Paeds · rheumatology-musculoskeletal-and-sports
Periodic fever and autoinflammatory syndromes
Also known as Hereditary recurrent fever · Periodic fever syndrome · Familial Mediterranean fever · PFAPA · TNF receptor-associated periodic syndrome · TRAPS · Cryopyrin-associated periodic syndrome · CAPS · Hyper-IgD syndrome · Mevalonate kinase deficiency · Inflammasomopathy
Fellowship guide to the periodic fever and autoinflammatory syndromes. Covers the distinction between autoinflammatory and autoimmune disease, the five syndromes a fellow must carry (familial Mediterranean fever, PFAPA, mevalonate kinase deficiency or hyper-IgD syndrome, tumour necrosis factor receptor-associated periodic syndrome, and the cryopyrin-associated periodic syndromes), the inflammasome and interleukin-one pathophysiology, the Livneh criteria for familial Mediterranean fever, the Gattorno 2019 Eurofever classification of the hereditary recurrent fevers, the colchicine-first principle for familial Mediterranean fever with its amyloidosis prevention, the interleukin-one blockade for the cryopyrin-associated periodic syndromes and mevalonate kinase deficiency, the corticosteroid-abort and tonsillectomy pathway for PFAPA, and the family and transition issues across a lifelong illness.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
Overview & Definition
A periodic fever syndrome is the body turning its innate immune system on and off in a faulty rhythm, so the child runs a predictable fever and inflammation for a few days, then returns to complete health, over and over again. The fault is not in the adaptive immune system that makes antibodies. It is in the innate system, the fast-responding first line, which over-activates in a self-limited burst because a single regulatory protein is mis-coded by a single gene. There is no infection to fight and no self-antigen to attack. The attack arises, burns itself out, and leaves the child well until the next one comes, on a clockwork rhythm that a careful parent can almost set a watch by. [1][2]
This is the core idea the fellowship candidate must hold. Autoinflammation is innate and antigen-independent, while autoimmunity is adaptive and self-antigen-driven. The periodic fever syndromes are the cleanest example of autoinflammation in all of paediatrics, and they teach the principle that a disease can be driven by a cytokine surge without any external trigger and without any antibody. The five syndromes a fellow must carry are familial Mediterranean fever, PFAPA, mevalonate kinase deficiency (also called hyper-IgD syndrome), tumour necrosis factor receptor-associated periodic syndrome, and the cryopyrin-associated periodic syndromes. The first four recur in attacks, while the cryopyrin-associated spectrum ranges from cold-triggered attacks to a continuous multisystem disease of the infant. [1][9]
Classification
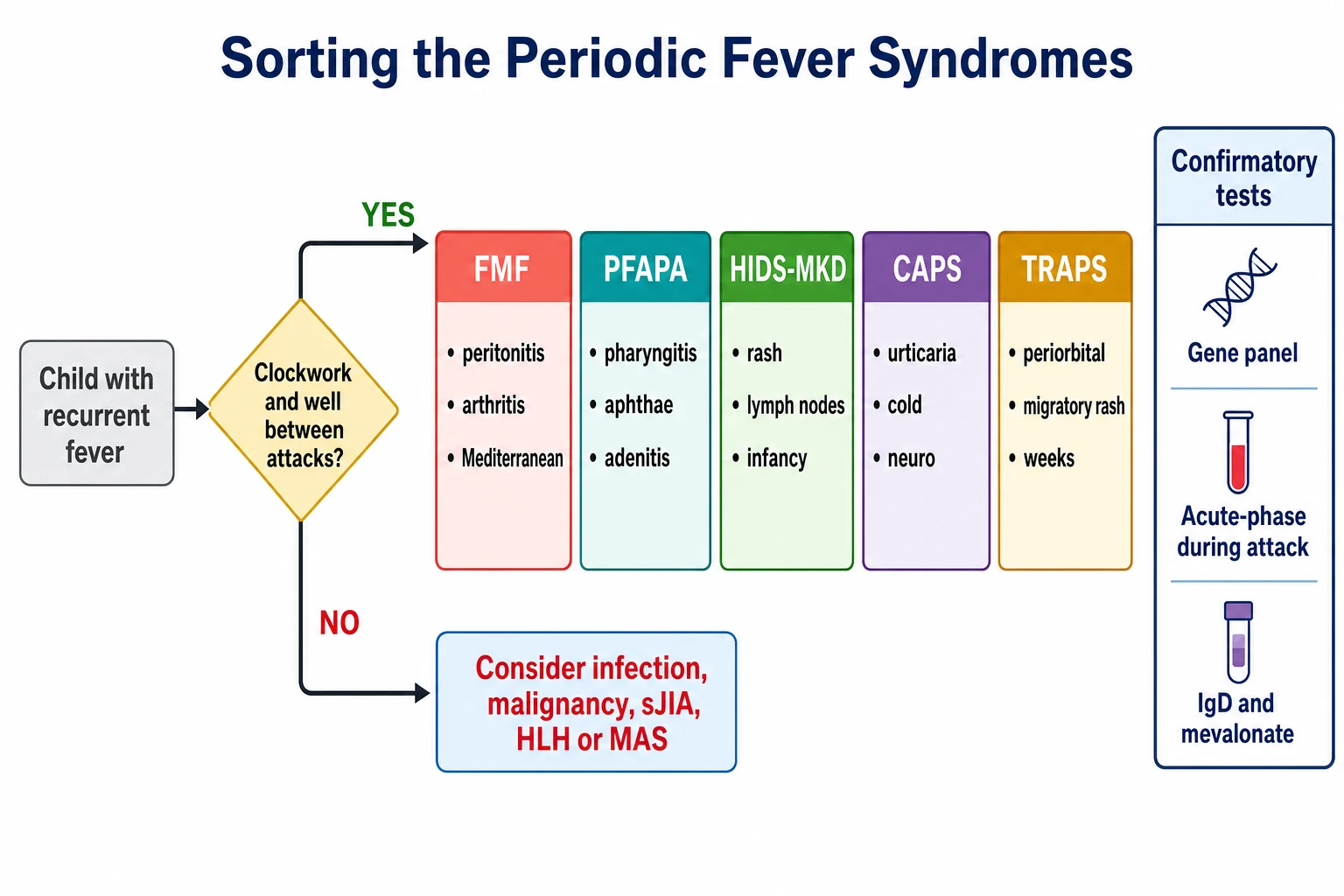
Hold the periodic fever syndromes as a single family of diseases, then sort them by the gene that fails and the attack pattern that results. Four of the five are hereditary, monogenic, and classifiable by the Eurofever and PRINTO collaboration criteria published by Gattorno and colleagues in 2019, which separated the hereditary recurrent fevers on the discriminating clinical features of each. PFAPA is the exception, because it is polygenic or clinically defined rather than monogenic, and it carries its own diagnostic criteria. The unifying thread is that all of them are autoinflammatory, innate, and antigen-independent. [1]
Familial Mediterranean fever is the most common and the most important, because it is the one whose untreated course leads to kidney failure from amyloidosis. It is autosomal recessive, caused by mutations in the MEFV gene that encodes the pyrin protein, and it affects the Mediterranean populations, the Armenian, Turkish, Sephardic and Ashkenazi Jewish, and Arab families. The attacks are short, lasting one to three days, and they feature fever with serositis, the peritonitis, pleuritis and arthritis that can mimic an acute abdomen. The erysipelas-like erythema on the lower leg is the pathognomonic skin sign. [3][4]
Mevalonate kinase deficiency, historically called hyper-IgD syndrome, is the second autosomal recessive syndrome, caused by mutations in the MVK gene. It declares itself in the first year of life, often triggered by a vaccination, with attacks lasting three to seven days of fever, a maculopapular rash, cervical lymphadenopathy, abdominal pain with diarrhoea, and oral ulcers. The serum immunoglobulin D is often but not always raised, and the urinary mevalonic acid rises during an attack, which is why a single test between attacks can mislead. [11]
