Paeds · rheumatology-musculoskeletal-and-sports
IgA vasculitis
Also known as Henoch-Schonlein purpura · HSP · IgA vasculitis · Henoch-Schonlein purpura nephritis · Anaphylactoid purpura
Fellowship guide to IgA vasculitis (Henoch-Schonlein purpura), the commonest childhood small-vessel vasculitis. Covers the EULAR/PRINTO/PRES Ankara 2008 classification criteria of mandatory palpable purpura plus at least one of diffuse abdominal pain, IgA deposition on biopsy, arthritis or arthralgia, or renal involvement. Details the clinical tetrad, the pathogenesis centred on galactose-deficient IgA1 immune complex deposition with leukocytoclastic vasculitis, the differentiation from meningococcaemia and other mimics, and the selective corticosteroid indications for severe gastrointestinal, joint, scrotal and renal disease. Reproduces the evidence from the Ronkainen and Jauhola trials that prednisone does not reliably prevent nephropathy, the Cochrane review conclusions, and the renal monitoring schedule of blood pressure and urinalysis for at least six months. Addresses the intussusception, the scrotal involvement, the nephritic and nephrotic presentations, the relapse rate, and the long-term prognosis determined by the renal outcome.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
Overview & Definition
A five-year-old boy is brought in three days after a sore throat, with a raised purplish rash over his ankles and shins, swollen painful knees, and crampy abdominal pain. His mother is frightened by the rash. The rash is the clue. These are not bruises and not a clotting problem; they are palpable purpura, raised spots you can feel with your fingertip, sitting on the lower legs because gravity has dropped the leaking vessels lowest. Together with the joints and the belly, this is the classic face of IgA vasculitis, once called Henoch-Schonlein purpura, and it is the commonest vasculitis of childhood. [5][2]
IgA vasculitis is a small-vessel vasculitis driven by the deposition of IgA-dominated immune complexes in the walls of the capillaries, venules and arterioles of the skin, the gut, the joints and the kidneys. The four features that define it clinically are the palpable purpura on the dependent areas, the arthritis or arthralgia, the colicky abdominal pain, and the renal involvement. The rash is the constant feature and the one the diagnosis hangs on. The other three vary in their presence and their severity, and it is the renal involvement that determines the long-term outcome, which is why the management is built around detecting and watching it. [5][6]
The name changed in 2012, when the Chapel Hill Consensus Conference renamed Henoch-Schonlein purpura as IgA vasculitis, because the new name captures the pathogenesis. The disease is driven by immunoglobulin A, the antibody of the mucosal surfaces, and it is the same immunoglobulin A that drives IgA nephropathy, the commonest glomerulonephritis in the world. The two diseases share the mesangial IgA deposition and the galactose-deficient IgA1, and they sit at the two ends of a spectrum of IgA-driven disease, with IgA nephropathy confined to the kidney and IgA vasculitis spreading across the small vessels of the skin, the gut and the joints as well. [6][9]
[1] [10]Classification
The classification that changes what you do at the bedside is the EULAR/PRINTO/PRES set, agreed at Ankara in 2008 and published in 2010. It was built to separate IgA vasculitis from the other childhood vasculitides in research and in clinical practice, and it gives the diagnosis a reproducible structure. The single mandatory criterion is the palpable purpura, defined as the raised, non-thrombocytopenic purpuric rash typically on the lower limbs and buttocks. On its own the purpura is not enough; the child must also have at least one of four additional features. [1][2]

The four additional criteria, of which at least one must be present, are diffuse abdominal pain, IgA deposition on biopsy, arthritis or arthralgia, and renal involvement. The renal involvement is defined as the presence of proteinuria and or haematuria, and it is the feature that carries the long-term weight. The abdominal pain is the diffuse, colicky pain that may precede the rash and that can herald the intussusception. The biopsy criterion is the skin or renal biopsy showing the dominant IgA deposition in the vessel wall or the glomerulus, and it is the confirmatory test for the atypical presentation. Against the other childhood vasculitides, this criterion set carries a sensitivity of 100 percent and a specificity of 87 percent, which is why the diagnosis is primarily clinical in the classic case and biopsy-confirmed only when the picture is unclear. [1][2]
A second axis runs alongside the criteria, and it is the severity of the organ involvement, because the severity sets the treatment. The abdominal involvement ranges from the mild self-limiting cramp to the intussusception and the gastrointestinal bleeding. The joint involvement ranges from the mild arthralgia to the painful arthritis that prevents walking. The renal involvement ranges from the isolated microscopic haematuria to the nephrotic and nephritic syndrome with hypertension and a falling glomerular filtration rate. It is this severity axis, not the classification axis, that sets whether the child is managed at home with paracetamol or admitted for the corticosteroid and the nephrology referral, and the candidate who names both axes shows they understand the disease. [5][10]
