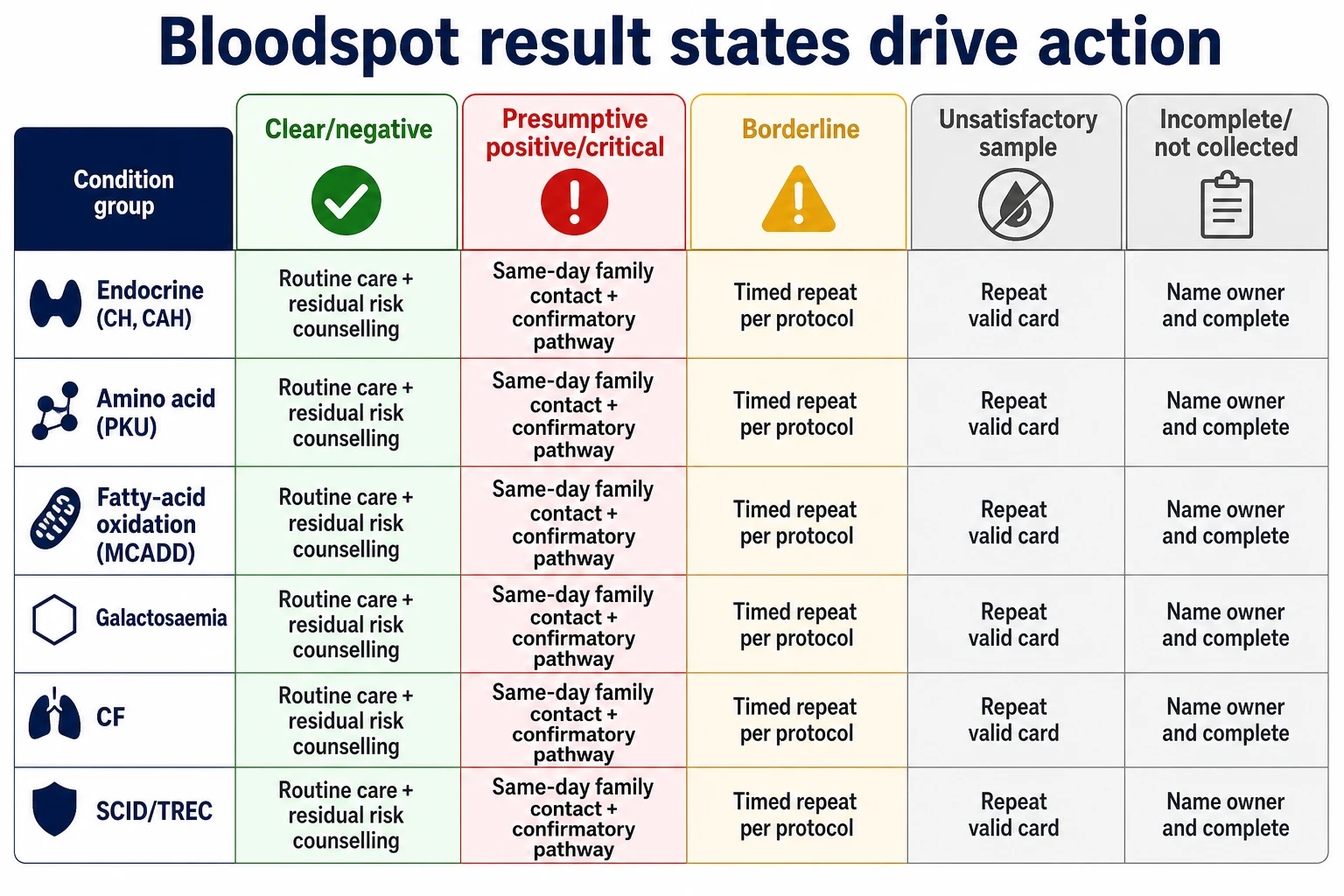
Read the figure like this: the exam trap is treating “probably done” as done, or treating “screen positive” as a final diagnosis in front of parents. [1] [2]
Clear
Screen negative on panel
- Continue routine newborn care
- Still investigate if clinical concern rises
- Does not exclude all future disease
- Document that the card was valid
Presumptive positive
Needs confirmation
- Same-day contact if critical
- Confirmatory labs and specialist path
- Well appearance does not delay action
- Use screen-not-diagnosis language
Borderline
Protocol-driven grey zone
- Timed repeat or second-tier test
- Do not invent your own cut-off
- Safety-net feeding and unwell advice
- Track until resolved
Unsatisfactory / incomplete
System problem
- Repeat valid collection
- Name an owner and date
- Early discharge is high risk
- Refusal needs documented residual risk
Epidemiology & Risk Factors
Bloodspot programmes exist because a few conditions are rare individually but costly when missed. Untreated CH risks preventable neurodevelopmental injury. Untreated PKU risks irreversible cognitive impairment. Untreated MCADD risks hypoketotic hypoglycaemia, encephalopathy and sudden death during fasting or infection. Classic galactosaemia can present with liver failure and E. coli sepsis after milk exposure. Salt-losing CAH can collapse in the second week of life. Missed CF delays airway and nutrition care. Missed SCID delays infection precautions and definitive immunology pathways. [3] [5] [7] [9] [11] [15]
Missed or delayed cards cluster where systems are fragile. Early discharge, home birth, interhospital transfer, weekend staffing, incomplete identifiers, wrong address and rural distance all raise the chance that a sample is never collected, never analysed, or never acted on. [17]
Infant factors change validity. Prematurity, total parenteral nutrition (TPN), transfusion, critical illness and steroids can distort markers or force delayed and repeated sampling. Maternal thyroid disease and antithyroid drugs change how you think about borderline newborn TSH. Family history, consanguinity and ethnicity can raise pre-test probability, but universal programmes still aim to catch first cases in families without a known story. [3] [13] [20]
Equity gaps matter. Indigenous, migrant, refugee, out-of-home-care and remote families face higher risk that follow-up calls fail. False-positive notifications create parental anxiety and extra tests. That harm is real and must be counselled, not dismissed as “just a screen.” [1] [12] [17]
Pathophysiology
Many screened conditions are silent at birth because the fetus was supported by placental function or residual maternal hormone. After birth, the infant’s own pathway must work. Screening buys time only if confirmation and treatment follow. [2] [19]