Tumour necrosis factor receptor-associated periodic syndrome is autosomal dominant, caused by mutations in TNFRSF1A, the gene for the tumour necrosis factor receptor. It has the longest attacks of the group, lasting from one week to several weeks, and it carries a distinctive triad of a migratory rash over the trunk and limbs, periorbital swelling with conjunctivitis, and severe migratory myalgia. The cryopyrin-associated periodic syndromes form a spectrum caused by gain-of-function mutations in NLRP3, the gene for cryopyrin, ranging from the mildest familial cold autoinflammatory syndrome at one end, through Muckle-Wells syndrome in the middle, to neonatal-onset multisystem inflammatory disease, also called CINCA, at the severe end. [1][2]
PFAPA, the periodic fever with aphthous stomatitis, pharyngitis and adenitis, is the most common periodic fever of childhood, and it is the one most fellows will actually meet. It is not hereditary in the single-gene sense, and its gene, if there is one, is not yet pinned down. Marshall described the syndrome in 1987 and Thomas refined the clinical criteria in 1999, and the diagnosis is made clinically after the hereditary syndromes are excluded. The cardinal features are a clockwork interval, the triad of pharyngitis, aphthous stomatitis and cervical adenitis, a high fever for three to six days, and complete wellness with normal growth between attacks. [6][7]
Epidemiology & Risk Factors
Familial Mediterranean fever is the most prevalent monogenic autoinflammatory disease worldwide, with the highest carrier rates in the eastern Mediterranean, where up to one in five people in some Armenian, Turkish and Jewish communities carry a single MEFV mutation. The disease shows itself in childhood or early adulthood, and roughly two-thirds of affected children declare their first attack before the age of ten years. The risk that the fellow must not miss is the AA amyloidosis, which in the pre-colchicine era caused renal failure in a large fraction of untreated patients, particularly those with the more severe M694V homozygous genotype. [4]
PFAPA is the most common periodic fever syndrome in children presenting to general paediatric clinics, with an estimated incidence of around two per ten thousand children under five. It begins before the age of five years in the large majority, with a slight male predominance, and it has no clear ethnic or genetic concentration. The natural history is favourable, with spontaneous resolution over years in most children, so the prognosis is excellent and the reassurance of the family is part of the treatment. [7]
Mevalonate kinase deficiency is rare, with an estimated prevalence that clusters in the Dutch and other northern European populations from a founder effect, though it is reported worldwide. The long-term follow-up of one hundred and three patients published by van der Hilst and colleagues showed a wide severity spectrum, from mild recurrent attacks to a severe neurological and growth-limiting disease, and the amyloidosis risk is lower than in familial Mediterranean fever but not absent. [11]
The tumour necrosis factor receptor-associated periodic syndrome and the cryopyrin-associated periodic syndromes are rare, with the latter carrying a spectrum of severity that tracks with the mutation. The Muckle-Wells form of the cryopyrin spectrum adds progressive sensorineural deafness and AA amyloidosis, while the neonatal-onset multisystem inflammatory disease adds a chronic aseptic meningitis, a characteristic arthropathy, and intellectual disability. The risk that defines these syndromes is that untreated disease causes irreversible organ damage, which is why early interleukin-one blockade changes the outcome. [2][9]
Pathophysiology
The pathophysiology of the periodic fever syndromes is the inflammasome, and the candidate who holds the inflammasome holds the whole topic. The inflammasome is a multiprotein complex inside the innate immune cells, the monocytes and macrophages, that senses danger and activates caspase-one. The activated caspase-one cleaves pro-interleukin-one-beta into its active form, releasing interleukin-one-beta, the master cytokine of fever and inflammation. Interleukin-one-beta drives the fever through the hypothalamic prostaglandin pathway, activates the endothelium, and triggers the acute-phase response that raises the C-reactive protein. When the inflammasome is over-active, this whole cascade fires without an infection, and the child runs an inflammatory attack that stops only when the cytokine surge exhausts itself. [5][2]
Each hereditary syndrome traces to a single gene that feeds this cascade. In the cryopyrin-associated periodic syndromes, the fault is in the NLRP3 gene itself, which encodes cryopyrin, the scaffold protein that builds the inflammasome. A gain-of-function mutation keeps the inflammasome assembled and firing, so the interleukin-one-beta pours out, which is why the interleukin-one blockade produces a dramatic response. In familial Mediterranean fever, the MEFV gene encodes pyrin, a related scaffold, and its mutation produces a pyrin inflammasome that fires recurrently, driving the serositis attacks and the long-term amyloidosis. [1][2]