Epidemiology & Risk Factors
IgA vasculitis is the commonest vasculitis of childhood. The annual incidence in children sits around 3 to 27 per 100,000, varying with the population and the season, and the disease accounts for nearly half of all childhood vasculitides. The peak age is four to six years, and the great majority of cases occur in children under ten, though the disease does occur in adolescence and in adults, where it tends to run a more severe renal course. There is a slight male predominance. [5][6]
The seasonal pattern is one of the most consistent epidemiological features, and it points to the trigger. The disease is commonest in the autumn, the winter and the spring, the seasons of the upper respiratory infections, and around half of the cases follow an upper respiratory or other infection in the preceding one to three weeks. The organisms implicated include the group A streptococcus, a range of viral pathogens, and occasionally a gastrointestinal or a drug trigger, and the mucosal origin of the trigger is the link to the IgA-driven pathogenesis. [5][9]
The risk of a more severe course, and especially of a more severe renal course, rises with a small set of features. Older age at onset, especially the adolescent, carries a higher renal risk than the young child. A nephrotic-range proteinuria or a nephritic syndrome at presentation carries a worse renal prognosis than the isolated microscopic haematuria. Persistent proteinuria, hypertension, and a declining renal function at the follow-up are the markers of the renal disease that will progress, and the family history of IgA nephropathy and the complement dysregulation syndromes are the inherited factors that tilt the course. [6][8]
Pathophysiology
The way to understand IgA vasculitis is to picture a mucosal trigger lighting a fuse that ends with the small vessels leaking. The fuse starts at the mucosal surface, where an upper respiratory or other infection drives the production of an abnormal immunoglobulin A. The antibody in question is the galactose-deficient IgA1, an immunoglobulin A1 molecule whose hinge-region O-glycans are under-galactosylated, and this structural abnormality is the shared root of IgA vasculitis and IgA nephropathy. The under-galactosylated IgA1 is recognised as foreign, and the body produces the autoantibodies, chiefly the IgG and the IgA directed against the galactose-deficient hinge. [6][9]
The autoantibodies bind the galactose-deficient IgA1 to form the circulating immune complexes, and these complexes are the cargo that damages the vessels. They are large, they are poorly cleared, and they deposit in the walls of the small vessels of the skin, the gut, the joints and the glomerular mesangium. The deposition activates the complement cascade, predominantly through the alternative and the lectin pathways rather than the classical pathway, and the activation recruits the neutrophils. The neutrophils arrive, release their enzymes, and generate the leukocytoclastic vasculitis, the pattern of vessel-wall injury marked by the fibrinoid necrosis, the neutrophil infiltration, and the nuclear dust that is the fragmented debris of the dead neutrophils. [6][9]
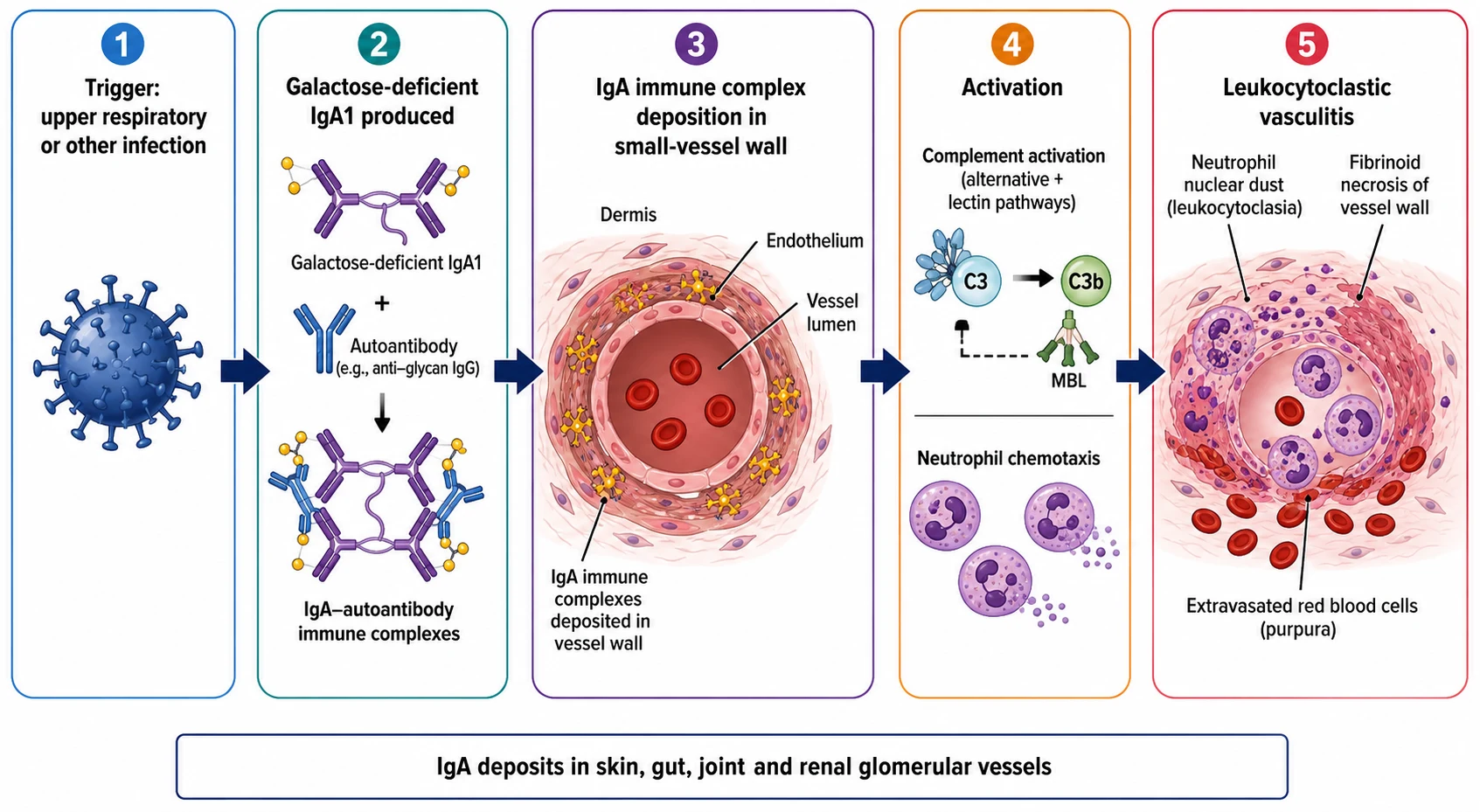
The clinical consequence of this mechanism is the four features the disease is named for, and each maps to a vascular bed. The skin leak produces the palpable purpura, raised because the vessel-wall injury has let the red cells escape into the dermis. The gut leak produces the colicky abdominal pain and occasionally the bleeding and the intussusception. The joint leak produces the periarticular swelling and the pain, which is periarticular rather than intra-articular and which leaves no erosion. The glomerular deposition produces the mesangial IgA nephritis, with the haematuria and the proteinuria that range from the microscopic trace to the nephrotic and the nephritic syndrome. Understanding the single mechanism across the four vascular beds is what lets the candidate hold the whole disease in one model. [5][6]
Clinical Presentation
The presentation is most often a child between four and six years who arrives one to three weeks after a respiratory infection with the palpable purpura, and the rash is the feature that brings them in. The purpura appears in crops on the dependent areas, the lower legs and the ankles, the buttocks, and less often the arms and the face, and it is symmetric. Each lesion is a raised, palpable, non-blanching purpuric spot, a few millimetres to a centimetre across, and it may progress through the colour change from red to purple to brown before fading over one to two weeks. New crops can appear over several weeks, especially with activity. The rash is the constant feature, present in essentially every case, and it is the one the diagnosis hangs on. [5][2]
The arthritis or the arthralgia accompanies the rash in around three quarters of cases, and it often precedes it. The large joints, the knees and the ankles, are most often involved, and the pain is periarticular, with the swelling of the soft tissues around the joint rather than the true intra-articular effusion. The arthritis is non-erosive and it leaves no deformity, but it can be painful enough to prevent walking, and it tends to resolve within days. It is the feature that most often brings the child to medical attention before the rash appears, and the trap is to attribute the limp to the joint alone and to miss the impending purpura. [5]
