Paeds · respiratory-sleep-and-airway
Primary ciliary dyskinesia
Also known as PCD · Immotile cilia syndrome · Kartagener syndrome · Ciliary dyskinesia · Motile ciliopathy
Fellowship guide to primary ciliary dyskinesia: why a genetic defect of motile cilia produces a lifelong triad of chronic wet cough, chronic rhinosinusitis and otitis media, how the same immotile cilia randomise left-right asymmetry to give situs inversus and Kartagener syndrome, the neonatal respiratory distress that is the earliest clue, the diagnostic pathway of nasal nitric oxide, high-speed video microscopy, electron microscopy and genetics, and the multidisciplinary airway-clearance-centred management that preserves lung function.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
Overview & Definition
Primary ciliary dyskinesia is an inherited disorder in which the tiny motile cilia that line the airways, the sinuses and the middle ear fail to beat normally, so mucus and trapped organisms are never cleared. It is usually autosomal recessive, and the failure of clearance produces a lifelong, progressive suppurative airway disease that begins in the newborn period rather than a disease that arrives later in childhood. The examiner wants the candidate to see it as a single genetic fault with consequences in every ciliated organ. [3] [6]
The reason the diagnosis matters is that it is common enough to meet in practice, it is chronically damaging, and it is repeatedly missed for years because each infection looks ordinary on its own. A child with PCD is often labelled with recurrent bronchitis, asthma or glue ear for a decade before the pattern is recognised, and every year of missed clearance costs lung function that does not come back. Recognising the phenotype early is the single most valuable thing a clinician does in this topic. [1] [4]
Classification
The most useful way to hold the disease in mind is by the underlying ciliary defect, because the defect determines which diagnostic test will detect it. Most cases are caused by absence or shortening of the outer dynein arms, the motor structures that power the ciliary beat, and these are readily seen on electron microscopy. Combined outer and inner dynein arm defects and inner dynein arm defects with microtubular disorganisation form the next groups, while central apparatus and radial spoke defects give subtle beating changes and can have normal-looking ultrastructure that only genetics or immunofluorescence will reveal. [3] [6]
The second axis is the position of the internal organs, and this is what makes PCD memorable at the bedside. Because the same motile cilia set up left-right asymmetry in the embryo, their failure leaves laterality to chance, so about half of children have normal situs and about half have situs inversus totalis. A smaller group has situs ambiguus or heterotaxy, which is important because these children carry a real risk of complex congenital heart disease and asplenia rather than a harmless mirror-image anatomy. [7] [4]
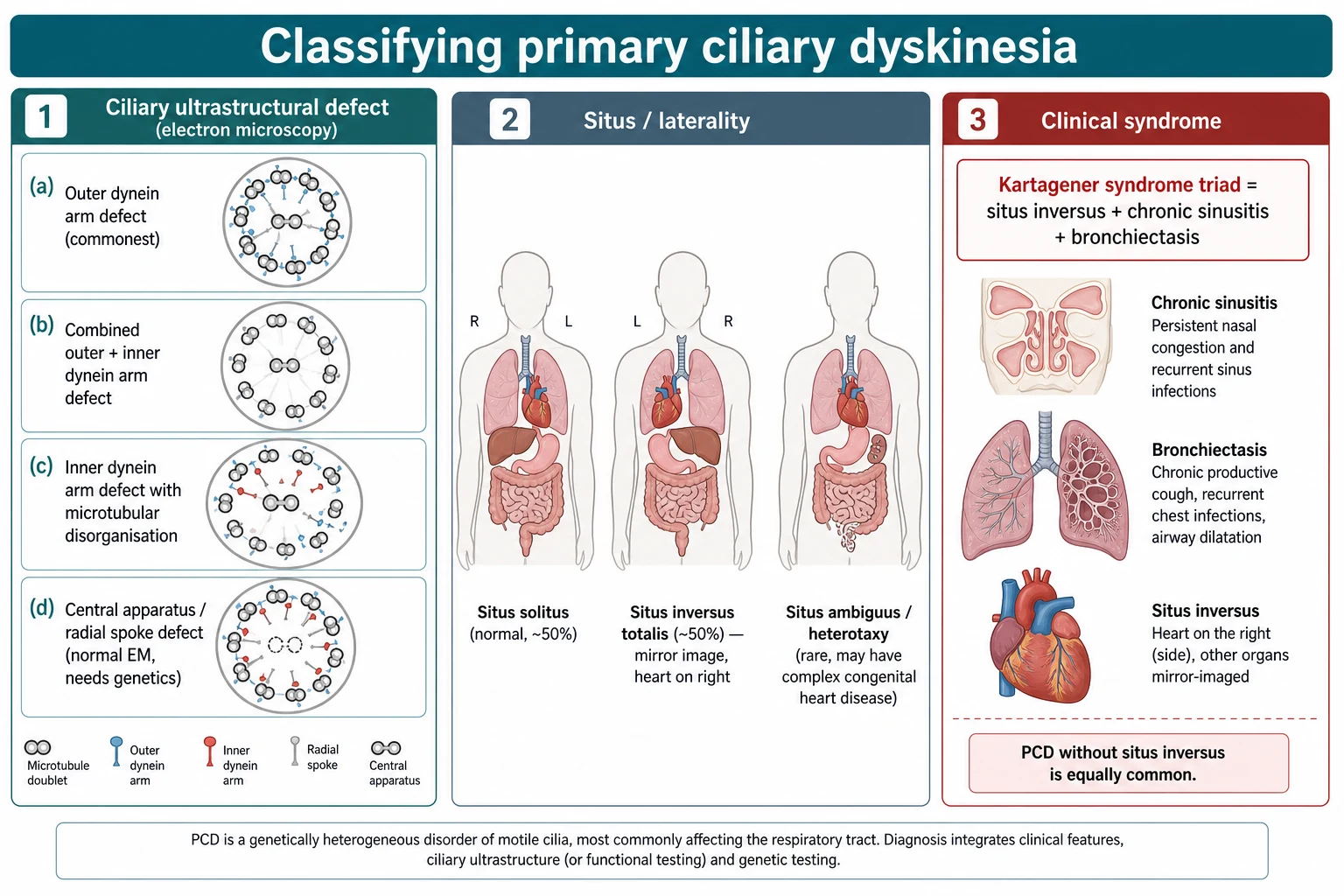
The clinical eponym completes the picture, because examiners still ask for it. Kartagener syndrome is the historical triad of situs inversus, chronic sinusitis and bronchiectasis, and it describes the roughly half of children with PCD who happen to have situs inversus. The key teaching point is that Kartagener syndrome is a subset of PCD, not a separate disease, and that a child with normal situs and identical airway disease has exactly the same condition and prognosis. [4] [3]