Mevalonate kinase deficiency sits a step upstream. The MVK gene encodes an enzyme in the cholesterol pathway, and its loss causes a build-up of mevalonic acid and a shortage of the geranylgeranyl products that the cell needs to regulate the inflammasome. The shortage removes a brake, the inflammasome fires, and the interleukin-one-beta rises, which is the same final pathway and the reason the interleukin-one blockade works here too. The tumour necrosis factor receptor-associated periodic syndrome is mechanistically distinct, because the TNFRSF1A mutation impairs the shedding of the tumour necrosis factor receptor, so the unbound receptor accumulates and the tumour necrosis factor signalling becomes dysregulated, though the interleukin-one pathway is secondarily involved and the interleukin-one blockade still helps. [2][9]
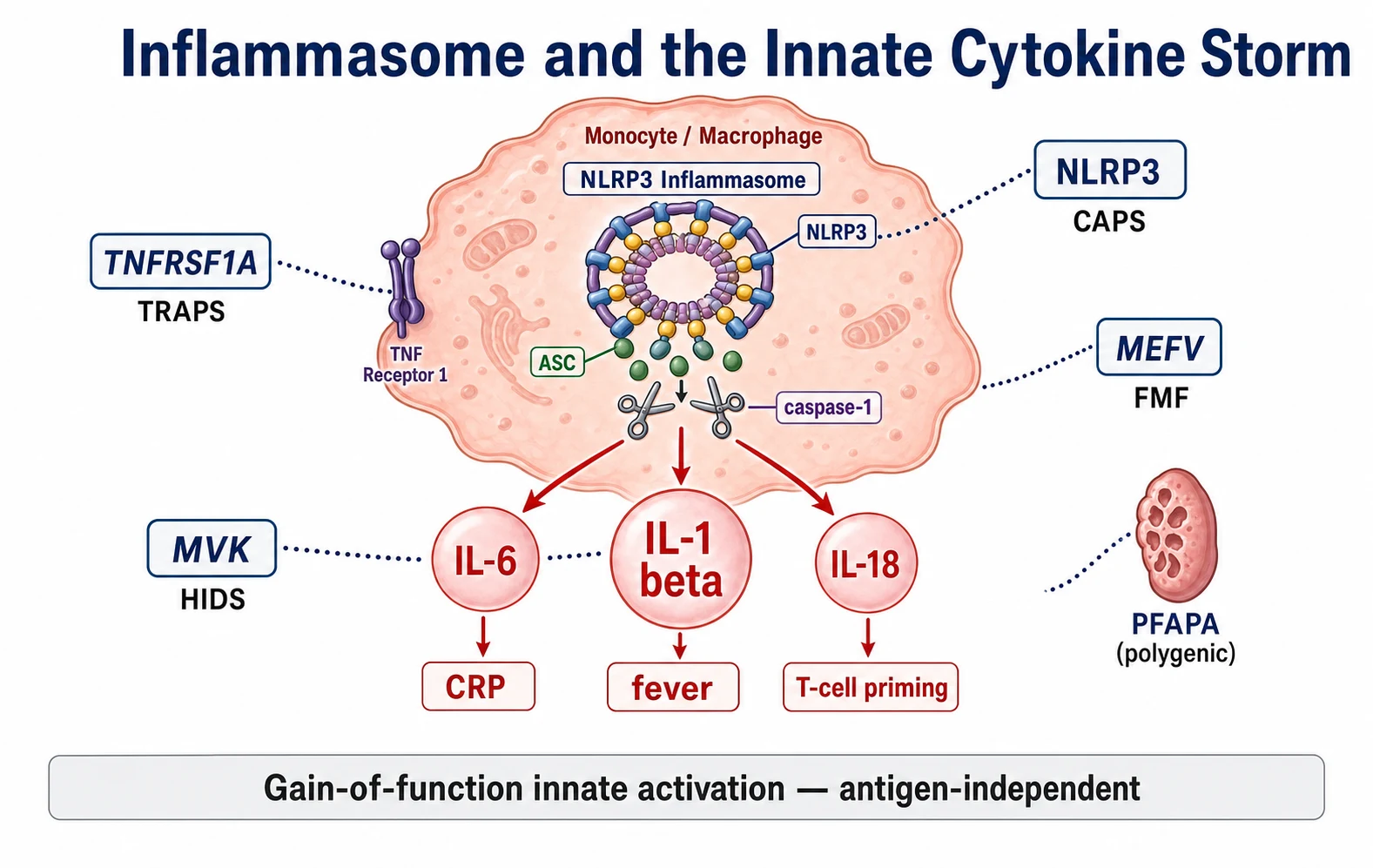
PFAPA is the outlier, because it is not monogenic and its mechanism is centred on the tonsil. Stojanov and colleagues showed that the PFAPA attack is a disorder of innate immunity and T-helper-one activation, with a surge of interleukin-one-beta and interleukin-eighteen during the attack, and that the interleukin-one blockade can abort it. The tonsil appears to be the source of the dysregulated cytokines, which is the biological explanation for why the tonsillectomy cures the syndrome in the refractory cases. The complete quiescence between attacks, with the normal acute-phase response, is the feature that sets PFAPA apart from the chronic inflammation of the other syndromes. [5]
Clinical Presentation
The clinical presentation of the periodic fever syndromes is a child with recurrent fever, and the discriminating features are the pattern of the fever and the features that accompany it. Begin with the fever itself, because the interval and the duration are the most powerful clues. A clockwork interval, with attacks coming every three to eight weeks and lasting three to six days, in a child who is entirely well and growing normally between them, is the signature of PFAPA. Short attacks of one to three days are familial Mediterranean fever. Longer attacks of weeks are the tumour necrosis factor receptor-associated periodic syndrome. Attacks from infancy, triggered by vaccination and lasting three to seven days, are mevalonate kinase deficiency. [7][1]
The features that accompany the fever complete the picture. Familial Mediterranean fever presents with fever and serositis, so the child has an acute abdominal pain that mimics appendicitis, a pleuritic chest pain, or a monoarthritis, often with the erysipelas-like rash on the shin. The attack resolves completely within three days, and the child is well between attacks. A family history of recurrent abdominal pain or kidney failure in a Mediterranean family is a powerful lead. The arthritis of familial Mediterranean fever is a distinctive non-erosive monoarthritis, often of the knee or hip, that resolves without damage. [3][4]
Mevalonate kinase deficiency presents in the first year of life with a fever that is accompanied by a maculopapular rash, a generalised lymphadenopathy, an abdominal pain with diarrhoea and vomiting, oral ulcers, and splenomegaly. The attacks are often triggered by a vaccination or a minor infection, and the trigger history is worth asking for directly. The cryopyrin-associated periodic syndromes present with a urticaria-like rash that is not histamine-mediated and does not itch like true urticaria, a fever, and a conjunctivitis, and the mildest form is triggered by cold. The Muckle-Wells form adds a progressive sensorineural deafness and a secondary amyloidosis, while the severe neonatal form adds a chronic aseptic meningitis, an arthropathy, and developmental delay. [11][2]
PFAPA presents with the clockwork interval and the triad. The fever is high, often over thirty-nine degrees, and it is accompanied by a pharyngitis with an inflamed throat, aphthous ulcers in the mouth, and tender cervical adenitis. The child looks miserable during the attack and entirely well between, with normal growth and development, and the parents can usually predict the next attack to the week. The attack does not respond to antibiotics, which is a useful negative, and a single dose of corticosteroid at the start of the attack aborts it within hours in most children. [7][5]
Differential Diagnosis
The first differential to clear is recurrent infection, because it is the most common cause of recurrent fever in a child and the most common misdiagnosis applied to the periodic fever syndromes. A child who is genuinely having one infection after another will not have a clockwork interval, will not be stereotyped in the features, and will usually have respiratory or gastrointestinal symptoms that point to the organism. The clockwork regularity, the stereotyped attack, and the complete wellness between episodes are the features that move the clinician away from infection and toward a periodic fever syndrome. [7]