The abdominal pain is the third feature, present in around half to three quarters of cases, and it is the one that carries the acute danger. It is typically a diffuse, colicky pain that may precede the rash by days, and it may be accompanied by the vomiting, the diarrhoea, and the gastrointestinal bleeding, which may be occult and detected only on the stool test or frank and melena-like. The serious complication is the intussusception, which in IgA vasculitis is often ileo-ileal rather than the classic ileo-caecal, which means the air or the barium enema that reduces the classic infant intussusception may fail, and the surgical reduction may be needed. Any severe or worsening abdominal pain, with a palpable mass, with the bile-stained vomiting, or with the significant bleeding, raises the intussusception and demands the ultrasound and the surgical review. [5][10]
[5] [6]The renal involvement is the fourth and the most consequential feature. It presents as the haematuria, microscopic or gross, with or without the proteinuria, and in a minority it progresses to the nephritic syndrome with the hypertension and the oedema, or the nephrotic syndrome with the heavy proteinuria and the low albumin. The renal involvement usually appears within four to six weeks of the onset, though it can declare as late as six months, which is why the monitoring extends across that whole window. The child who looks well and whose early urines are clear can still develop the nephritis weeks later, and this is the single most important point for the safety-netting. [6][8]
A set of less common but examinable features rounds out the picture. The scrotal involvement, with the swelling and the tenderness of the scrotum or the testis, occurs in up to a third of boys and can mimic the testicular torsion. The pulmonary involvement, with the diffuse alveolar haemorrhage, is rare but life-threatening. The central nervous system involvement, with the headache, the seizures, or the encephalopathy, is also rare. The acute haemorrhagic oedema of the face and the ears, the pancreatic involvement, and the myocardial involvement are the rarest of all. Naming these shows the breadth, but the candidate should remember that the four features carry the weight. [5][10]
Differential Diagnosis
The first differential is the life-threatening one, and it is the one that must never be missed. A febrile, ill child with rapidly progressive purpura or petechiae is meningococcaemia or sepsis until proven otherwise, not IgA vasculitis. The child with IgA vasculitis is typically afebrile or low-grade febrile and looks well, and the purpura is raised and confined to the dependent areas. The child with meningococcaemia is febrile, ill, and the lesions are petechial and rapidly progressive, spreading to the trunk and the palms and the soles. The bedside discipline is to treat the ill child with the empiric antibiotic within the first hour while the diagnosis is clarified, because the cost of treating the IgA vasculitis child with a dose of ceftriaxone is small and the cost of missing the meningococcaemia is fatal. [5]
Within the purpuric rashes, the distinction from the idiopathic thrombocytopenic purpura rests on the full blood count and the distribution. The idiopathic thrombocytopenic purpura shows the isolated thrombocytopenia with the otherwise normal full blood count, and the purpura is flat and scattered rather than raised and dependent, and there is no arthritis, no abdominal pain, and no renal involvement. The coagulation is normal in both, which is why the platelet count is the discriminator. The child with the sepsis and the disseminated intravascular coagulation shows the thrombocytopenia, the prolonged coagulation, and the ill appearance, and is managed as the emergency. [5]
The other vasculitides form the next ring of the differential, and each has a distinguishing feature. Kawasaki disease presents with the fever for five days or more, the conjunctivitis, the strawberry tongue, the extremity changes, and the coronary artery involvement, and it is a medium-vessel vasculitis rather than a small-vessel one. The ANCA-associated vasculitides present with the constitutional features, the pulmonary and the renal involvement, and the positive ANCA. The polyarteritis nodosa presents with the fever, the weight loss, the livedo, the nodules, and the medium-vessel involvement. The acute haemorrhagic oedema of infancy presents in the infant under two years with the large target-like purpuric lesions on the face, the ears and the limbs, and the child looks well despite the dramatic rash, and it resolves without sequelae. [5][10]
Clinical & Bedside Assessment
Begin with the rapid sick-or-well assessment, because the first question is whether this is the life-threatening mimic. A febrile, ill child with the rapidly progressive purpura is managed as the meningococcaemia or the sepsis, with the empiric antibiotic within the first hour, while the diagnosis is clarified. The child with IgA vasculitis is typically afebrile or low-grade febrile and looks well, and the assessment then proceeds to the four features and the severity. [5]
The focused history establishes the preceding infection, the onset and the progression of the rash, the joint pain, the abdominal pain, the urine colour, and the swelling. Ask about the sore throat, the upper respiratory symptoms, or the gastrointestinal illness in the preceding weeks, because the mucosal trigger is the link to the pathogenesis. Ask about the abdominal pain, its severity, the vomiting, and the blood in the stool, because these raise the intussusception. Ask about the urine colour and the urine output, because the dark or the bloody urine and the reduced output raise the renal involvement. Ask about the swelling of the face, the legs, and the scrotum, because the oedema and the scrotal swelling carry their own management implications. [5][10]