Epidemiology & Risk Factors
Primary ciliary dyskinesia is uncommon but not rare, with prevalence estimates usually quoted at around one in ten to twenty thousand people, and it is almost certainly under-diagnosed because milder phenotypes never reach a specialist. It occurs across all populations, and the numbers place it as a leading non-cystic-fibrosis cause of childhood bronchiectasis, which is the context an examiner expects the candidate to supply. [3] [6]
The dominant risk factor is genetic, because the disease is inherited in an autosomal recessive pattern and now involves defects in more than fifty genes that encode the proteins of the ciliary axoneme and its assembly. Consanguinity therefore raises the risk substantially, and a family history of a similarly affected sibling, of unexplained bronchiectasis, or of situs inversus should prompt testing. This genetic heterogeneity is also why a single test cannot diagnose every case and why gene panels have transformed the field. [6] [4]
Certain groups deserve a lower threshold for suspicion, and naming them shows clinical maturity. Children of consanguineous parents, those with an affected relative, and those with an otherwise unexplained laterality defect are all higher risk, and in the Australian and New Zealand setting Aboriginal and Torres Strait Islander and Maori and Pacific children carry a high burden of chronic suppurative lung disease and bronchiectasis in which PCD is an important and under-tested contributor. Access to specialised diagnostic testing is itself a risk factor for late diagnosis in remote communities. [1] [4]
Pathophysiology
The mechanism begins with the structure of a motile cilium, and holding this picture makes the whole disease predictable. A motile cilium has a nine-plus-two arrangement of microtubules, with nine outer doublets surrounding a central pair, and the outer and inner dynein arms are the molecular motors that make adjacent doublets slide and the cilium bend. When a genetic fault removes or disables these motors, the cilium cannot generate a normal coordinated beat, so the escalator that normally sweeps mucus upward stalls. [3] [6]
Once clearance fails, the rest of the disease follows as a vicious cycle. Stagnant mucus traps inhaled bacteria, which establish chronic infection with organisms such as Haemophilus influenzae, Staphylococcus aureus and, increasingly with age, Pseudomonas aeruginosa. The resulting neutrophilic inflammation damages the airway wall, which impairs clearance further and dilates the airway, so recurrent infection and inflammation drive the progression from a persistently wet cough in the toddler to established bronchiectasis in later childhood. [8] [3]
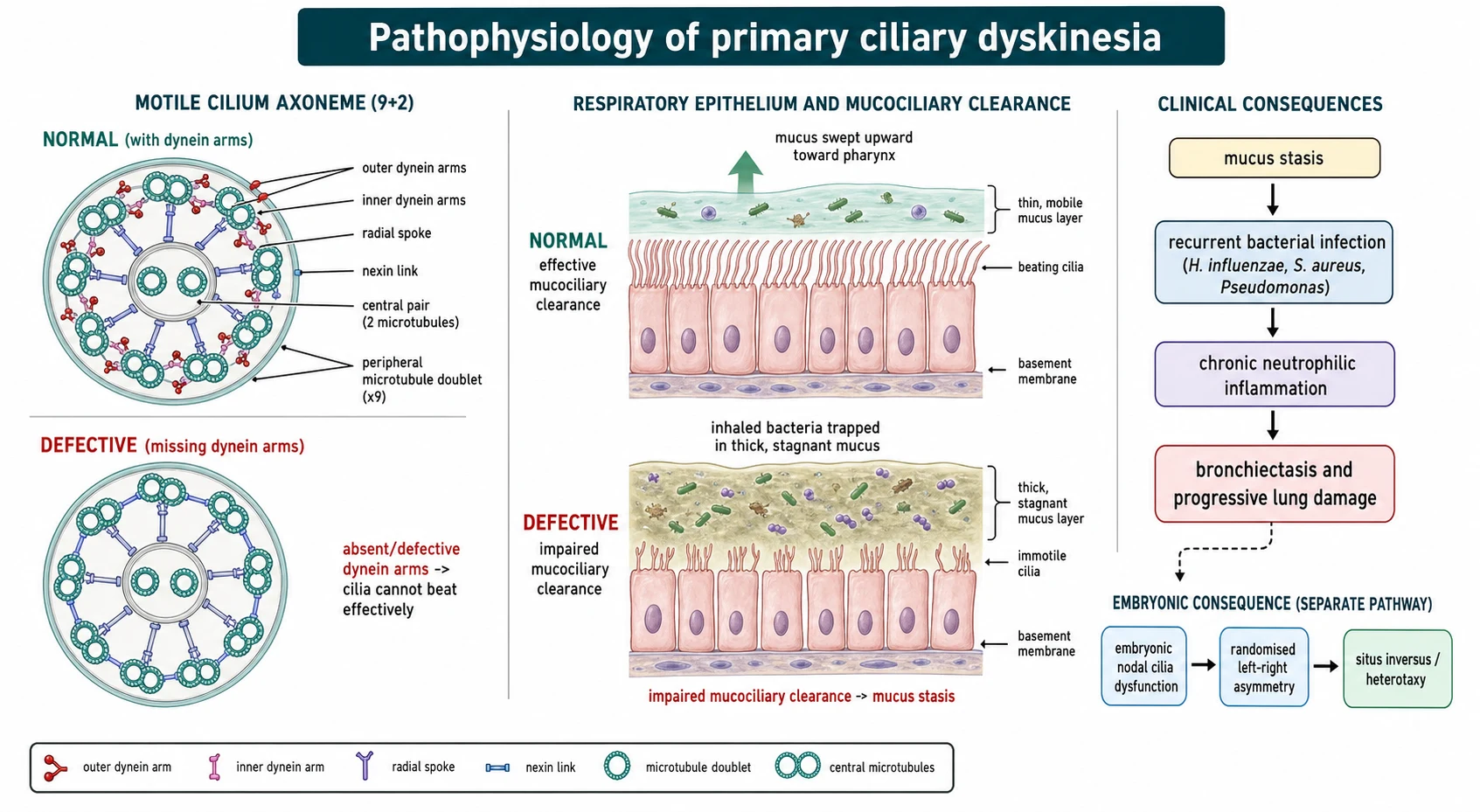
The laterality defect is the elegant second consequence and a favourite examiner probe. During early embryogenesis, motile cilia at the primitive node generate a leftward fluid flow that sets normal left-right asymmetry, and when these cilia are immotile the flow is absent and organ position is left to chance. That is why about half of children with PCD have situs inversus and a minority have heterotaxy with its associated cardiac risk, and why the same disease that fills the lungs with mucus also flips the heart to the right. [7] [4]
