Paeds · rheumatology-musculoskeletal-and-sports
Juvenile dermatomyositis
Also known as Juvenile dermatomyositis · JDM · Childhood dermatomyositis · Juvenile idiopathic inflammatory myopathy
Fellowship guide to juvenile dermatomyositis: the child with a heliotrope rash and Gottron papules who cannot climb stairs, the Bohan and Peter and 2017 EULAR and ACR classification criteria, the myositis-specific antibodies that define phenotype and prognosis, methotrexate with corticosteroids as first-line therapy, calcinosis and lipodystrophy as the long-term damage, and the CARRA, PRINTO and UK consensus treatment pathways.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
Overview & Definition
Picture the six-year-old girl who, over three weeks, has become unable to climb the school bus steps, needs both hands to push herself up from a chair, and has developed a violaceous flush around her eyes and scaly red patches over her knuckles. No single finding is unique, but the combination of that rash with progressive proximal weakness is juvenile dermatomyositis, and the window to prevent permanent damage is already open. [4]
Juvenile dermatomyositis is the commonest idiopathic inflammatory myopathy of childhood, an immune-mediated small-vessel vasculopathy that targets skin and skeletal muscle and, less often, the gut, lungs and heart. It differs from the adult disease in its vasculopathic biology, its high rate of calcinosis and its near-absence of malignancy association, and it replaces earlier labels such as childhood dermatomyositis. Modern classification leans on the 2017 EULAR and ACR criteria that fold in muscle biopsy, magnetic resonance imaging and myositis-specific antibodies, alongside the older Bohan and Peter framework that many examination questions still use. [1] [2]
Three ideas anchor the topic for a fellowship candidate. The diagnosis rests on pattern recognition of a characteristic rash with proximal weakness, supported by muscle enzymes and imaging rather than on any single blood test. Treatment is corticosteroid plus methotrexate given promptly, and the intensity of early therapy predicts whether the child escapes calcinosis and lipodystrophy. And the myositis-specific antibody defines a clinical subgroup that changes both the differential and the prognosis, from the steroid-responsive anti-Mi-2 child to the anti-MDA5 child at risk of interstitial lung disease. [5] [12]
Classification
Think about classification along two tracks that the examiners want you to hold in parallel: the diagnostic criteria that confirm the disease, and the antibody-defined subgroups that predict its behaviour. [1] [12]

The Bohan and Peter criteria, still taught for examinations, define definite juvenile dermatomyositis as the characteristic rash plus any three of four muscle features: symmetric proximal weakness, raised muscle enzymes, a myopathic electromyogram, and a characteristic muscle biopsy. Probable disease needs two of the four muscle features and possible disease needs one, and the framework says nothing about modern imaging or antibodies, which is its main weakness. The 2017 EULAR and ACR criteria fix this by weighting muscle biopsy, magnetic resonance imaging and myositis-specific antibodies into a probability score that classifies juvenile dermatomyositis without needing every old test. [2] [3]