The second differential is malignancy, because a child with a lymphoma or a leukaemia can present with recurrent fever, and a missed malignancy is a catastrophic error. The features that point to malignancy are the cytopenias, the persistent organomegaly, the weight loss, and the failure to return to complete wellness between the febrile episodes. A full blood count with a film, a lactate dehydrogenase, and a careful examination for lymphadenopathy and organomegaly are the first steps to exclude it, and any cytopenia or persistent finding warrants escalation to a bone marrow examination. [1]
The third group is the systemic inflammatory diseases. Systemic juvenile idiopathic arthritis presents with a quotidian fever that differs from the clockwork interval of the periodic fever syndromes, and it carries the evanescent rash and the arthritis that declare it. The macrophage activation syndrome and the haemophagocytic lymphohistiocytosis present with a persistent fever, splenomegaly, cytopenias and a very high ferritin, and they are the emergencies that must not be missed, because they progress to multi-organ failure within days. The inflammatory bowel disease and the Behcet disease can mimic the periodic fever syndromes with their oral ulcers and abdominal pain, but they declare themselves over time with their distinctive features. [1]
Cyclic neutropenia is the one mimic that is easy to miss and important to find, because it is a separate disease with a specific treatment. It is caused by mutations in the ELANE gene, and it produces a fever and an infectious episode every twenty-one days as the neutrophil count falls to its nadir. The discriminating feature is the twenty-one-day cycle and the infectious nature of the episodes, with mouth ulcers and a documented neutropenia at the nadir. A serial full blood count three times a week for six weeks will reveal the cyclic oscillation and separate cyclic neutropenia from the periodic fever syndromes. [1]
Clinical & Bedside Assessment
Begin the assessment with a fever diary, because the pattern of the fever is the single most powerful diagnostic tool, and no other investigation comes close. Ask the parents to record the dates, the peak temperatures, the duration, and the features of each attack over at least two or three cycles, and to note whether the child returns to complete wellness between them. The diary reveals the clockwork interval that defines PFAPA, the short attacks of familial Mediterranean fever, and the long attacks of the tumour necrosis factor receptor-associated periodic syndrome. A diary kept across at least two suspected PFAPA attacks is part of the diagnostic criteria, not an optional extra. [7]
Take the family history with purpose, because the hereditary periodic fevers are genetic and the family tree often carries the answer. Ask specifically about recurrent abdominal pain, kidney failure, recurrent fever, arthritis, deafness, and consanguinity, and ask about the ethnic origin, because the Mediterranean and the founder-population backgrounds raise the probability of familial Mediterranean fever and mevalonate kinase deficiency. A family history of a relative on dialysis from amyloidosis in a Mediterranean family is familial Mediterranean fever until proven otherwise. [3][4]
Examine the child during an attack if it is possible, because the attack features are the phenotype that sorts the syndromes. Look for the serositis of familial Mediterranean fever, the rash and the lymphadenopathy of mevalonate kinase deficiency, the periorbital oedema and the migratory rash of the tumour necrosis factor receptor-associated periodic syndrome, and the urticaria-like rash and the conjunctivitis of the cryopyrin-associated periodic syndromes. Examine the throat and the mouth for the aphthae and the pharyngitis of PFAPA, and the cervical nodes for the adenitis. Between attacks, examine for the organomegaly that points away from a periodic fever syndrome and toward a malignancy or a haemophagocytic disorder. [1][11]
Document the growth and the development, because the periodic fever syndromes, with the exception of the severe end of the cryopyrin spectrum, allow normal growth between the attacks, and a child who is failing to thrive is more likely to have a chronic inflammatory disease, a malignancy, or an immunodeficiency. The measurement of the growth over time is a cheap and powerful discriminator, and a child with a normal growth trajectory who is entirely well between attacks is the one who most likely has PFAPA or a hereditary periodic fever. [7]
Investigations
The investigations serve two purposes, to document the inflammation during the attack and to confirm the syndrome by its gene. Draw the acute-phase markers during an attack, because the raised C-reactive protein and serum amyloid A during the attack, with their return to normal between the attacks, is the signature of a periodic fever syndrome and a powerful point against a chronic inflammation or a malignancy. A single set of normal inflammatory markers between attacks does not exclude the diagnosis, and the markers must be paired, high during and normal between, to carry their full weight. [1][7]