Clinical Presentation
The history is where the diagnosis is made, and its hallmark is chronicity from the very beginning of life. The majority of affected term infants have unexplained neonatal respiratory distress, and beyond the newborn period the defining symptom is a daily wet or productive cough that has been present since the first weeks of life rather than one that comes and goes with viral seasons. This persistence, and the early onset, are what separate PCD from the ordinary run of childhood chest infections. [4] [8]
The upper airway is involved just as consistently as the lower airway, and the pattern is telling. Children have chronic year-round nasal congestion and rhinorrhoea often from the neonatal period, chronic rhinosinusitis, and persistent or recurrent otitis media with effusion that produces conductive hearing loss and often chronic ear discharge after grommet insertion. A child with the combination of a lifelong wet cough, a constantly runny nose and glue ear that never settles is displaying the full ciliary phenotype. [4] [8]
On examination the aim is to find the signs of chronic suppuration and to look actively for laterality. There may be digital clubbing, chest wall changes, coarse crackles over bronchiectatic areas, nasal polyps or a persistently discharging nose, and dull, immobile eardrums or grommets with discharge. A displaced apex beat or dextrocardia is a gift, because finding situs inversus in a child with chronic sinopulmonary disease points almost directly at the diagnosis. [8] [4]
Differential Diagnosis
The first and most important comparison is with cystic fibrosis, because both cause early-onset wet cough and bronchiectasis and the two are the leading genetic suppurative lung diseases of childhood. Cystic fibrosis is distinguished by an abnormal sweat chloride, pancreatic insufficiency with faltering growth and steatorrhoea, and its own genetics, whereas PCD is suggested by neonatal distress in a term baby, situs inversus and very low nasal nitric oxide with a normal sweat test. Both should be actively excluded in a child with unexplained bronchiectasis. [2] [3]
Beyond cystic fibrosis, the differential is the rest of the chronic wet cough and bronchiectasis workup. Protracted bacterial bronchitis, immunodeficiency such as antibody deficiency, recurrent aspiration, a retained foreign body causing localised disease, and post-infective bronchiectasis all belong on the list, and the discriminator is again the pattern. PCD is the diagnosis when the disease is diffuse, lifelong, and accompanied by the upper-airway and laterality features rather than being localised or acquired. [1] [8]
Clinical & Bedside Assessment
Bedside assessment is a targeted history that hunts for the pattern rather than the single symptom. The candidate should ask specifically about neonatal respiratory distress in a term infant, the age at which the cough began and whether it is wet and daily, year-round nasal symptoms from birth, recurrent ear disease and hearing concerns, and a family history of consanguinity, bronchiectasis or situs inversus. A validated clinical index such as the PICADAR tool built from these predictive features helps decide who to refer for definitive testing. [1] [4]