The antibody track matters because each myositis-specific antibody carves out a clinical subgroup. Anti-p155/140, which is anti-transcription intermediary factor 1-gamma, is the commonest antibody in juvenile disease and associates with calcinosis. Anti-Mi-2 carries the classic rash and a good, steroid-responsive prognosis. Anti-NXP-2 marks severe muscle disease and calcinosis. Anti-MDA5 defines the amyopathic or skin-ulcerating child at risk of rapidly progressive interstitial lung disease, and it is the one that should change your investigation panel and your urgency. [7] [12]
Anti-TIF1γ (p155/140)
commonest in JDM
- Most frequent antibody in children
- Characteristic rash, calcinosis risk
- Good overall survival
- Malignancy link only in adults
Anti-Mi-2
good prognosis
- Classic heliotrope and Gottron rash
- Strongly steroid-responsive
- Good functional outcome
- Lower calcinosis burden
Anti-NXP-2
severe muscle
- Severe proximal weakness
- High calcinosis rate
- Oedema and GI features
- Malignancy link only in adults
Anti-MDA5
ILD risk
- Amyopathic or cutaneous ulceration
- Rapidly progressive interstitial lung disease
- Oral ulcers and palmar papules
- High mortality — escalate early
Epidemiology & Risk Factors
Juvenile dermatomyositis is rare, with an incidence around three per million children per year in the United Kingdom and Ireland and a similar figure reported in North America. The peak age sits between five and ten years, with a smaller early-childhood cluster, and girls outnumber boys by roughly two to three to one. Seasonal and geographic variation in onset hints at an infectious trigger in a genetically susceptible host, but no single organism has been confirmed. [9] [5]
The risk factors that matter clinically are not inherited so much as they are modifiable. Delayed diagnosis and treatment, prolonged active disease, and a high early disease activity score all raise the risk of calcinosis, which is the dominant source of long-term disability. Anti-NXP-2 and anti-TIF1-gamma antibodies also associate with calcinosis, and anti-MDA5 marks the children at risk of interstitial lung disease and death. These are the variables that should lower your threshold to treat early and to escalate. [8] [11]
The numbers that anchor your viva
Pathophysiology
The driving biology is an immune-mediated small-vessel vasculopathy with a striking type-one interferon signature, in which complement deposition on capillaries produces ischaemic muscle injury and the characteristic skin inflammation. Inflamed muscle then releases enzymes and generates the perifascicular atrophy that is so suggestive on biopsy. [5] [4]
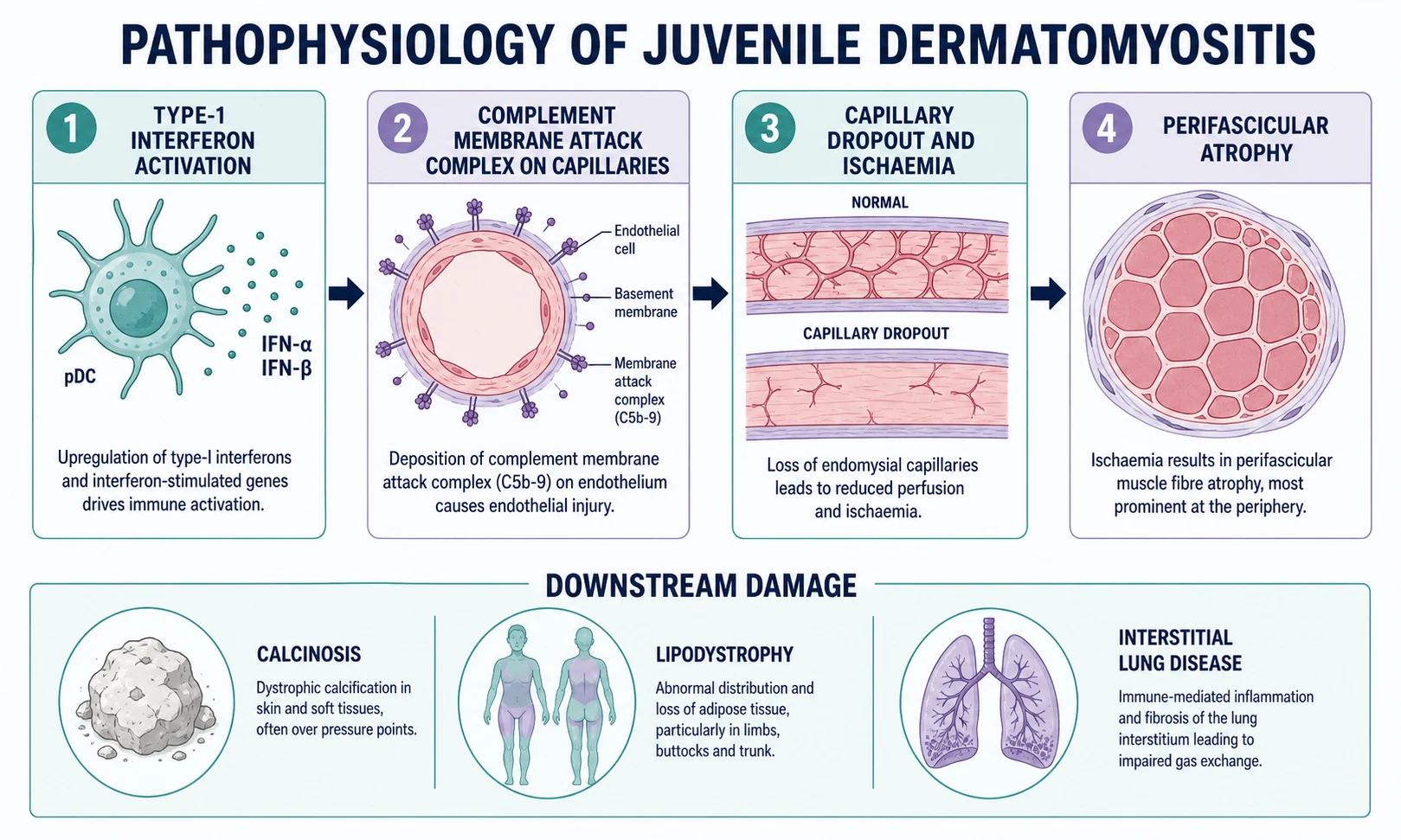
At the vessel level, immune complexes and complement fix on the endothelium of endomysial and dermal capillaries, and the membrane attack complex appears early and is visible on biopsy even before muscle necrosis. Capillaries are lost, the blood supply to the perifascicular region fails first because it sits at the watershed of perfusion, and the muscle fibres there atrophy. That perifascicular atrophy is close to pathognomonic for dermatomyositis and is the histological signature examiners ask for. [5]
The same vasculopathic process explains the calcinosis, lipodystrophy and gut ulceration that define the long-term burden of juvenile disease. Dystrophic calcification follows chronic tissue injury and inflammation in subcutaneous tissue and muscle, and it can erode through skin or limit joint movement. Partial or generalised lipodystrophy, sometimes with insulin resistance and hypertriglyceridaemia, reflects chronic inflammation and typically declares itself months to years after onset. [8] [5]