Thyroid hormone is essential for brain development. In primary CH, low thyroxine with high TSH appears after the maternal contribution falls. The baby may still feed and gain weight while the brain pays the price of delay. That is why a thriving neonate with a critical TSH result is a systems emergency. [3] [4] [19]
In PKU, deficient phenylalanine hydroxylase activity allows phenylalanine to accumulate. High levels injure the developing brain. Early dietary treatment transforms the historical natural history. [5] [6]
In MCADD, the infant cannot efficiently oxidise medium-chain fatty acids during fasting or intercurrent illness. Energy failure and hypoketotic hypoglycaemia can appear suddenly in a previously well child. Screening aims to identify the infant before the first dangerous fast. [7] [8]
Classic galactosaemia impairs galactose-1-phosphate uridylyltransferase activity. Once milk feeds start, galactose-1-phosphate and related metabolites accumulate. Liver injury, coagulopathy, cataracts and gram-negative sepsis can follow. Continuing milk while classic disease is likely is unsafe. [9] [10]
CAH from 21-hydroxylase deficiency elevates 17-hydroxyprogesterone. Salt-losing infants can present with hyponatraemia, hyperkalaemia and shock after the first days of life. Prematurity raises false-positive 17-OHP rates, which is why second-tier steroid profiling and gestational-age-aware cut-offs matter where used. [13] [14]
CF screening commonly uses IRT, sometimes with DNA panels. Elevated IRT is not CF. It is a ticket to a defined algorithm that may include mutation analysis and sweat testing. SCID screening uses TREC as a marker of T-cell production. Low TREC triggers immunology pathways before opportunistic infection declares. [11] [15] [16]
MS/MS detects characteristic amino-acid and acylcarnitine signatures of many intermediary metabolism defects from a single dried spot. Abnormal profiles need disciplined interpretation with clinical context, not pattern-matching from memory alone. [13] [14]
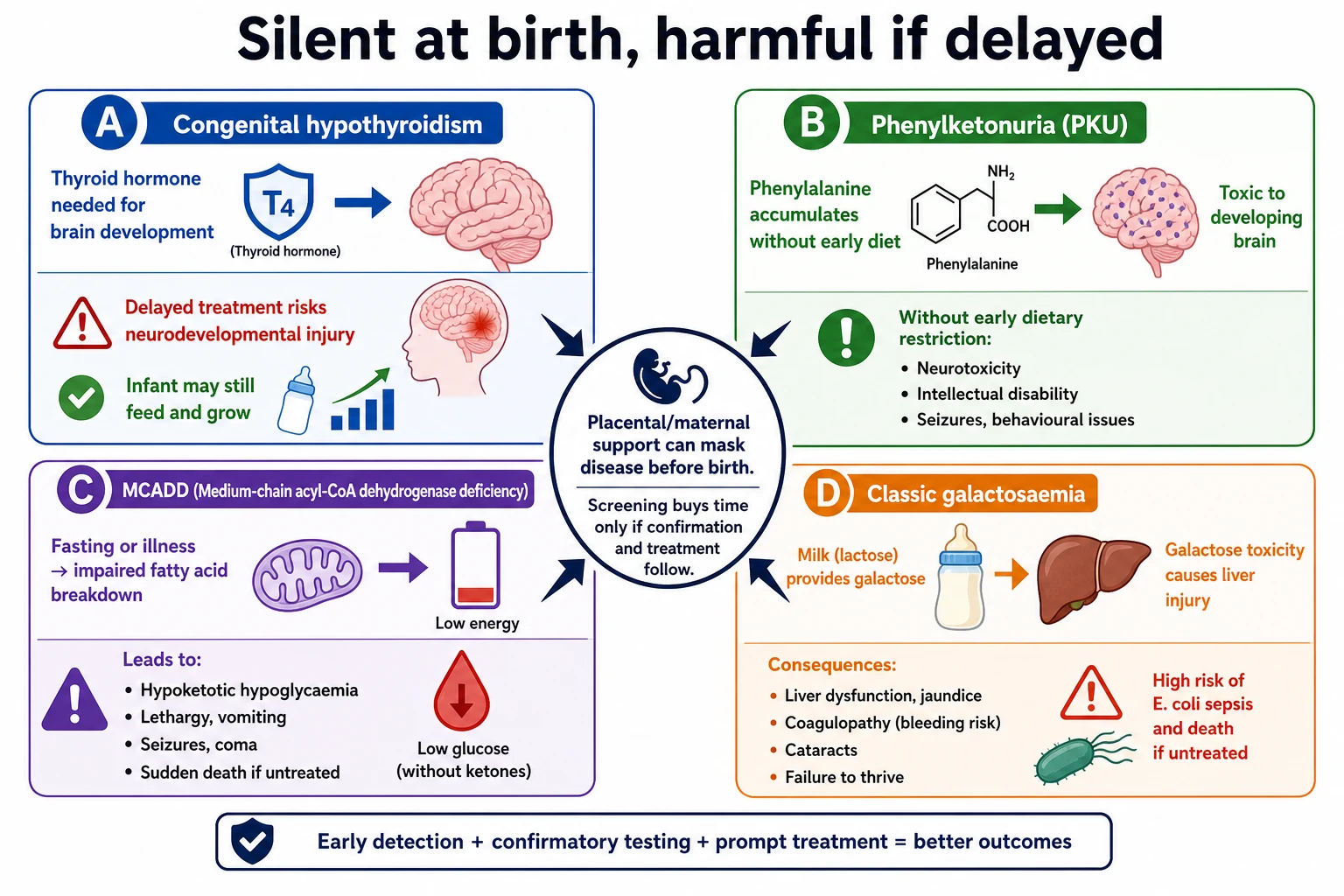
Clinical Presentation
Most screen-positive newborns look well. That is the design of the programme. Train yourself to fear the calm baby with a critical laboratory call more than the slightly unsettled baby with a complete clear package. [2] [19]
If CH is missed, later infancy may show prolonged jaundice, large fontanelle, macroglossia, hypotonia, constipation, poor growth and developmental delay. If PKU is missed, developmental impairment accumulates without dramatic neonatal collapse. MCADD crisis looks like lethargy, vomiting, encephalopathy, seizures or sudden death risk during illness or prolonged fasting. Classic galactosaemia after milk may show poor feeding, jaundice, hepatomegaly, coagulopathy, E. coli sepsis and cataracts. Salt-losing CAH may show poor feeding, vomiting, dehydration, virilisation in some infants, and shock with electrolyte crisis. Missed CF may present with meconium ileus, failure to thrive, steatorrhoea or recurrent respiratory symptoms. Missed SCID presents with severe or opportunistic infection, chronic diarrhoea or failure to thrive. [5] [7] [9] [11] [15] [19]