The examination then documents the burden of disease and searches deliberately for laterality. Growth is plotted, the chest is examined for clubbing, deformity and crackles, the nose and ears are inspected for polyps, discharge and effusions, and the position of the apex beat and liver edge is checked because dextrocardia transforms the probability of the diagnosis. Hearing should be assessed formally because conductive loss is common and educationally important. [8] [4]
Investigations
There is no single perfect test, so the diagnosis rests on a combination performed in an experienced centre, and stating this explicitly is expected. Nasal nitric oxide is the practical screening test in cooperative children, because levels are characteristically very low in PCD, and a low result in a compatible clinical picture strongly supports the diagnosis while a normal result makes it much less likely. It is a screen rather than a stand-alone confirmatory test, and cystic fibrosis must be excluded because it too can lower nasal nitric oxide. [2] [1]
The confirmatory tests examine ciliary function and structure directly. High-speed video microscopy of a nasal or bronchial brushing assesses the ciliary beat frequency and, crucially, the beat pattern, while transmission electron microscopy identifies the classic ultrastructural defects such as absent dynein arms. Genetic testing with panels covering the many known PCD genes confirms the diagnosis when a biallelic disease-causing variant is found, and immunofluorescence for ciliary proteins is a further adjunct, so the modern pathway integrates several of these rather than relying on one. [1] [6]
The remaining investigations characterise the disease and guide treatment rather than make the diagnosis. Regular airway cultures identify the colonising organisms and detect Pseudomonas early, spirometry tracks lung function over time, and cross-sectional imaging with computed tomography defines the extent of bronchiectasis when clinically warranted while balancing radiation exposure in a child who will need lifelong monitoring. Echocardiography is important in the child with heterotaxy to detect complex congenital heart disease. [11] [7]
Management — Resuscitation
Acute deterioration in PCD is usually an infective exacerbation rather than a sudden collapse, but it must be treated promptly and thoroughly. A child with increased cough, changed sputum, breathlessness or falling saturations needs an assessment of severity, an airway culture, and prompt antibiotics guided by previous and current isolates, intensified airway clearance, and admission for intravenous antibiotics and oxygen when the exacerbation is severe or fails to respond to oral treatment. Undertreating an exacerbation is how lung damage accumulates. [5] [8]
The genuinely urgent scenarios cluster around the neonate and the heterotaxy child. The term newborn with respiratory distress needs standard neonatal stabilisation with oxygen and respiratory support while the cause is investigated, and the child with situs ambiguus and complex congenital heart disease may present with the emergencies of that cardiac lesion, so recognising heterotaxy changes the acute plan entirely. Asplenia associated with heterotaxy also mandates urgent sepsis precautions. [7] [4]