[8] [11]Clinical Presentation
The presentation is a child with a rash and weakness, and the skill is in putting the two together when each could be dismissed on its own. Weakness comes on over weeks, is symmetric and proximal, and shows itself as difficulty climbing stairs, rising from the floor, lifting the arms to comb hair or wash, and, in severe disease, swallowing and phonation. The child may also be tired, febrile and irritable, and a smaller group presents with arthritis, abdominal pain or dyspnoea. [4] [11]
The skin signs are the hallmark and the examination favourites. The heliotrope rash is a violaceous, lilac discolouration of the upper eyelids, often with periorbital oedema, and Gottron papules are erythematous, scaly papules over the metacarpophalangeal, proximal and distal interphalangeal joints; the same erythema over the elbows and knees is the Gottron sign. Look also for the V-sign over the anterior chest, the shawl-sign across the shoulders, the holster-sign on the lateral thighs, periungual erythema with abnormal nailfold capillaries, and the cutaneous ulceration and palmar papules that should make you think of anti-MDA5 disease. [11] [7]
| Clinical picture | What it implies | Act |
|---|
Muscle involvement can extend to the pharynx and the respiratory muscles, and these are the features that change a stable child into an unwell one. Dysphagia, nasal regurgitation, a weak cough and a soft voice signal bulbar and respiratory weakness, with a real risk of aspiration and ventilatory failure. Lip and mouth ulceration, abdominal pain and melaena point to gut vasculopathy, and dyspnoea with crackles on the chest points to interstitial lung disease, especially in the anti-MDA5 subgroup. [5] [11]
Differential Diagnosis
Build the differential in layers, because a raised creatine kinase and proximal weakness have many causes and the rash can mimic several dermatoses. The first layer is the acquired myopathy: post-infectious myositis such as influenza and enterovirus, which is acute and self-limited; drug-induced myopathy from steroids or statins; and endocrine myopathy, particularly hypothyroidism. [4]
The second layer is the inherited and neuromuscular disease that progresses over months to years rather than weeks. Duchenne and Becker muscular dystrophy, limb-girdle muscular dystrophy and spinal muscular atrophy all cause progressive proximal weakness, but they lack the inflammatory rash and the acute muscle enzyme rise, and they follow a slower, painless course. A creatine kinase that is astronomically high in a boy with calf pseudohypertrophy should redirect you to the muscular dystrophy pathway. [4]
Acquired myopathy
- Viral myositis (influenza, enterovirus) — acute, self-limited
- Drug-induced (steroids, statins)
- Endocrine (hypothyroid myopathy)
- Post-infectious and transient
Inherited neuromuscular
- Duchenne and Becker muscular dystrophy
- Limb-girdle muscular dystrophy
- Spinal muscular atrophy
- Slow, painless, no inflammatory rash
Other rheumatic
- Systemic lupus erythematosus
- Mixed connective tissue disease and overlap
- Scleroderma
- Systemic juvenile idiopathic arthritis
Cutaneous mimics
- Atopic and contact eczema
- Psoriasis on extensor surfaces
- Allergic and photosensitive dermatitis
- Lupus tumidus and subacute cutaneous lupus