False-positive notifications present as family distress: sleepless parents, bonding disruption and repeated questions about whether the baby is “really sick.” Incomplete cards present as a system problem: early discharge, lost paperwork, or “we thought the hospital did it.” [1] [17]
Differential Diagnosis
Elevated TSH may reflect true primary CH, delayed TSH rise in preterm infants, iodine issues, maternal factors or laboratory artefact. Confirm with free T4 and TSH on a quality venous or protocol sample and act on the clinical laboratory pathway. [3] [20]
Elevated phenylalanine may reflect classic PKU, milder hyperphenylalaninaemia or, less often, disorders of biopterin metabolism that need different treatment. Do not assume every high phenylalanine is simple PKU. [5] [6]
Abnormal acylcarnitine profiles may reflect MCADD, other fatty-acid oxidation disorders, secondary carnitine changes or sample factors. Specialist interpretation and confirmatory testing matter. [7] [13]
Elevated 17-OHP may reflect classic CAH, prematurity-related false positives or illness. Second-tier testing helps where available, but a sick infant with salt-losing features is a clinical emergency regardless of the card. [14]
Elevated IRT may reflect CF, heterozygote states, prematurity or perinatal stress. Low TREC may reflect SCID, other T-cell lymphopenias, preterm effects or sample failure. Galactosaemia markers need distinction between classic disease, variants and timing issues. [9] [11] [15]
When a neonate is acidotic, hypoglycaemic or encephalopathic and the card is pending or missing, keep sepsis, cardiac disease and inborn errors on the same list. Do not wait for the screening laboratory to finish before resuscitating. [7] [14]
Clinical & Bedside Assessment
Start with the history that changes bloodspot care: gestation, day of life, milk or TPN feeds, transfusions, maternal thyroid disease, family metabolic or CF history, consanguinity, early discharge plan, working phone number and who holds parental responsibility. [3] [17]
Examine a recalled well neonate carefully. Check alertness, hydration, jaundice pattern, hepatomegaly, ambiguous genitalia, virilisation, dysmorphology, tone and feeding effectiveness. A normal examination never cancels a critical result. [4] [19]
Assess parental understanding without jargon. Ask them to teach back what the screen means, what happens today, and when to return if the baby feeds poorly, vomits, becomes sleepy, or looks different. [1] [17]
If the infant is already unwell, use a structured ABCD assessment first. Bloodspot status is part of the problem representation, not a reason to skip glucose, gas, perfusion and sepsis care. [7] [14]