Management — Definitive & Stepwise
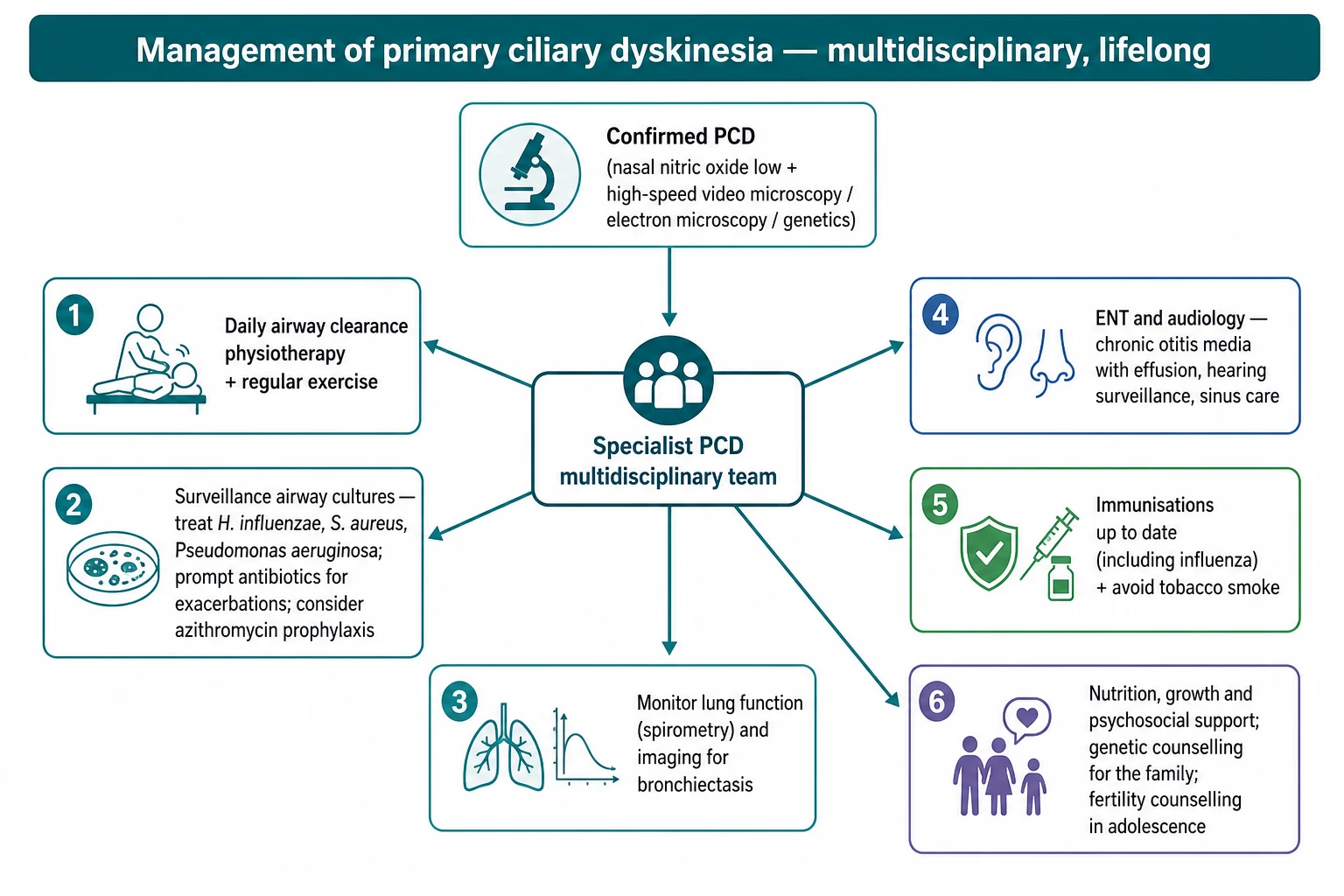
There is no cure, so definitive management is a lifelong programme built on daily airway clearance, and the candidate should lead with this. Regular chest physiotherapy and airway clearance techniques, supported by physical exercise, are the cornerstone because they substitute mechanically for the clearance the cilia cannot provide, and adherence to daily clearance is the single most important determinant of preserved lung function over decades. Everything else supports this foundation rather than replacing it. [5] [1]
Infection control is the second pillar and follows the cystic fibrosis model of surveillance and aggressive treatment. Airway cultures are taken regularly, exacerbations are treated promptly with antibiotics chosen for the isolated organisms, and new Pseudomonas is treated with eradication rather than left to become chronic. Maintenance macrolide therapy has been tested directly in PCD: the BESTCILIA randomised controlled trial showed that regular azithromycin roughly halved the rate of respiratory exacerbations, which supports its use as prophylaxis in selected children. [9] [5]
Stepwise management of the child with PCD
Confirm the diagnosis in a specialist centre and exclude cystic fibrosis
Establish daily airway clearance physiotherapy and encourage regular exercise
Take surveillance airway cultures and monitor spirometry and growth
Treat every exacerbation promptly with culture-directed antibiotics
Eradicate new Pseudomonas aeruginosa rather than allowing chronic colonisation
Consider maintenance azithromycin to reduce exacerbation frequency (BESTCILIA)
Provide ENT and audiology care and formal hearing support
Keep immunisations current, avoid tobacco smoke, and support nutrition and mental health
The supporting measures complete the plan and are heavily examined. Immunisations including influenza should be kept current, exposure to tobacco smoke and other pollutants must be avoided, nutrition and growth are monitored, and the ear disease is managed jointly with ENT and audiology because chronic otitis media and hearing loss affect language and schooling. Genetic counselling for the family and, in adolescence, counselling about the subfertility that accompanies the disease round out comprehensive care. [1] [4]
Azithromycin (maintenance prophylaxis to reduce exacerbations)
Loading dose
No loading dose; used as regular maintenance, not for acute exacerbation treatment
Maintenance dose
10 mg/kg three times weekly (or a weight-banded regimen) as used in the BESTCILIA trial, reviewed periodically
Specific Subtypes & Scenarios
The neonate with respiratory distress is the earliest scenario and the one most often missed in real life. A term baby with unexplained distress, an oxygen requirement or lobar collapse in the first days of life, especially alongside situs inversus, should prompt the question of PCD and a plan to follow the respiratory course and refer if the pattern continues. Recognising this presentation is how the diagnosis is brought forward by years rather than made after bronchiectasis is established. [4] [2]