The focused examination searches the four vascular beds systematically. Examine the skin for the palpable purpura on the lower legs, the ankles, and the buttocks, confirming that the lesions are raised, non-blanching, and not associated with the thrombocytopenia. Examine the large joints for the periarticular swelling and the tenderness, and the abdomen for the tenderness, the guarding, and the palpable mass that raises the intussusception. Examine the scrotum in the boy for the swelling and the tenderness, and distinguish the IgA vasculitis involvement from the torsion, recognising that both can coexist and that the Doppler ultrasound and the surgical review are needed when the torsion cannot be excluded. Measure the blood pressure, look for the oedema, and examine the urine, because the renal involvement is the feature that carries the long-term weight. [5]
[1] [5]Investigations
The diagnosis of IgA vasculitis is primarily clinical, and the investigations serve to exclude the mimics, to grade the organ involvement, and to establish the baseline for the monitoring. The full blood count confirms the normal platelet count that excludes the idiopathic thrombocytopenic purpura and the disseminated intravascular coagulation, and the coagulation screen confirms the normal clotting that excludes the coagulopathy. The urea, the electrolytes and the creatinine assess the renal function, the albumin grades the nephrotic component, and the C-reactive protein and the blood culture are sent if the sepsis is in the differential. [5][10]
The urinalysis is the single most important investigation for the renal monitoring, and it is performed at the diagnosis and then repeated at the scheduled intervals. The presence of the blood or the protein on the dipstick raises the renal involvement, and the degree is quantified by the urine protein-to-creatinine ratio on the early-morning sample. The microscopic haematuria alone is the mildest form, and the proteinuria with or without the haematuria is the marker that escalates the monitoring and the referral. The serum complement is usually normal in IgA vasculitis, which helps distinguish it from the lupus and the post-streptococcal glomerulonephritis, though the C3 may occasionally be low. [6][8]
The skin biopsy is reserved for the atypical presentation, and it confirms the diagnosis by showing the leukocytoclastic vasculitis and the dominant IgA deposition on the immunofluorescence. The biopsy is not needed for the classic case, where the clinical criteria settle the diagnosis, but it is the confirmatory test when the rash is atypical, when the platelet count is borderline, or when the diagnosis is genuinely unclear. The renal biopsy is indicated when the renal involvement is significant, defined as the nephrotic syndrome, the nephritic syndrome, the persistent proteinuria, or the declining renal function, and it grades the injury by the International Study of Kidney Disease in Children classification, which guides the intensity of the immunosuppression. [6][9]
The imaging is directed by the presentation. The abdominal ultrasound is the first-line investigation for the suspected intussusception, and it shows the target sign and the length of the involved segment, with the ileo-ileal location that is typical of the IgA vasculitis and that may not reduce with the enema. The scrotal Doppler ultrasound is the investigation for the suspected torsion in the boy with the scrotal swelling, and it confirms the blood flow and excludes the surgical emergency. The renal ultrasound is performed when the renal involvement is significant, to exclude the obstruction and the structural anomaly. [5][10]
Management — Resuscitation
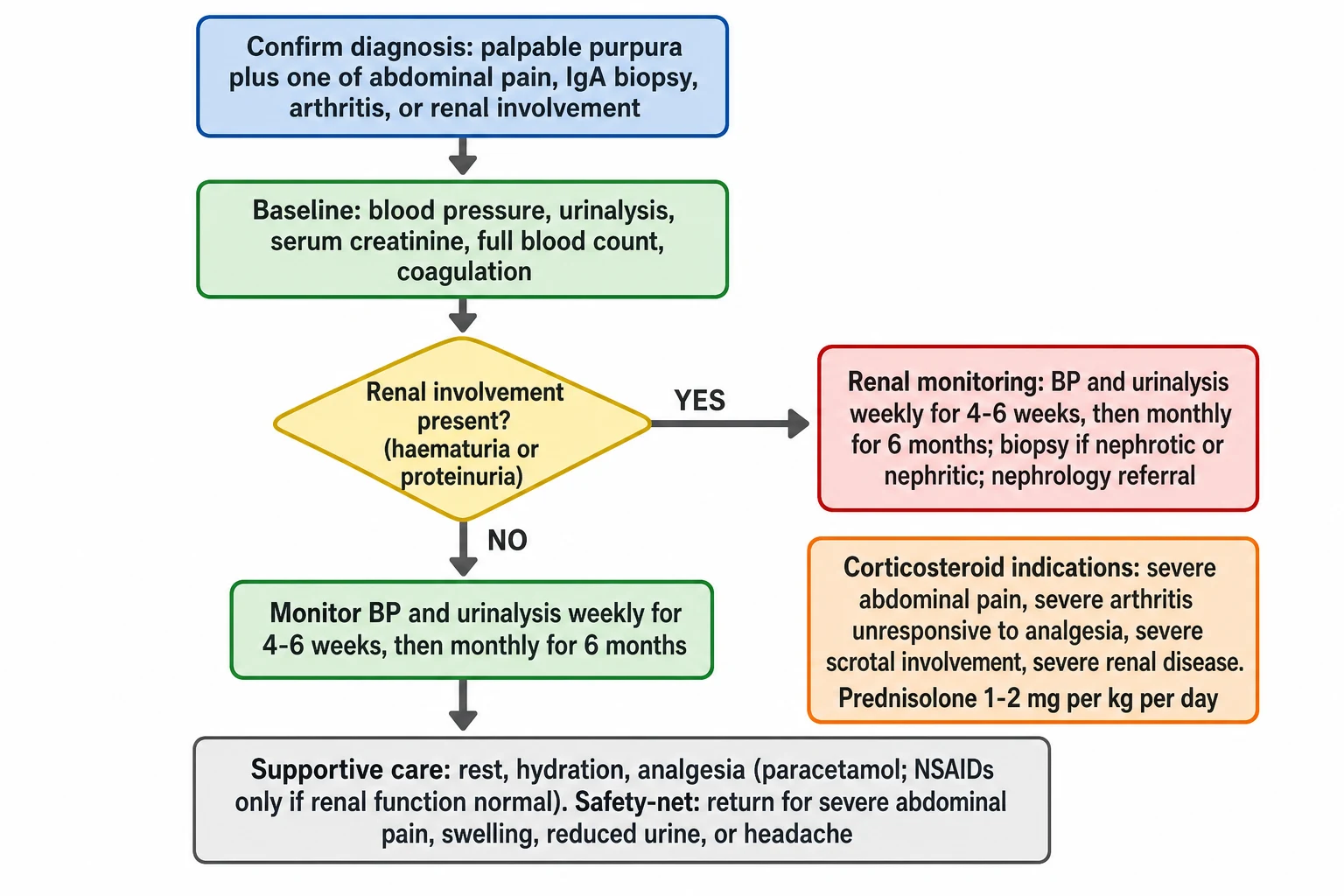
The first action in the resuscitation is the exclusion of the life-threatening mimic. A febrile, ill child with the rapidly progressive purpura receives the empiric antibiotic, typically the ceftriaxone, within the first hour, while the diagnosis is clarified, because the cost of the empirical treatment is small and the cost of the missed meningococcaemia is fatal. The child with the shock receives the intravenous fluids and the escalation to the intensive care by the paediatric sepsis pathway. Once the life-threatening mimic is excluded, the management of the IgA vasculitis itself is largely supportive, with the corticosteroid reserved for the specific severe indications. [5]
The child with the severe abdominal pain is the first to draw the corticosteroid decision, because the pain can be severe, and because the intussusception must be excluded first. The abdominal ultrasound is performed for the severe or the worsening pain, the palpable mass, the bile-stained vomiting, or the significant bleeding, and the surgical review is sought for the confirmed intussusception. The ileo-ileal intussusception of the IgA vasculitis often fails the enema reduction and may need the surgical reduction. The corticosteroid, given as the prednisolone at 1 to 2 milligrams per kilogram per day, relieves the severe gastrointestinal inflammation and the pain, and it is the first-line indication for the corticosteroid in the disease. [3][10]