Investigations
Valid collection needs a warm heel, correct fill of circles without layering or squeezing that damages quality, air drying, and identifiers that match the right infant. Sampling too early can miss some conditions. Sampling after transfusion can mask others. TPN and prematurity need programme-specific schedules, often with repeat cards. Follow local hour windows rather than inventing a universal number. [2] [17]
Confirmatory testing is condition-specific. CH pathways use free T4 and TSH. PKU pathways use quantitative amino acids and specialist input for biopterin disorders when indicated. MCADD pathways use acylcarnitine profiles, urine organic acids and genetic or enzymatic confirmation per service. Galactosaemia pathways need urgent enzyme or metabolite confirmation while feeds are made safe. CAH pathways need electrolytes and endocrine-directed steroid work-up. CF pathways follow IRT/DNA algorithms to sweat testing and CF centre care. SCID pathways need flow cytometry and immunology review after abnormal TREC. [3] [6] [7] [10] [11] [16]
In a decompensating infant, do not wait for perfect metabolic panels before treating hypoglycaemia and shock. Useful emergency labs often include glucose, blood gas, ammonia, lactate, electrolytes, liver enzymes, coagulation and cultures, guided by the presentation and specialist advice. [7] [14]
A clear screen does not cancel investigation when the clinical picture says inborn error, CH or CF. Screens have false negatives. Clinical judgement still leads. [2] [13]
Management — Resuscitation
Treat the collapsed or encephalopathic neonate with ABCDE care first. Support airway and breathing. Check and correct glucose. Obtain access. Stop milk feeds when classic galactosaemia or a feed-triggered metabolic crisis is plausible, and use intravenous dextrose according to neonatal/metabolic guidance while you call for senior and specialist help. Avoid prolonged fasting in suspected fatty-acid oxidation disorders. [7] [10] [14]
Suspected salt-losing CAH crisis needs senior review, fluid resuscitation, electrolyte management and urgent endocrine involvement with stress-dose steroid pathways as per local protocol. Suspected SCID with infection needs infection control precautions and early immunology input, not repeated live vaccines or casual community observation. [15] [16]
Rural services should prepare retrieval early when confirmatory testing or intensive care is not available on site. Do not observe overnight without a destination after a critical pathway is opened. [17]
Management — Definitive & Stepwise
Close the loop for every card
Document collection time, quality, despatch and result state. Incomplete and unsatisfactory cards get a named clinician, a date and a family communication path. [17]
Critical positive in a well neonate