The child with heterotaxy is the high-stakes scenario because the laterality defect carries cardiac and splenic risk. Unlike the child with straightforward situs inversus, the child with situs ambiguus needs cardiac assessment for complex congenital heart disease and evaluation of splenic function, and the coexistence of a motile ciliopathy with a cardiac lesion complicates both diagnosis and surgery. This subtype shows why laterality must always be characterised rather than assumed benign. [7] [4]
Across Australia, New Zealand and the United Kingdom, PCD is managed through specialist paediatric respiratory centres that concentrate the diagnostic expertise, because nasal nitric oxide, high-speed video microscopy, electron microscopy and genetic testing are not available in every hospital. National networks and diagnostic hubs, together with telehealth links, are used so that children in regional and remote areas can access testing and specialist review. In the ANZ context Aboriginal and Torres Strait Islander and Maori and Pacific children carry a high burden of chronic suppurative lung disease and bronchiectasis in which PCD is under-tested, so equitable access to the diagnostic pathway is a genuine clinical priority rather than an afterthought. [1] [4]
The adolescent facing transition and fertility questions is the third scenario worth rehearsing. As children with PCD reach adolescence they move toward adult services, and the conversation broadens to include subfertility, which affects many men because sperm flagella share the affected machinery and can affect women through impaired fallopian tube cilia. Framing fertility honestly and early, and planning a structured transition to adult respiratory care, are marks of thorough management. [4] [6]
Complications & Pitfalls
The complications of PCD are the natural history of untreated suppurative airway disease, and they should be recited by system. Progressive bronchiectasis with declining lung function is the central respiratory complication, chronic Pseudomonas infection accelerates that decline, chronic rhinosinusitis and nasal polyps persist, and chronic otitis media with effusion causes conductive hearing loss with its effects on speech and learning. Subfertility and, in the heterotaxy subgroup, congenital heart disease complete the list. [8] [11]
The pitfalls are almost all failures of recognition and of clearance. The dominant pitfall is diagnostic delay from mislabelling chronic wet cough as asthma or recurrent infection; the second is assuming normal organ position excludes the disease when only half of children have situs inversus; and the third is under-treating airway clearance and infection so that reversible early disease progresses to fixed bronchiectasis. Failing to characterise a laterality defect and missing dangerous heterotaxy is a further trap. [1] [7]
Prognosis & Disposition
The prognosis is genuinely better than it once was and hinges on early diagnosis and disciplined care, which is the optimistic message to convey. Longitudinal studies show that lung function in PCD can remain relatively stable or decline slowly when children are managed in specialist centres with consistent airway clearance and infection control, in contrast to the steeper decline seen with neglect or late diagnosis. Life expectancy is usually near normal, though morbidity from chronic airway and ear disease is lifelong. [11] [10]