The third layer is the other rheumatic and cutaneous diseases. Systemic lupus erythematosus, mixed connective tissue disease and overlap syndromes can produce myositis and rashes that overlap with juvenile dermatomyositis, and they are separated by their antibody profiles — anti-double-stranded-DNA, anti-Smith and anti-U1-ribonucleoprotein — and by their patterns of organ involvement. Cutaneous mimics such as atopic eczema, psoriasis and photosensitive dermatitis can be confused with the Gottron distribution, but they lack the muscle disease and the nailfold capillary changes. The decisive step is to combine the clinical pattern, the muscle enzymes, the imaging and the antibody panel rather than to rely on any single test. [5] [1]
Clinical & Bedside Assessment
Assessment runs in parallel with the decision to treat, because disease activity accumulates damage and a weak child can deteriorate. Take a focused history of the pace of the weakness, the rash and its photosensitivity, dysphagia and breathing, recent infections, and school attendance, while you examine the skin, the muscles, the swallow and the chest. Document the duration of untreated disease, because that number drives the risk of calcinosis and the urgency of treatment. [4]
The muscle examination assesses symmetric proximal strength with a validated tool such as the Childhood Myositis Assessment Scale or manual muscle testing of eight muscle groups, and it looks for Gowers sign and a waddling gait. The skin examination looks for heliotrope rash, Gottron papules and sign, the photo-distributed V and shawl rashes, periungual erythema, ulceration and abnormal nailfold capillaries — dilated, tortuous, dropout — best seen with a capillaroscope or ophthalmoscope oil drop. Examine the swallow and the chest, and screen for calcinosis as firm subcutaneous nodules around joints, the buttocks and the trunk. [11] [8]
Investigations
There is no single diagnostic blood test for juvenile dermatomyositis, so the panel is built to confirm muscle inflammation, to exclude mimics and to define the antibody phenotype. Measure the creatine kinase first, because it is the most useful muscle enzyme, and add aldolase, lactate dehydrogenase and the transaminases; remember that alanine and aspartate aminotransferase are released by inflamed muscle as well as liver, so a transaminitis in a weak child is often myogenic rather than hepatic. Add a full blood count, inflammatory markers and a biochemistry screen to stage systemic involvement. [4] [5]
The myositis-specific antibody panel is now central to classification and prognosis. Send it early, because anti-Mi-2, anti-TIF1-gamma, anti-NXP-2 and anti-MDA5 each define a phenotype, a calcinosis risk and a prognosis, and anti-MDA5 in particular should redirect you to a high-resolution chest scan and lung function. Add a myositis-associated antibody panel and an antinuclear antibody to capture overlap syndromes. [7] [12]
Imaging has replaced electromyography as the preferred non-invasive test for muscle inflammation. Magnetic resonance imaging of the proximal thigh muscles, with fat-suppressed or short-tau inversion recovery sequences, shows oedema in actively inflamed muscle, guides the site of biopsy and monitors the response to treatment. A muscle biopsy remains the gold standard where imaging and antibody panels are inconclusive, and it shows perivascular and perimysial inflammation, perifascicular atrophy and membrane attack complex deposition on capillaries. For the anti-MDA5 child or anyone dyspnoeic, add spirometry with lung volumes, diffusing capacity and a high-resolution chest scan to detect interstitial lung disease. [1] [5]
The investigation set — JDM WORKUP