Phone the family the same day. Explain that screening is not diagnosis. Bring the infant for confirmatory tests. Start treatment when the protocol and specialist pathway say treatment should not wait for every confirmatory nuance. Arrange transport help if the family cannot return. [2] [14]
Congenital hypothyroidism
Urgent free T4/TSH confirmation. Early levothyroxine under current national or consensus dosing protocols, with endocrine follow-up. Do not invent microgram-per-kilogram figures in an exam if you cannot source the protocol you use; state that you would use the current guideline-backed dosing pathway and monitor free T4/TSH. Neurodevelopment is the reason for speed. [3] [4] [19]
PKU
Urgent metabolic and dietetic referral. Phenylalanine-restricted diet with tyrosine and micronutrient support as directed by the metabolic service. European guidelines emphasise early, sustained control and structured follow-up. [5] [6]
MCADD
Confirm on pathway. Educate families to avoid prolonged fasting, use age-appropriate feeding intervals, and follow an emergency regimen during illness with early presentation if intake fails. Screening improves outcomes when education is real, not a leaflet alone. [7] [8]
Classic galactosaemia
Treat as a newborn screening emergency when classic disease is likely. Stop lactose/galactose-containing feeds promptly and use a suitable non-galactose formula while confirming the diagnosis. Manage sepsis and liver failure supportively with neonatal intensive care as needed. [9] [10]
CAH, CF and SCID
CAH: electrolytes, endocrine review, glucocorticoid and mineralocorticoid replacement as indicated, and crisis education. CF: complete the IRT/DNA/sweat algorithm and refer early to a CF centre. SCID: abnormal TREC means immunology referral, infection precautions, and avoidance of live vaccines until cleared. [11] [15] [16]
Refusal or incomplete screen
Explore reasons, correct specific myths, offer lawful process, document residual risk, and ensure the infant is not lost. Safeguarding escalation is for clear risk of significant harm with avoidance of critical care, not for every values-based refusal. [1] [17]
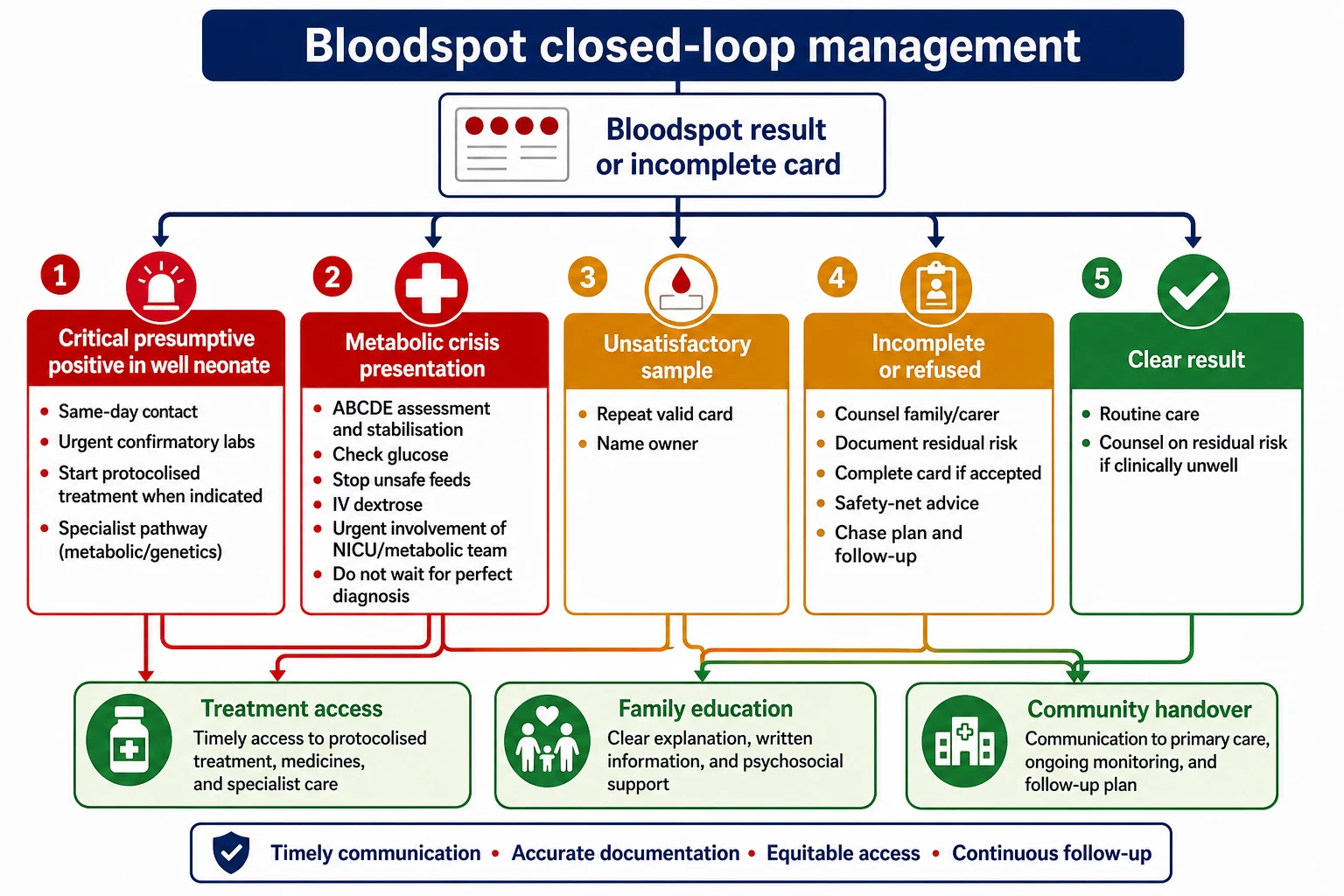
Specific Subtypes & Scenarios
Day 5–10 thriving neonate, critical TSH. Same-day contact, confirmatory free T4/TSH, start levothyroxine on protocol, endocrine follow-up. Do not wait for developmental signs. [3] [19]
Preterm infant with high 17-OHP. Use gestational-age-aware interpretation and second-tier testing where available. Still treat clinical salt-losing features as emergency. [14]
Possible classic galactosaemia on milk feeds. Stop galactose-containing feeds now while you confirm. Do not wait for the perfect enzyme report if the infant is unwell or the screen is highly concerning. [10]
MCADD positive before first illness. Confirm, educate, written emergency regimen, ensure carers understand fasting limits. [7] [8]