The targeted syndrome-specific tests confirm the phenotype. For familial Mediterranean fever, apply the Livneh clinical criteria, which combine the typical attack features with the ethnic predisposition and the family history, and the colchicine response supports the diagnosis. For mevalonate kinase deficiency, measure the serum immunoglobulin D and immunoglobulin A, and the urinary mevalonic acid during an attack, while bearing in mind that the immunoglobulin D is neither sensitive nor specific on its own. For the cryopyrin-associated periodic syndromes, the audiology and the ophthalmology assessment and the magnetic resonance imaging of the brain define the severity along the spectrum. [3][11]
The gene panel is the confirmatory test for the hereditary syndromes, and it should be sent as a targeted panel of the MEFV, MVK, TNFRSF1A and NLRP3 genes, guided by the phenotype. Send the panel after the clinical phenotype is established, not as a fishing expedition, because a variant of uncertain significance in a gene that does not match the phenotype will create more confusion than clarity. The Gattorno 2019 Eurofever classification criteria, validated in the independent cohort published by Dingulu and colleagues in 2020, allow the hereditary recurrent fevers to be classified on the clinical features alone when the genetics are equivocal. [1][10]
PFAPA has no confirmatory laboratory test, and this is a point the examiner will test. The diagnosis is clinical, made after the hereditary periodic fevers and the cyclic neutropenia are excluded, supported by the fever diary and the rise and fall of the C-reactive protein. A single dose of corticosteroid that aborts the attack within hours is a supporting feature, and the tonsillectomy that cures the refractory case is both a treatment and a retrospective confirmation. The mistake to avoid is to label a child with PFAPA before the hereditary syndromes are excluded, because the corticosteroid will mask and delay the diagnosis of a monogenic disease. [7][6]
Management — Resuscitation
The periodic fever syndromes rarely require resuscitation, because the attacks are self-limited and the child is well between them, but two emergencies demand an immediate response. The first is the acute severe attack that mimics a surgical abdomen, most often the peritonitis of familial Mediterranean fever, which can be indistinguishable from appendicitis at the first presentation. The management is analgesia and careful observation, with the surgical review if the diagnosis is not yet established, and the realisation that the attack will resolve within three days once the syndrome is recognised. [3]
The second is the sepsis risk in the child on biologic therapy, because the interleukin-one blockade and the tumour-necrosis-factor blockade impair the innate response to infection. A febrile child on a biologic must be assessed for sepsis without delay, with the empiric broad-spectrum antibiotics given after the cultures while the autoinflammatory flare and the infection are distinguished. Never assume that a fever in a child on a biologic is an autoinflammatory attack until the sepsis has been excluded, because the consequence of a missed sepsis is far worse than a brief course of unnecessary antibiotics. [9]
The AA amyloidosis of familial Mediterranean fever is the third emergency, and it is the one that the long-term management must prevent rather than resuscitate. A child with familial Mediterranean fever who develops new proteinuria or a rising creatinine has amyloidosis until proven otherwise, and the response is the urgent reinforcement of the colchicine adherence, the assessment of the renal function, and the referral to nephrology. The colchicine prevents the amyloidosis, and the adherence to it is the single most important determinant of the long-term renal outcome. [4]
Management — Definitive & Stepwise
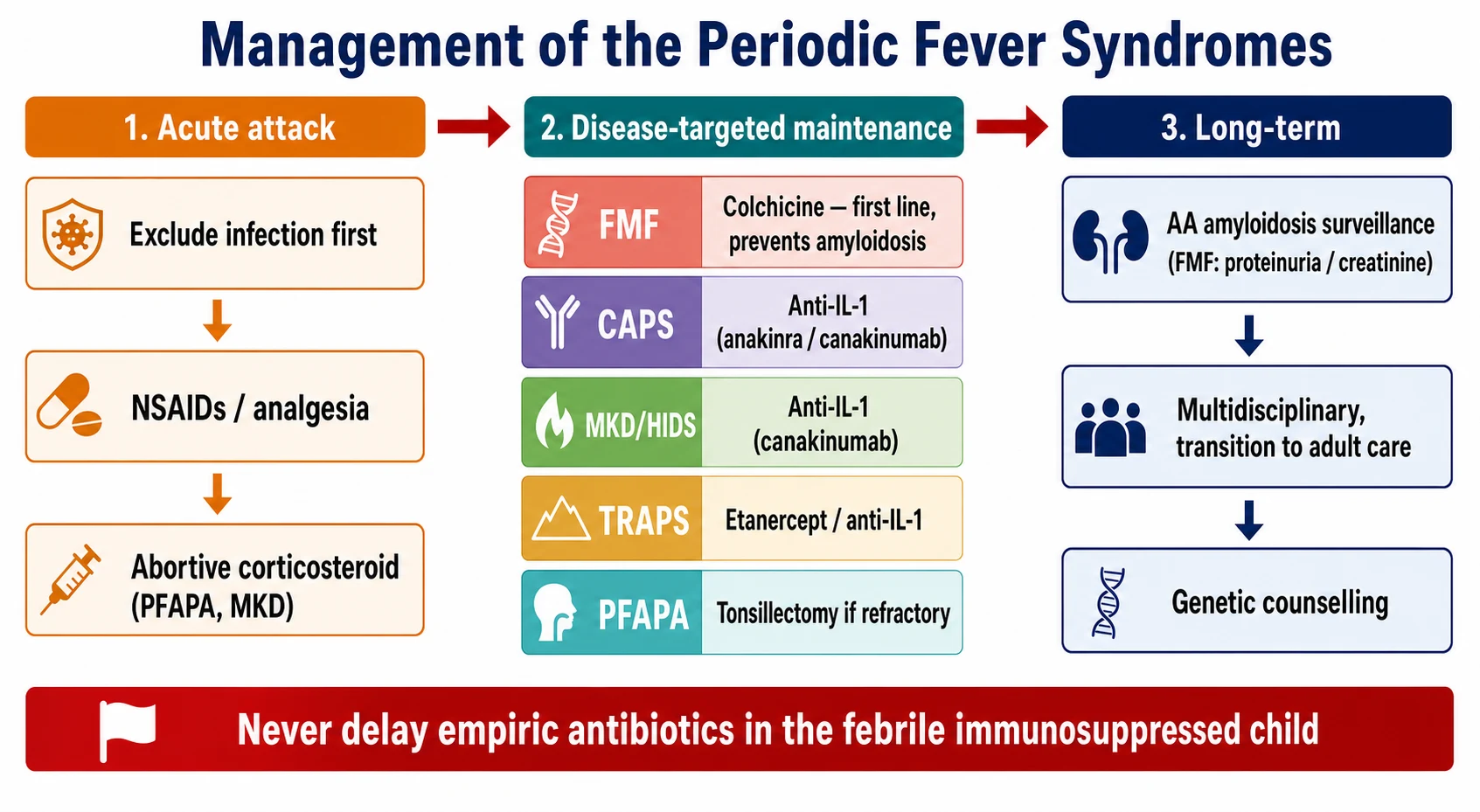
The definitive management of familial Mediterranean fever is colchicine, and it is the one treatment in this topic that every fellow must know. Colchicine is the first-line therapy, it reduces both the frequency and the severity of the attacks, and it is the only treatment proven to prevent the AA amyloidosis. The EULAR recommendations for the management of familial Mediterranean fever, published by Ozen and colleagues in 2016, establish colchicine as the cornerstone, with the dose titrated to the response, and the adherence to it sustained for life. The child who does not tolerate or respond to colchicine moves to the interleukin-one blockade, which is effective for the colchicine-resistant disease. [4][9]
Colchicine
Dose
Titrated to response, typically a maintenance of one to two milligrams daily in divided doses