The first steps in the child with IgA vasculitis
Exclude the life-threatening mimic: a febrile ill child with rapidly progressive purpura receives the empiric ceftriaxone within the first hour while the diagnosis is clarified
Establish the diagnosis clinically with the palpable purpura plus at least one of the EULAR PRINTO PRES criteria, and confirm the normal platelet count and coagulation
Take the baseline: blood pressure, urinalysis, serum creatinine, albumin, full blood count, coagulation, and the urine protein-to-creatinine ratio if the protein is present
Assess the severity of the four features: the rash extent, the joint pain, the abdominal pain with the intussusception exclusion, and the renal involvement
Start the supportive care: rest, hydration, and the analgesia with paracetamol, reserving the non-steroidal anti-inflammatory drugs for when the renal function is normal
Consider the corticosteroid for the severe abdominal pain, the severe arthritis unresponsive to the analgesia, the severe scrotal involvement, or the severe renal disease, at prednisolone 1 to 2 milligrams per kilogram per day
Set the monitoring schedule: blood pressure and urinalysis weekly for 4 to 6 weeks, then monthly for 6 months, and arrange the safety-net for the return features
The child with the severe arthritis that prevents walking, and that has not responded to the paracetamol, is the second indication for the corticosteroid. The non-steroidal anti-inflammatory drugs are used with caution and only when the renal function is normal, because they can worsen the renal perfusion in the child with the active nephritis. The child with the severe scrotal involvement is the third indication, and the corticosteroid relieves the swelling and the pain, though the torsion must be excluded first by the Doppler ultrasound and the surgical review. The child with the acute nephritic syndrome and the hypertensive emergency is managed with the antihypertensive agents, the fluid and the sodium restriction, and the escalation to the nephrology and the intensive care. [3][10]
Management — Definitive & Stepwise
The definitive management runs as a stepwise pathway from the supportive care to the selective corticosteroid to the renal surveillance, and the cleanest way to hold it is as a ladder from the mild to the severe. At the top is the supportive care that is the mainstay for the great majority. Below that is the corticosteroid for the specific severe indications. Below that is the immunosuppression for the severe nephritis, and at the bottom is the long-term renal surveillance that is the safety-net for the disease. [5][10]
[5] [10]The supportive care is the foundation, and it is all that the great majority of children need. The rest and the hydration are advised during the acute phase, and the analgesia with the paracetamol at 15 milligrams per kilogram per dose manages the joint and the abdominal pain. The non-steroidal anti-inflammatory drugs are used only when the renal function is normal, because they can worsen the renal perfusion in the child with the active nephritis. The elevation and the comfortable positioning relieve the joint and the scrotal swelling. The family is given the clear safety-net to return for the severe abdominal pain, the reduced urine, the swelling, and the headache, because these are the features that herald the complications and the renal involvement. [5]
The corticosteroid is the selective therapy for the specific severe indications, and the evidence base for its use is one of the most-tested points in the topic. The indications are the severe abdominal pain, the severe arthritis unresponsive to the simple analgesia, the severe scrotal involvement, and the severe renal disease. The agent is the prednisolone at 1 to 2 milligrams per kilogram per day, given orally or intravenously, tapered as the symptoms resolve. The corticosteroid is effective for the relief of the severe gastrointestinal, joint and scrotal symptoms, and the evidence is clear on that. [3][10]
The question of whether the corticosteroid prevents the nephropathy is the point that separates the informed candidate from the rest, and the evidence says it does not reliably. The randomised, double-blind, placebo-controlled trial of Ronkainen and colleagues, the foundational trial in the field, found that the early prednisone reduced the intensity of the abdominal and the joint pain but did not prevent the renal involvement. The eight-year follow-up of Jauhola and colleagues confirmed that the renal outcome was no different between the prednisone and the placebo groups. The Cochrane review of Chartapisak and colleagues concluded that the evidence does not support the routine corticosteroid for the prevention of the nephropathy. This is why the routine prednisone for the renal prevention alone is not recommended, and why the renal monitoring is mandatory regardless of the corticosteroid use. [3][4][7]
Ronkainen 2006 and Jauhola 2012: prednisone does not prevent the nephropathy
Population: Children with newly diagnosed IgA vasculitis
Key finding
The randomised, double-blind, placebo-controlled trial of early prednisone in IgA vasculitis found that the corticosteroid relieved the severe abdominal and joint pain but did not prevent the renal involvement. The eight-year follow-up confirmed no difference in the renal outcome between the prednisone and the placebo groups. The conclusion: the corticosteroid is for the symptomatic severe disease, not for the routine renal prevention.