Meconium ileus or early CF signs. Clinical CF pathways run in parallel with screening algorithms; do not delay care because the IRT result is pending. [11]
Abnormal TREC in a well outpatient. Urgent immunology pathway and infection precautions; do not give live vaccines until cleared. [15] [16]
Home birth or rural early discharge. Treat first clinical contact as day zero for the package. Book collection with a realistic transport plan. Critical positives need destination planning, not hope. [17]
Out-of-home care. Clarify who can consent and who receives results. Document legal caregivers. [17]
Complications & Pitfalls
False positives cause anxiety, extra venepuncture and possible overtreatment. Mitigate with clear “screen, not diagnosis” language and timely resolution. False negatives create false confidence. Clinical disease can still appear after a clear card. [1] [2] [12]
Early sampling, poor card quality, heat damage, contamination, mislabelling, transfusion and TPN create programme errors. Delaying CH treatment because the baby looks fine is a classic harm. Continuing milk in likely classic galactosaemia is a classic harm. Incomplete MCADD illness plans leave families unprotected at the first viral illness. Treating another country’s panel as local truth fails viva and clinical care. [3] [7] [10] [18]
Prognosis & Disposition
Safe disposition is not “feeding well.” It is “card complete or card owned, critical results actioned.” A well infant with a critical positive does not go home without a same-day plan. A well infant with a non-critical pending result may go home only if contact details work and the family understands how results return. [2] [17]
Early-treated CH and PKU have far better neurodevelopmental outlooks than historical late presentations. MCADD outcomes improve when crises are prevented. CF and SCID benefit from earlier organised care. Missed screens still carry death and disability risk. [5] [8] [11] [19]
Handover language should fit one sentence: collection status, result state, open actions, appointments and family contacts. [17]
Special Populations
Preterm and NICU infants need modified timing, sometimes repeated cards, and careful interpretation of TSH, 17-OHP, IRT and TREC. Indigenous, rural and remote families need culturally safe communication and geography-real return plans. Migrant and refugee families need interpreters and verified contact paths. Out-of-home-care infants need clarity on consent and result recipients. Infants of mothers with thyroid disease need adapted CH thinking. Technology-dependent newborns still need the card completed. [3] [15] [17] [20]
Evidence, Guidelines & Regional Differences