Management — Resuscitation
Frame the resuscitation around the threats that can hurt a child today rather than around airway and circulation in every case. The genuinely unwell child with juvenile dermatomyositis is usually in one of three situations: pharyngeal and respiratory muscle weakness with aspiration or ventilatory failure, rapidly progressive interstitial lung disease in anti-MDA5 disease, or gut vasculopathy with ulceration and bleeding. [5]
For pharyngeal and respiratory weakness, assess the swallow and cough, arrange speech and language review and a videofluoroscopy, and protect the airway with positioning, thickened fluids or, in ventilatory failure, non-invasive or invasive respiratory support. For suspected interstitial lung disease, request a high-resolution chest scan and lung function early and involve respiratory and rheumatology together, because anti-MDA5 disease can decline fast. For gut vasculopathy with bleeding or perforation, resuscitate with fluids and blood, give intravenous corticosteroids and involve surgery. [4] [5]
Management — Definitive & Stepwise
The backbone of treatment is early, aggressive immunosuppression with corticosteroids and methotrexate, with escalation to intravenous immunoglobulin, rituximab and other agents for refractory, skin-predominant or antibody-defined disease. The aim is not merely to suppress inflammation but to prevent the calcinosis, contractures and lipodystrophy that follow uncontrolled disease. [6] [10]
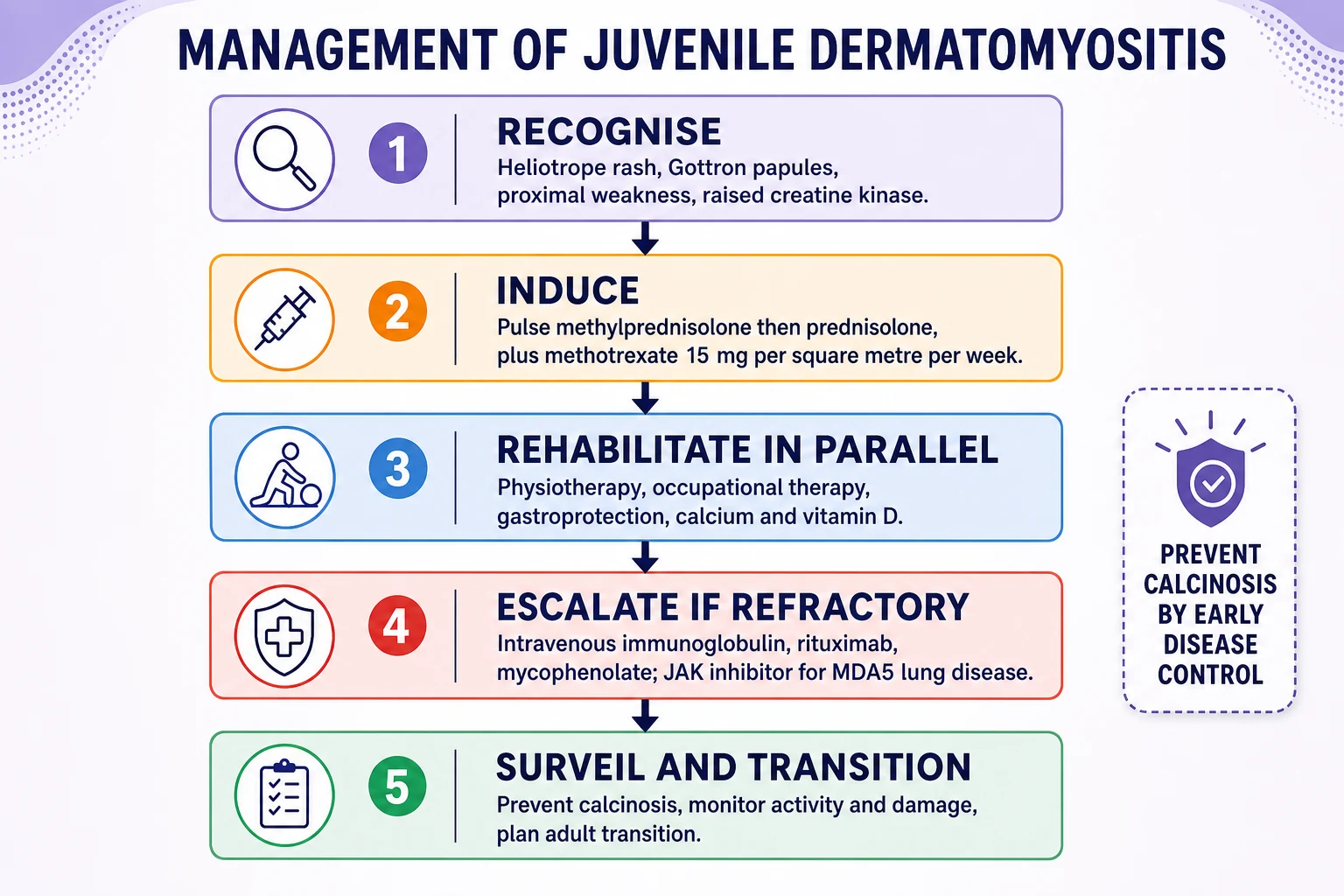
For moderate to severe new-onset disease, give intravenous pulse methylprednisolone at 10 to 30 mg/kg per day to a maximum of 1 g, usually for three days, followed by oral prednisolone at 1 to 2 mg/kg per day to a maximum of 60 to 80 mg per day, and introduce methotrexate at 15 mg per square metre per week, subcutaneous or oral, with folic acid. The PRINTO randomised trial showed that adding methotrexate to prednisone was as effective as adding ciclosporin with a better safety and tolerability profile, and both combinations allowed faster and safer steroid tapering than prednisone alone. [6] [10]
Methotrexate (first-line steroid-sparing agent)
Dose
15 mg/m² once weekly (subcutaneous or oral)
First-line induction — the standard sequence
Pulse methylprednisolone 10–30 mg/kg/day (max 1 g) for ~3 days, then oral prednisolone 1–2 mg/kg/day (max 60–80 mg/day)
Add methotrexate 15 mg/m²/week with folic acid as the steroid-sparing backbone
Add gastroprotection, calcium and vitamin D, and bone-density baseline; start physiotherapy and occupational therapy early
Taper corticosteroid slowly on clinical and enzyme response over months, aiming for methotrexate monotherapy
Escalate for refractory disease — persistent weakness, rash or enzymes after three months — and for skin-predominant or antibody-defined disease. Intravenous immunoglobulin at 2 g/kg is useful for refractory skin disease and dysphagia, rituximab is used for refractory myositis, and mycophenolate mofetil, calcineurin inhibitors such as ciclosporin and tacrolimus, and cyclophosphamide all have a place in severe or vasculopathic disease. For anti-MDA5 interstitial lung disease, early combination immunosuppression and a JAK inhibitor such as ruxolitinib or tofacitinib may be considered with respiratory input, and hydroxychloroquine, topical corticosteroids and rigorous sun protection help the skin. [5] [12]