The severe nephritis with the nephrotic or the nephritic syndrome is the scenario that demands the nephrology referral and the biopsy, and the management is the high-dose corticosteroid with or without the immunosuppression. The angiotensin-converting-enzyme inhibitor is used for the persistent proteinuria, because it reduces the intraglomerular pressure and the protein leak, and it is the long-term renoprotective agent. The immunosuppressants, including the cyclophosphamide, the azathioprine, the mycophenolate mofetil, and the rituximab, are used for the severe or the refractory nephritis, under the nephrology guidance, and the evidence base is less robust than for the corticosteroid. The European consensus SHARE recommendations of Ozen and colleagues provide the contemporary framework for the diagnosis and the treatment, and they are the reference the candidate should cite. [10][8]
Specific Subtypes & Scenarios
The child with the significant renal involvement is the subtype that the viva most often presents, because it is the one that carries the long-term weight. The significant involvement is defined as the nephrotic syndrome, the nephritic syndrome, the persistent proteinuria, or the declining renal function, and it demands the renal biopsy, the nephrology referral, and the immunosuppressive management. The biopsy grades the injury by the International Study of Kidney Disease in Children classification, and the histological severity, including the crescents and the sclerosis, guides the intensity of the treatment. The candidate who names the biopsy indication, the classification, and the immunosuppressive framework holds the renal subtype. [6][9]
The child with the intussusception is the subtype that tests the candidate on the surgical interface. The intussusception in IgA vasculitis is often ileo-ileal, unlike the classic ileo-caecal intussusception of the infant, and the ileo-ileal location means that the air or the barium enema, which reduces the classic form, often fails. The presentation is the severe or the worsening abdominal pain, the palpable mass, the bile-stained vomiting, and the significant bleeding, and the ultrasound is the first-line investigation. The management is the surgical review and the surgical reduction when the enema fails, with the corticosteroid for the concurrent vasculitic inflammation. [5][10]
The boy with the scrotal involvement is the subtype that tests the candidate on the torsion differential. The scrotal swelling and the tenderness occur in up to a third of boys with IgA vasculitis, and the IgA vasculitis involvement can mimic the testicular torsion. The trap is twofold: the torsion can be missed in the child attributed to the vasculitis, and the vasculitis can be over-investigated as the torsion. The discipline is to assess both, to perform the Doppler ultrasound when the torsion cannot be excluded clinically, and to involve the surgical team early. The corticosteroid relieves the vasculitic swelling and the pain, once the torsion is excluded. [5]
The rare pulmonary, the central nervous system, and the pancreatic involvement form the last set of subtypes, and they are the ones that test the breadth. The diffuse alveolar haemorrhage presents with the haemoptysis, the dyspnoea, and the infiltrates, and it is managed with the high-dose corticosteroid and the intensive care. The central nervous system involvement presents with the headache, the seizures, or the encephalopathy, and it is managed with the blood pressure control and the corticosteroid. The pancreatic involvement presents with the abdominal pain and the raised amylase. Naming these shows the candidate has read widely, but the four features carry the weight. [5][10]
Complications & Pitfalls
The acute complications are the intussusception, the gastrointestinal bleeding and the perforation, the testicular involvement, and the acute nephritic syndrome. The intussusception is the most common surgical complication, and its ileo-ileal location is the reason the enema may fail. The gastrointestinal bleeding is usually occult but can be significant, and the perforation is the rarest and the most dangerous. The testicular involvement can rarely progress to the infarction if the torsion is missed. The acute nephritic syndrome, with the hypertension, the oedema, and the oliguria, is the renal emergency that demands the antihypertensive and the fluid management. [5][10]
The long-term complications are the chronic kidney disease, the hypertension, and the end-stage renal failure, and they are the ones that determine the lifelong outcome. The proportion of children who develop the chronic kidney disease is around 1 to 3 per cent, concentrated in those with the nephrotic or the nephritic presentation and the severe biopsy findings. The hypertension may persist as the legacy of the renal injury. The end-stage renal failure is the worst outcome, and it is the reason the monitoring extends across the six months and the nephrology follow-up is arranged for the significant involvement. [6][8]
The principal pitfall, and the one that costs the most, is the failure to maintain the blood pressure and the urinalysis monitoring for the full six months. The renal involvement can declare weeks after the rash has faded, in a child who looks completely well, and the only way to catch it is the scheduled monitoring. The child who is lost to the follow-up after the acute episode is the child whose nephritis is missed, and the nephritis that is missed is the one that progresses. The second pitfall is the over-reliance on the corticosteroid for the renal prevention, because the evidence does not support it, and the false reassurance that the prednisone has been given can replace the proper monitoring. [7][8]
[5] [10]The third pitfall is attributing the palpable purpura to the trauma, the abdominal pain to the simple gastroenteritis, and the scrotal swelling to the torsion alone, without recognising the unified vasculitic diagnosis. The fourth is missing the meningococcaemia by anchoring on the IgA vasculitis in the febrile, ill child. The fifth is using the non-steroidal anti-inflammatory drug in the child with the active nephritis, which worsens the renal perfusion. The sixth is failing to exclude the intussusception in the child with the severe abdominal pain, and the seventh is failing to involve the surgical team for the scrotal involvement when the torsion cannot be excluded. [5]
Prognosis & Disposition