Andermann’s review keeps Wilson and Jungner usable as panels expand. Wilcken’s overview frames newborn screening as a public-health system. CH consensus guidance from Léger and the later van Trotsenburg update set modern diagnosis and treatment expectations, reinforced by contemporary clinical reviews. European PKU guidelines and disease primers define diagnosis and dietary care standards. MCADD screening literature links early identification to prevention of metabolic decompensation. Galactosaemia reviews mark classic disease as a time-critical feed-management problem. CF and SCID screening reviews support organised algorithms and early specialist care. MS/MS and laboratory follow-up guidelines structure expanded panel interpretation. Australian policy frameworks and ANZ panel-counting work remind clinicians that local lists differ and must be checked. [1] [2] [3] [4] [6] [8] [10] [11] [14] [15] [17] [18]
Genomic sequencing as primary newborn screening remains controversial. Benefits, consent, variants of uncertain significance and equity are unsettled. Mild variants, carrier reporting and residual risk after a clear screen also need careful programme design. [1] [12]
Australia and Aotearoa New Zealand run state, territory or national bloodspot panels that are not identical. Collection timing and condition lists must be read from the local programme. Culturally safe follow-up for Aboriginal, Torres Strait Islander, Māori and Pacific families includes realistic geography and whānau participation. [17] [18]
United Kingdom uses the NHS newborn blood spot programme with a defined core condition set and national handbooks for operational detail. Quote the current handbook rather than memory. [2]
United States uses state-based panels often aligned to a recommended uniform screening panel concept, with ACT sheets and short-term follow-up structures after positives. Condition lists still differ by state. [14] [16]
Canada uses provincial programmes with panel and logistics variation; remote geography often dominates follow-up design. [2]
State only differences you have checked against current official programme pages. Do not invent a universal analyte list or hour cut-off. [1] [18]
Exam Pearls
- Screen positive ≠ diagnosis. Clear ≠ lifelong metabolic immunity. [1]
- CH urgency is the brain, not how settled the neonate looks. [3] [19]
- Stop galactose-containing feeds when classic galactosaemia is a live possibility. [10]
- MCADD education is fasting safety plus an illness emergency regimen. [7]
- Unsatisfactory cards are incomplete screens until repeated. [17]
- Panel lists are jurisdiction-specific. [18]
- False positives are programme harms you must counsel through. [1]
- Clinical concern overrides a clear screen. [2]
- Rural critical positives need destination planning. [17]
- Boards test systems thinking more than memorised cut-offs. [1] [14]
SPOT CARE loop
References
- [1]Andermann A Revisiting Wilson and Jungner in the genomic age: a review of screening criteria over the past 40 years. Bulletin of the World Health Organization, 2008.PMID 18438522