[8]Rehabilitation runs in parallel from the start. Physiotherapy preserves range of movement and prevents contractures, occupational therapy supports function and fatigue, and speech and language therapy manages dysphagia. Give gastroprotection, calcium and vitamin D with the steroids, baseline bone density, and sun protection and psychology support for the child and family throughout a long illness. [4] [10]
Specific Subtypes & Scenarios
Three subgroups change the conversation and the management. The amyopathic or clinically amyopathic child has the characteristic rash with little or no muscle weakness and normal muscle enzymes, and is defined by the antibody panel; the anti-MDA5 amyopathic child is the one at risk of interstitial lung disease, so screen the lungs even when the muscles are quiet. [7] [5]
The anti-MDA5 child sits at the severe end of the spectrum. Cutaneous and oral ulceration, palmar papules, arthritis and amyopathy combine with a risk of rapidly progressive interstitial lung disease that drives the mortality of juvenile disease. Request a high-resolution chest scan and lung function early, treat with combination immunosuppression and consider a JAK inhibitor, and involve respiratory and intensive care before the child deteriorates. [12] [5]
Calcinosis is the third and most common scenario, affecting up to four in ten children and presenting as painful, firm nodules or plaques under the skin that can ulcerate, drain and limit movement. Risk factors are delayed diagnosis and treatment, prolonged active disease, and the anti-NXP-2 and anti-TIF1-gamma antibodies. The priority is prevention through early disease control, because once calcinosis is established the options — bisphosphonates, diltiazem, warfarin, sodium thiosulfate, intravenous immunoglobulin and surgery — are inconsistent and mostly guided by expert opinion. [8] [11]
Amyopathic JDM
rash, quiet muscle
- Characteristic rash, normal strength and enzymes
- Defined by myositis-specific antibody panel
- Screen for MDA5 and interstitial lung disease
- Skin-directed therapy and surveillance
Anti-MDA5
ILD and ulceration
- Cutaneous and oral ulceration, palmar papules
- Rapidly progressive interstitial lung disease
- Early combination immunosuppression
- Highest mortality — escalate early
Calcinosis
≈20–40%
- Painful nodules, ulceration, contractures
- Risk: delay, prolonged activity, NXP-2 and TIF1γ
- Prevention beats treatment
- Inconsistent drug options; surgery for focal disease
Refractory disease
after 3 months
- Persistent weakness, rash or enzymes
- Intravenous immunoglobulin, rituximab
- Mycophenolate, calcineurin inhibitors
- Repeat imaging and biopsy if needed
Complications & Pitfalls
The complications fall into damage from the disease and harm from its treatment. Calcinosis is the dominant disease-related complication, and lipodystrophy — partial or generalised, sometimes with insulin resistance and hypertriglyceridaemia — is a late and disfiguring consequence of chronic inflammation. Interstitial lung disease drives mortality, especially in anti-MDA5 disease, and contractures, muscle atrophy and growth failure follow prolonged active disease. [8] [5]
Treatment-related harm is real and predictable. Corticosteroids bring growth suppression, osteoporosis and vertebral fracture, hypertension, hyperglycaemia, cataracts and mood change, and the broad immunosuppression carries infection risk including opportunists. Methotrexate needs full blood count, liver and renal monitoring, and nausea and mucositis are common and improved by route change and folic acid. Hydroxychloroquine adds a retinopathy screen. The pitfall to avoid is a transaminitis assumed to be hepatic when it is myogenic, leading to unnecessary interruption of methotrexate or liver investigations. [4] [10]