The determinants of a good trajectory are exactly the things treatment targets. A large longitudinal analysis of lung function from school age into adulthood confirmed that a substantial proportion of patients maintain reasonable function while others progress, and that outcomes vary with adherence, infection control and, to some extent, the underlying genotype. This variability is the reason follow-up is lifelong and intensive rather than occasional. [10] [11]
Disposition is therefore lifelong specialist follow-up woven into ordinary childhood. The child is reviewed regularly by a paediatric respiratory team with physiotherapy, microbiology surveillance and spirometry, jointly with ENT and audiology, with structured transition to adult services in adolescence. The uncomplicated child lives a full life with daily treatment built into the routine, while the child with heterotaxy or rapidly progressive disease needs closer, more complex care. [5] [1]
Special Populations
The neonate is the first special population because the disease can present before it is suspected. A term infant with unexplained respiratory distress or persistent atelectasis, particularly with situs inversus, should be flagged for respiratory follow-up and later diagnostic testing, since acting on this earliest presentation is the clearest opportunity to diagnose PCD before lung damage accrues. Neonatal recognition genuinely changes the long-term trajectory. [4] [2]
The child with heterotaxy is the second special population and needs a broader team. Cardiac assessment for complex congenital heart disease, evaluation of splenic function with the associated infection precautions, and coordination between respiratory, cardiology and surgical teams are all required, because in this group the ciliary disease is only part of the problem. Treating these children as though they simply had situs inversus is unsafe. [7] [4]
Aboriginal and Torres Strait Islander, Maori and Pacific children, and children in rural and remote communities, are the third population who need tailored attention. They carry a disproportionate burden of chronic suppurative lung disease and bronchiectasis, they face real barriers to the concentrated diagnostic testing PCD requires, and they benefit from outreach, telehealth and culturally safe care pathways that bring diagnosis and specialist follow-up closer to home. Matching the intensity of case-finding to this higher risk is an equity imperative. [1] [4]
Evidence, Guidelines & Regional Differences
The guideline backbone of this topic is the pair of major diagnostic statements, and naming them earns marks. The European Respiratory Society guideline sets out the diagnostic pathway combining nasal nitric oxide, high-speed video microscopy, electron microscopy and genetics, and the American Thoracic Society clinical practice guideline provides a complementary evidence-graded diagnostic framework. Both agree that no single test suffices and that diagnosis belongs in experienced centres, while differing in emphasis on the order and weighting of tests. [1] [2]