- [2]Wilcken B Newborn screening. Pathology, 2008.PMID 18203033
- [3]van Trotsenburg P Congenital Hypothyroidism: A 2020-2021 Consensus Guidelines Update-An ENDO-European Reference Network Initiative Endorsed by the European Society for Pediatric Endocrinology and the European Society for Endocrinology. Thyroid, 2021.PMID 33272083
- [4]Léger J European Society for Paediatric Endocrinology consensus guidelines on screening, diagnosis, and management of congenital hypothyroidism. The Journal of clinical endocrinology and metabolism, 2014.PMID 24446653
- [5]van Spronsen FJ Phenylketonuria. Nature reviews. Disease primers, 2021.PMID 34017006
- [6]van Wegberg AMJ The complete European guidelines on phenylketonuria: diagnosis and treatment. Orphanet journal of rare diseases, 2017.PMID 29025426
- [7]Mason E Medium-chain Acyl-COA dehydrogenase deficiency: Pathogenesis, diagnosis, and treatment. Endocrinology, diabetes & metabolism, 2023.PMID 36300606
- [8]Dezateux C Newborn screening for medium chain acyl-CoA dehydrogenase deficiency: evaluating the effects on outcome. European journal of pediatrics, 2003.PMID 14628139
- [9]Succoio M Galactosemia: Biochemistry, Molecular Genetics, Newborn Screening, and Treatment. Biomolecules, 2022.PMID 35883524
- [10]Berry GT Galactosemia: when is it a newborn screening emergency? Molecular genetics and metabolism, 2012.PMID 22483615
- [11]Shenoy A Cystic Fibrosis Newborn Screening. Clinics in perinatology, 2025.PMID 40850711
- [12]Course CW Newborn screening for cystic fibrosis: Is there benefit for everyone? Paediatric respiratory reviews, 2019.PMID 30956155
- [13]Fernández-Lainez C Tandem mass spectrometry newborn screening for inborn errors of intermediary metabolism: abnormal profile interpretation. Current medicinal chemistry, 2012.PMID 22934775
- [14]Dietzen DJ National academy of clinical biochemistry laboratory medicine practice guidelines: follow-up testing for metabolic disease identified by expanded newborn screening using tandem mass spectrometry; executive summary. Clinical chemistry, 2009.PMID 19574465
- [15]van der Spek J TREC Based Newborn Screening for Severe Combined Immunodeficiency Disease: A Systematic Review. Journal of clinical immunology, 2015.PMID 25893636
- [16]Routes J Newborn Screening for Severe Combined Immunodeficiency. Current allergy and asthma reports, 2018.PMID 29749587
- [17]O'Leary P Newborn bloodspot screening policy framework for Australia. Australasian medical journal, 2015.PMID 26464586
- [18]Heather N Counting Conditions on Newborn Bloodspot Screening Panels in Australia and New Zealand. International journal of neonatal screening, 2024.PMID 39051403
- [19]Cavarzere P Primary congenital hypothyroidism: a clinical review. Frontiers in endocrinology, 2025.PMID 40862110
- [20]Grob F Newborn screening for primary congenital hypothyroidism: past, present and future. European thyroid journal, 2025.PMID 40029014