[4]Prognosis & Disposition
Mortality in the modern era sits around one to three per cent, and it is driven by anti-MDA5 interstitial lung disease, severe vasculopathy with gut ulceration and sepsis from immunosuppression, not by the muscle disease itself. Before corticosteroids the mortality approached a third, which is why the history of treatment is taught as a caution against undertreatment. [9] [5]
The course runs in one of three patterns. Monocyclic disease settles into remission within two years and does not relapse, polycyclic disease relapses and remits, and chronic continuous disease stays active and drives the calcinosis and damage burden. Most children achieve remission with modern therapy, but functional outcome depends on the speed of control, the prevention of calcinosis and contractures, and the durability of multidisciplinary rehabilitation. [11] [4]
Disposition is a tertiary paediatric rheumatology service with shared care, regular disease activity scoring, and surveillance for calcinosis, lipodystrophy, interstitial lung disease and treatment toxicity. Plan transition early to adult rheumatology, because the disease and its antibody phenotype, the calcinosis and the cumulative treatment burden all travel with the patient into adult life. [5] [12]
Course of juvenile dermatomyositis
Chronic continuous
persistent activity, calcinosis
Special Populations
Adolescents face the dual burden of a disfiguring rash and the transition into adult care, and they need explicit planning for adherence, contraception, alcohol and methotrexate, and the psychological impact of chronic illness. Start transition early, rehearse the adult appointment, and hand over a clear summary of the antibody phenotype, the treatment history and the surveillance plan. [5]
The very young child, under four years, often presents with irritability, refusal to walk and abdominal pain rather than a clear history of weakness, and carries a higher rate of calcinosis and lipodystrophy because the disease is harder to recognise and treat early. Lower the threshold to check muscle enzymes and to image in the irritable toddler who stops walking. [11] [8]
Evidence, Guidelines & Regional Differences
The classification evidence is the 2017 EULAR and ACR criteria, which folded muscle biopsy, magnetic resonance imaging and myositis-specific antibodies into a probability-based system and displaced the older Bohan and Peter framework as the research and trial standard while leaving it in the examinations. The treatment evidence is the PRINTO randomised trial of prednisone against prednisone plus ciclosporin against prednisone plus methotrexate, which established methotrexate as the preferred steroid-sparing partner. [1] [6]
The consensus pathways are the CARRA treatment plans from North America, which offer three defined initial pathways for moderate disease and a pathway for skin-predominant disease, and the UK and European SHARE recommendations. In Australia and New Zealand, practice follows these international consensus documents delivered through paediatric rheumatology networks, with rituximab, intravenous immunoglobulin and JAK inhibitors available for refractory or antibody-defined disease. [10] [5]
Exam Pearls