The treatment evidence is thinner than in cystic fibrosis and is largely extrapolated, with one important exception. Much of management is adapted from cystic fibrosis and bronchiectasis care and codified in the PCD Foundation consensus recommendations, but the BESTCILIA trial is the landmark PCD-specific randomised controlled trial, showing that maintenance azithromycin reduces respiratory exacerbations and providing the strongest direct treatment evidence in the field. The genomics-era reviews frame how expanding gene discovery is reshaping diagnosis and phenotype prediction. [5] [9] [6]
The regional differences are practical rather than conceptual, and they revolve around access. High-resource systems agree on the principles, but the specialised diagnostics are concentrated in a small number of centres, so networks, referral pathways and telehealth determine how quickly a child is actually diagnosed. In Australia, New Zealand and comparable systems the added dimension is the higher burden of bronchiectasis among Indigenous and remote populations and the equity challenge of delivering concentrated diagnostic testing to them. [1] [4]
Exam Pearls
Suspect PCD — 'CILIA'
References
- [1]Lucas JS; Barbato A; Collins SA; Goutaki M; Behan L; Caudri D; et al European Respiratory Society guidelines for the diagnosis of primary ciliary dyskinesia. Eur Respir J, 2017.PMID 27836958
- [2]Shapiro AJ; Davis SD; Polineni D; Manion M; Rosenfeld M; Dell SD; et al Diagnosis of Primary Ciliary Dyskinesia. An Official American Thoracic Society Clinical Practice Guideline. Am J Respir Crit Care Med, 2018.PMID 29905515
- [3]Knowles MR; Daniels LA; Davis SD; Zariwala MA; Leigh MW Primary ciliary dyskinesia. Recent advances in diagnostics, genetics, and characterization of clinical disease. Am J Respir Crit Care Med, 2013.PMID 23796196
- [4]Leigh MW; Pittman JE; Carson JL; Ferkol TW; Dell SD; Davis SD; et al Clinical and genetic aspects of primary ciliary dyskinesia/Kartagener syndrome. Genet Med, 2009.PMID 19606528
- [5]Shapiro AJ; Zariwala MA; Ferkol T; Davis SD; Sagel SD; Dell SD; et al Diagnosis, monitoring, and treatment of primary ciliary dyskinesia: PCD foundation consensus recommendations based on state of the art review. Pediatr Pulmonol, 2016.PMID 26418604
- [6]Lucas JS; Davis SD; Omran H; Shoemark A Primary ciliary dyskinesia in the genomics age. Lancet Respir Med, 2020.PMID 31624012
- [7]Shapiro AJ; Davis SD; Ferkol T; Dell SD; Rosenfeld M; Olivier KN; et al Laterality defects other than situs inversus totalis in primary ciliary dyskinesia: insights into situs ambiguus and heterotaxy. Chest, 2014.PMID 24577564
- [8]Noone PG; Leigh MW; Sannuti A; Minnix SL; Carson JL; Hazucha M; et al Primary ciliary dyskinesia: diagnostic and phenotypic features. Am J Respir Crit Care Med, 2004.PMID 14656747
- [9]Kobbernagel HE; Buchvald FF; Haarman EG; Casaulta C; Collins SA; Hogg C; et al Efficacy and safety of azithromycin maintenance therapy in primary ciliary dyskinesia (BESTCILIA): a multicentre, double-blind, randomised, placebo-controlled phase 3 trial. Lancet Respir Med, 2020.PMID 32380069
- [10]Halbeisen FS; Pedersen ESL; Goutaki M; Spycher BD; Amirav I; Boon M; et al Lung function from school age to adulthood in primary ciliary dyskinesia. Eur Respir J, 2022.PMID 35301251
- [11]Marthin JK; Petersen N; Skovgaard LT; Nielsen KG Lung function in patients with primary ciliary dyskinesia: a cross-sectional and 3-decade longitudinal study. Am J Respir Crit Care Med, 2010.PMID 20167855