The overall prognosis of IgA vasculitis is excellent, and this is the reassurance the family needs to hear. The great majority of children recover fully within four to six weeks, with the supportive care alone, and the disease is self-limiting in the uncomplicated case. The purpura fades, the joints settle, the abdominal pain resolves, and the child returns to the normal activity. The prognosis is set by the renal involvement, and the child without the significant renal involvement has an excellent long-term outlook. [5][6]
About one third of children relapse, usually within the first year, and the relapse is typically milder than the initial episode. The relapse most often presents with the recurrent purpura, with or without the abdominal and the joint features, and it is managed with the supportive care and the renewed monitoring. The relapse rate is higher in the child with the more severe initial presentation, and the family is counselled that a recurrence is possible and that the monitoring resumes with the relapse. [5][6]
The prognosis of the renal involvement is the one that carries the weight, and it is stratified by the presentation. The child with the isolated microscopic haematuria has an excellent renal outlook. The child with the proteinuria has a slightly higher risk, and the child with the nephrotic or the nephritic syndrome has the highest risk of the chronic kidney disease and the end-stage renal failure. The proportion of children who develop the chronic kidney disease is around 1 to 3 per cent, and the persistent proteinuria, the hypertension, and the severe biopsy findings are the markers of the progression. [6][8]
The disposition follows the severity. The uncomplicated child is managed as an outpatient, with the supportive care and the scheduled monitoring. The child with the severe abdominal pain, the significant bleeding, the suspected intussusception, the significant renal involvement, or the inability to maintain the oral intake is admitted for the investigation, the corticosteroid, and the surgical or the nephrology review. The child with the significant renal involvement is followed by the paediatric nephrology, with the blood pressure, the urinalysis, and the serum creatinine monitored for the long term, because the renal disease can progress even after the acute episode has resolved. [5][10]
Special Populations
The older child and the adolescent with IgA vasculitis carry a higher risk of the severe renal course than the younger child, and the monitoring and the threshold for the biopsy are correspondingly tighter. The adolescent is engaged directly in the adherence to the monitoring schedule, the activity restriction during the acute phase, and the return-to-activity plan, because the failure mode in this age group is often the loss to the follow-up. The disease in the adult runs a more severe renal course than in the child, which is the reason the adolescent is counselled on the long-term surveillance. [6][9]
The child with the complement dysregulation, including the complement factor H or I abnormality, carries a more severe renal course, and the complement testing is considered in the child with the significant or the atypical nephritis. The child from the socioeconomic disadvantage or the rural and the remote setting needs the monitoring adapted to the access, with the telehealth and the local general practitioner enlisted for the scheduled blood pressure and urinalysis, because the loss to the follow-up in this population is the barrier that misses the nephritis. The family and the school are engaged in the activity restriction during the acute phase, the gradual return to the activity, and the understanding of the safety-net, because the child who returns too early to the full activity can flare the purpura. [5][10]
The child with the recurrent or the relapsing disease is managed with the renewed monitoring and the corticosteroid for the severe relapse, and the family is counselled that the relapse is typically milder than the initial episode. The child with the persistent proteinuria is managed with the angiotensin-converting-enzyme inhibitor and the long-term nephrology follow-up, because the persistent proteinuria is the marker of the renal disease that may progress. The transition to the adult care is planned for the adolescent with the significant renal involvement, to ensure the continuity of the surveillance across the lifespan. [6][8]
Evidence, Guidelines & Regional Differences
The evidence base for IgA vasculitis is anchored by the EULAR/PRINTO/PRES classification criteria and a set of trials and reviews. The EULAR/PRINTO/PRES Ankara 2008 criteria, published in 2010 by Ozen and colleagues in Part II and Ruperto and colleagues in Part I, give the diagnostic structure that unifies the disease across the centres, with the sensitivity of 100 percent and the specificity of 87 percent. The European consensus SHARE recommendations of Ozen and colleagues in 2019 provide the contemporary framework for the diagnosis and the treatment, and they are the primary reference the candidate should cite. [1][2][10]
The corticosteroid evidence rests on the randomised, double-blind, placebo-controlled trial of Ronkainen and colleagues, the foundational trial in the field, and its eight-year follow-up by Jauhola and colleagues. These established that the early prednisone relieves the severe abdominal and joint pain but does not prevent the renal involvement. The Cochrane review of Chartapisak and colleagues, and its companion systematic review in the Archives of Disease in Childhood, synthesised the interventions for the kidney disease and concluded that the evidence does not support the routine corticosteroid for the renal prevention. The pathogenesis and the nephritis framework rest on the reviews of Davin and Coppo and of Davin alone. [3][4][7][6][9]
In Australia and Aotearoa New Zealand, IgA vasculitis is managed in the general paediatric and the paediatric rheumatology and nephrology services under protocols aligned with the EULAR/PRINTO/PRES criteria and the SHARE recommendations. The diagnosis is clinical in the classic case, with the skin biopsy reserved for the atypical presentation. The supportive care is the mainstay, the corticosteroid is reserved for the severe gastrointestinal, joint, scrotal and renal indications, and the routine prednisone for the renal prevention is not recommended. The blood pressure and the urinalysis monitoring extends for at least six months. The child with the significant renal involvement is referred to the paediatric nephrology for the biopsy and the long-term surveillance. The rural and the remote child has the monitoring adapted to the access, with the telehealth and the local general practitioner enlisted for the scheduled checks.