The high-yield facts for the fellowship examination cluster around the rash, the antibodies and the calcinosis. Gottron papules over the knuckles and a heliotrope rash around the eyes are pathognomonic, and proximal weakness with a raised creatine kinase is the clinical core. The 2017 EULAR and ACR criteria and the older Bohan and Peter criteria are both examinable, so be ready to recite both. [2] [1]
The antibody associations are a favourite written question. Anti-Mi-2 carries the classic rash and a good prognosis, anti-TIF1-gamma is the commonest in children and links with calcinosis, anti-NXP-2 marks severe muscle and calcinosis, and anti-MDA5 defines the amyopathic child at risk of rapidly progressive interstitial lung disease. Remember that malignancy associates with the adult, not the juvenile, disease. [7] [12]
The antibody pairs — MINT
On treatment, name corticosteroids plus methotrexate as first line, cite the PRINTO trial for the methotrexate choice, and escalate with intravenous immunoglobulin, rituximab or other agents for refractory or skin-predominant disease. On calcinosis, the examiner wants prevention through early disease control, not a list of drugs that do not work. And on prognosis, give the modern mortality of one to three per cent and name anti-MDA5 lung disease and vasculopathy as the drivers. [6] [8]
References
- [1]Lundberg IE, Tjärnlund A, Bottai M, et al. 2017 European League Against Rheumatism/American College of Rheumatology classification criteria for adult and juvenile idiopathic inflammatory myopathies and their major subgroups. Ann Rheum Dis, 2017.PMID 29079590
- [2]Bohan A, Peter JB Polymyositis and dermatomyositis (first of two parts). N Engl J Med, 1975.PMID 1090839
- [3]Bohan A, Peter JB Polymyositis and dermatomyositis (second of two parts). N Engl J Med, 1975.PMID 1089199
- [4]Huber AM Juvenile dermatomyositis: advances in pathogenesis, evaluation, and treatment. Paediatr Drugs, 2009.PMID 19877722
- [5]Rider LG, Nistala K The juvenile idiopathic inflammatory myopathies: pathogenesis, clinical and autoantibody phenotypes, and outcomes. J Intern Med, 2016.PMID 27028907
- [6]Ruperto N, Pistorio A, Oliveira S, et al. Prednisone versus prednisone plus ciclosporin versus prednisone plus methotrexate in new-onset juvenile dermatomyositis: a randomised trial. Lancet, 2016.PMID 26645190
- [7]Tansley SL, Simou S, Shaddick G, et al. Autoantibodies in juvenile-onset myositis: their diagnostic value and associated clinical phenotype in a large UK cohort. J Autoimmun, 2017.PMID 28663002
- [8]Hoeltzel MF, Oberle EJ, Robinson AB, et al. The presentation, assessment, pathogenesis, and treatment of calcinosis in juvenile dermatomyositis. Curr Rheumatol Rep, 2014.PMID 25366934
- [9]McCann LJ, Juggins AD, Maillard SM, et al. The Juvenile Dermatomyositis National Registry and Repository (UK and Ireland)—clinical characteristics of children recruited within the first 5 yr. Rheumatology (Oxford), 2006.PMID 16567354
- [10]Huber AM, Giannini EH, Bowyer SL, et al. Protocols for the initial treatment of moderately severe juvenile dermatomyositis: results of a Children's Arthritis and Rheumatology Research Alliance Consensus Conference. Arthritis Care Res (Hoboken), 2010.PMID 20191521
- [11]Shah M, Mamyrova G, Targoff IN, et al. The clinical phenotypes of the juvenile idiopathic inflammatory myopathies. Medicine (Baltimore), 2013.PMID 23263716
- [12]Rider LG, Shah M, Mamyrova G, et al. The myositis autoantibody phenotypes of the juvenile idiopathic inflammatory myopathies. Medicine (Baltimore), 2013.PMID 23877355