The cryopyrin-associated periodic syndromes and mevalonate kinase deficiency are treated with the interleukin-one blockade, because the interleukin-one-beta is the final mediator of both, and the response is dramatic. The canakinumab, a monoclonal antibody to the interleukin-one-beta, was demonstrated in the cluster randomised trial published by De Benedetti and colleagues in 2018 to produce a sustained response across the autoinflammatory recurrent fever syndromes, including the cryopyrin-associated periodic syndromes, the mevalonate kinase deficiency, and the colchicine-resistant familial Mediterranean fever. The anakinra, the interleukin-one receptor antagonist, is the alternative, and it is fast, titratable and useful for the acute attack and the diagnostic confirmation. [8][2]
The tumour necrosis factor receptor-associated periodic syndrome is treated with the etanercept, the soluble tumour necrosis factor receptor, because the fault is in the tumour necrosis factor pathway, and the interleukin-one blockade is added for the refractory disease. The PFAPA is managed differently, because it is not monogenic and it resolves spontaneously. The abortive management is a single dose of corticosteroid at the very start of the attack, which aborts it within hours in most children, and the tonsillectomy is reserved for the severe and refractory cases. The natural history is one of spontaneous resolution over years, and the reassurance of this favourable prognosis is part of the treatment. [9][5]
The stepwise management of a suspected periodic fever syndrome
Confirm the clockwork pattern and the wellness between attacks with a fever diary across at least two cycles
Exclude infection, malignancy and the haemophagocytic disorders with the acute-phase markers, the full blood count and the film
Apply the Livneh criteria for familial Mediterranean fever and the Gattorno Eurofever criteria for the hereditary recurrent fevers
Send the targeted gene panel of MEFV, MVK, TNFRSF1A and NLRP3 guided by the phenotype
Start the pathway-targeted therapy, colchicine for familial Mediterranean fever and interleukin-one blockade for the cryopyrin-associated periodic syndromes and mevalonate kinase deficiency
Arrange the amyloidosis surveillance for familial Mediterranean fever and the multidisciplinary and transition planning for the lifelong disease
Specific Subtypes & Scenarios
The severe end of the cryopyrin-associated periodic spectrum, the neonatal-onset multisystem inflammatory disease or CINCA, is the subtype that demands the most aggressive and the earliest treatment. It presents in the first weeks of life with a chronic urticaria-like rash, a chronic aseptic meningitis, a characteristic arthropathy of the knees and the elbows, and a developmental delay. Untreated, it causes a progressive neurological damage and a severe joint destruction, and the early interleukin-one blockade changes the outcome. The fellow must recognise that a chronically unwell infant with a persistent rash and a developmental delay has a cryopyrin spectrum disease, not eczema or a chronic infection, and escalate to the paediatric rheumatologist without delay. [2]
The colchicine-resistant familial Mediterranean fever is the scenario that tests the fellow's knowledge of the modern management. Roughly five to ten percent of the patients with familial Mediterranean fever do not respond adequately to the colchicine, and they are at the highest risk of the amyloidosis. The management is the addition of the interleukin-one blockade, with the canakinumab demonstrated in the randomised trial to control the colchicine-resistant disease, and the ongoing amyloidosis surveillance with the regular urinalysis for the proteinuria and the serum creatinine. The adherence to the colchicine must be checked before the disease is declared resistant, because the non-adherence is the most common cause of the apparent failure. [4][8]
The child with PFAPA who is offered the tonsillectomy is the scenario that requires the careful counselling. The tonsillectomy is effective for the refractory PFAPA, with the evidence from the randomised trials, but it is not without the surgical risk, and it is reserved for the children whose attacks are frequent, severe, and disruptive to the family life and the schooling despite the corticosteroid. The fellow must weigh the natural history of the spontaneous resolution against the immediate benefit of the surgery, and present the options honestly to the family. The tonsillectomy is the treatment of the refractory disease, not the first-line, and the corticosteroid-abort strategy is tried first. [9][7]
Complications & Pitfalls
The AA amyloidosis is the defining complication of the untreated familial Mediterranean fever, and it is the reason the colchicine is lifelong and non-negotiable. The amyloid deposits in the kidneys, the gut, and the heart, and it presents first as a proteinuria and a falling creatinine clearance, progressing to the end-stage renal failure. The amyloidosis is preventable with the colchicine, and the fellow who understands this understands the central importance of the adherence. The serum amyloid A can be used to monitor the inflammatory burden and to titrate the colchicine, and a persistently raised serum amyloid A between the attacks is a warning that the disease is not controlled and the amyloidosis risk is rising. [4]