The regional differences are modest, because the consensus is international. The United Kingdom and the Europe follow the EULAR/PRINTO/PRES and the SHARE framework. The United States and the Canada follow the same criteria and the same corticosteroid evidence. The live controversy, and the one the viva may probe, is the role of the routine corticosteroid for the renal prevention, where the trial evidence is negative but the clinical practice still varies. The other controversy is the immunosuppressive regimen for the severe nephritis, where the evidence base is less robust and the practice ranges from the corticosteroid alone to the corticosteroid with the cyclophosphamide, the mycophenolate, or the rituximab. The candidate who names the controversy and the evidence that anchors it shows the depth. [7][8][10]
Exam Pearls
IgA vasculitis (Henoch-Schonlein purpura) is the commonest childhood small-vessel vasculitis, presenting with the tetrad of palpable purpura on the dependent areas, arthritis or arthralgia, colicky abdominal pain, and renal involvement. The EULAR/PRINTO/PRES Ankara 2008 criteria require the mandatory palpable purpura plus at least one of diffuse abdominal pain, IgA deposition on biopsy, arthritis or arthralgia, or renal involvement, and they carry a sensitivity of 100 percent and a specificity of 87 percent. The diagnosis is primarily clinical in the classic case, and the skin biopsy showing the IgA deposition is reserved for the atypical presentation. [1][5]
IGAV
The pathogenesis centres on the galactose-deficient IgA1, the immune complex deposition, the alternative and lectin complement activation, and the neutrophil-driven leukocytoclastic vasculitis. The long-term prognosis is set by the renal involvement, and every child needs the blood pressure and the urinalysis monitored for at least six months, regardless of the corticosteroid use. The corticosteroid at prednisolone 1 to 2 milligrams per kilogram per day is reserved for the severe abdominal pain, the severe arthritis unresponsive to the analgesia, the severe scrotal involvement, and the severe renal disease. The Ronkainen 2006 and the Jauhola 2012 trials established that the prednisone does not reliably prevent the nephropathy, and the Cochrane review of Chartapisak confirmed the conclusion. [3][4][7]
The intussusception in IgA vasculitis is often ileo-ileal and may need the surgical rather than the enema reduction. The scrotal involvement mimics the torsion, and both can coexist, so the Doppler ultrasound and the surgical review are needed when the torsion cannot be excluded. About one third of children relapse, usually within the first year, and the relapse is typically milder. The chronic kidney disease occurs in around 1 to 3 per cent, concentrated in those with the nephrotic or the nephritic presentation. The acute haemorrhagic oedema of infancy is the mimic in the infant under two years, with the large target-like lesions and the well child. [5][6][10]
[5] [8] [5] [7]References
- [1]Ozen S, Pistorio A, Iusan SM, et al EULAR/PRINTO/PRES criteria for Henoch-Schonlein purpura, childhood polyarteritis nodosa, childhood Wegener granulomatosis and childhood Takayasu arteritis: Ankara 2008. Part II: Final classification criteria. Ann Rheum Dis, 2010.PMID 20413568
- [2]Ruperto N, Ozen S, Pistorio A, et al EULAR/PRINTO/PRES criteria for Henoch-Schonlein purpura, childhood polyarteritis nodosa, childhood Wegener granulomatosis and childhood Takayasu arteritis: Ankara 2008. Part I: Overall methodology and clinical characterisation. Ann Rheum Dis, 2010.PMID 20388738
- [3]Ronkainen J, Koskimies O, Ala-Houhala M, et al Early prednisone therapy in Henoch-Schonlein purpura: a randomized, double-blind, placebo-controlled trial. J Pediatr, 2006.PMID 16887443
- [4]Jauhola O, Ronkainen J, Koskimies O, et al Outcome of Henoch-Schonlein purpura 8 years after treatment with a placebo or prednisone at disease onset. Pediatr Nephrol, 2012.PMID 22311342
- [5]McCarthy HJ, Tizard EJ Clinical practice: Diagnosis and management of Henoch-Schonlein purpura. Eur J Pediatr, 2010.PMID 20012647
- [6]Davin JC, Coppo R Henoch-Schönlein purpura nephritis in children. Nat Rev Nephrol, 2014.PMID 25072122
- [7]Chartapisak W, Opastirakul S, Hodson EM, et al Interventions for preventing and treating kidney disease in Henoch-Schonlein Purpura (HSP). Cochrane Database Syst Rev, 2009.PMID 19588365
- [8]Chartapisak W, Sirisuthana S, Hutapruk S, et al Prevention and treatment of renal disease in Henoch-Schonlein purpura: a systematic review. Arch Dis Child, 2009.PMID 18701559
- [9]Davin JC Henoch-Schonlein purpura nephritis: pathophysiology, treatment, and future strategy. Clin J Am Soc Nephrol, 2011.PMID 21393485
- [10]Ozen S, Marks SD, Brogan P, et al European consensus-based recommendations for diagnosis and treatment of immunoglobulin A vasculitis-the SHARE initiative. Rheumatology (Oxford), 2019.PMID 30879080