The irreversible organ damage of the untreated cryopyrin-associated periodic syndromes is the second defining complication. The sensorineural deafness of the Muckle-Wells syndrome, the chronic meningitis and the developmental delay of the neonatal form, and the amyloidosis are all preventable with the early interleukin-one blockade. The fellow who delays the diagnosis of a cryopyrin spectrum disease in an infant allows an irreversible damage to accumulate, and the recognition of the neutrophilic urticaria plus fever in the first months of life is the bedside skill that prevents it. [2]
What is the single most common pitfall in the diagnosis of the periodic fever syndromes?
The mislabelling of a monogenic syndrome as PFAPA, because the corticosteroid that aborts a PFAPA attack will also transiently quieten a hereditary periodic fever and mask its phenotype. The defence is to exclude the hereditary syndromes, with the fever diary, the targeted gene panel, and the ethnic and family history, before the diagnosis of PFAPA is made. PFAPA is a diagnosis of exclusion, supported by the complete wellness between the attacks and the normal growth.
[7][1]The steroid masking is the pitfall that links to the misdiagnosis. A child with a hereditary periodic fever who is given the corticosteroid for a presumed PFAPA may improve, but the underlying gene defect is untreated, the amyloidosis risk of familial Mediterranean fever is unchecked, and the diagnosis is delayed. The fellow must hold the rule that the corticosteroid-abort is tried only after the hereditary syndromes are excluded, and that a response to the corticosteroid does not confirm the PFAPA. [7]
Prognosis & Disposition
The prognosis of the periodic fever syndromes is, with two important exceptions, excellent, because the treatments are effective and the child returns to a normal life. Familial Mediterranean fever is controlled with the colchicine in over ninety percent of the patients, and the amyloidosis is preventable with the adherence. The mevalonate kinase deficiency and the tumour necrosis factor receptor-associated periodic syndrome are controlled with the biologic therapy, and the cryopyrin-associated periodic syndromes respond dramatically to the interleukin-one blockade. The two exceptions are the amyloidosis of the untreated disease and the irreversible organ damage of the severe cryopyrin spectrum, and both are preventable with the early and the sustained treatment. [4][8]
PFAPA has the most favourable prognosis of the group, because it resolves spontaneously over years in most children, with no long-term sequelae and no amyloidosis. The attacks become less frequent and less severe over time, and they typically resolve before or during adolescence. The family can be reassured that the child will outgrow the syndrome, and the role of the paediatrician is to manage the attacks in the meantime and to exclude the hereditary syndromes. [7]
The disposition is shared between the general paediatrician and the specialist. The general paediatrician confirms the clockwork pattern, excludes the mimics, and starts the pathway-targeted therapy in consultation with the paediatric rheumatologist or the clinical immunologist. The specialist centre confirms the gene, titrates the colchicine or the biologic, and coordinates the amyloidosis surveillance and the transition. The child with a cryopyrin spectrum disease is managed in a tertiary centre with the paediatric rheumatology, the ophthalmology, the audiology and the neurology, because the multisystem involvement demands the coordinated care. [2][9]
Special Populations
The migrant and the refugee families from the Mediterranean and the Middle East carry the highest prevalence of familial Mediterranean fever, and the fellow must hold a high index of suspicion in any child of the Armenian, Turkish, Sephardic Jewish, or Arab background who presents with the recurrent abdominal pain or the serositis. The cultural and the language considerations matter, because the adherence to the lifelong colchicine depends on the family's understanding, and the interpreter and the culturally appropriate education are part of the standard of care. The consanguinity in some of these communities raises the probability of the autosomal recessive syndromes and warrants the explicit family history. [4]
The adolescent and the young person in transition face the twin challenges of the adherence and the reproductive planning. The adherence to the colchicine often falters in the adolescence, and the fellow must anticipate this and build the transition plan early, with the education of the young person about the amyloidosis risk and the consequences of the non-adherence. The genetic counselling is part of the transition, because the hereditary periodic fevers carry the reproductive implications, and the young woman with familial Mediterranean fever needs to know the inheritance and the option of the genetic testing of her partner. The pregnancy in the woman on the colchicine is manageable, and the fellow must coordinate the rheumatology and the obstetric care. [4]
The indigenous children of the Aboriginal, Torres Strait Islander, Maori and Pacific peoples may carry a higher burden of the recurrent infection and the post-streptococcal disease that can mimic the periodic fever syndromes, and the fellow must distinguish the recurrent infection from the autoinflammatory pattern with the fever diary and the acute-phase markers. The access to the specialist care may be limited in the rural and the remote settings, and the telehealth and the shared care with the regional centre are part of the standard of care. [9]
Evidence, Guidelines & Regional Differences
The evidence base for the periodic fever syndromes has matured rapidly over the last decade. The Gattorno 2019 Eurofever and PRINTO classification criteria formalised the hereditary recurrent fevers, and the validation by Dingulu and colleagues in 2020 confirmed their performance in an independent cohort. The Livneh 1997 criteria remain the standard for the clinical diagnosis of familial Mediterranean fever, and the EULAR 2016 management recommendations established the colchicine as the cornerstone. The Romano 2022 EULAR and American College of Rheumatology points to consider set out the diagnosis and the management of the interleukin-one-mediated diseases, and the De Benedetti 2018 trial demonstrated the canakinumab across the cluster of the autoinflammatory recurrent fever syndromes. [1][2][8]
The Australian and Aotearoa New Zealand practice concentrates the specialist care in the tertiary children's hospitals, with the paediatric rheumatology and the clinical immunology shared in the multidisciplinary clinic. The access to the biologics is subsidised through the Pharmaceutical Benefits Scheme or Pharmac, but it usually requires the specialist authorisation and the genetic confirmation. The migrant and the refugee families from the Mediterranean and the Middle East are the highest-prevalence group for familial Mediterranean fever, and the culturally safe care is mandated. [4][9]
The regional differences are concordant in the principles, with the colchicine for familial Mediterranean fever and the interleukin-one blockade for the cryopyrin-associated periodic syndromes and mevalonate kinase deficiency held universally. The differences lie in the funding and the access to the biologics, the genetic testing infrastructure, and the organisation of the specialist care. The EULAR, the PRINTO and the Eurofever collaboration drive the classification and the care standards, and the ANZ and the UK practice is broadly concordant with the European framework. [2][9]
Exam Pearls
FEVER
The Gattorno 2019 Eurofever classification criteria are the modern standard for the hereditary recurrent fevers, and the fellow who reproduces the principle that the criteria separate the syndromes on the discriminating clinical features carries the core of the examination. The criteria are built to be applied after the clinical phenotype is established, not as a fishing expedition, and they are validated by the independent cohort. The Livneh 1997 criteria for familial Mediterranean fever remain the standard for the clinical diagnosis, and the colchicine response supports it. [1][10]
The colchicine-first principle for familial Mediterranean fever is the examination favourite, because it tests the understanding of the amyloidosis prevention. The colchicine is the only treatment proven to prevent the AA amyloidosis, and it is the cornerstone of the lifelong management. The fellow who carries the principle that the adherence to the colchicine is the single most important determinant of the long-term renal outcome demonstrates the understanding of the modern management that the examination rewards. [4]
References
- [1]Gattorno M, Hofer M, Federici S, et al Classification criteria for autoinflammatory recurrent fevers Ann Rheum Dis, 2019.PMID 31018962
- [2]Romano M, Arici ZS, Piskin D, et al The 2021 EULAR/American College of Rheumatology points to consider for diagnosis, management and monitoring of the interleukin-1 mediated autoinflammatory diseases Ann Rheum Dis, 2022.PMID 35623638
- [3]Livneh A, Langevitz P, Zemer D, et al Criteria for the diagnosis of familial Mediterranean fever Arthritis Rheum, 1997.PMID 9336425
- [4]Ozen S, Demirkaya E, Erer B, et al EULAR recommendations for the management of familial Mediterranean fever Ann Rheum Dis, 2016.PMID 26802180
- [5]Stojanov S, Lapidus S, Chitkara P, et al Periodic fever, aphthous stomatitis, pharyngitis, and adenitis (PFAPA) is a disorder of innate immunity and Th1 activation responsive to IL-1 blockade Proc Natl Acad Sci U S A, 2011.PMID 21478439
- [6]Marshall GS, Edwards KM, Butler J, et al Syndrome of periodic fever, pharyngitis, and aphthous stomatitis J Pediatr, 1987.PMID 3794885
- [7]Thomas KT, Feder HM Jr, Lawton AR, et al Periodic fever syndrome in children J Pediatr, 1999.PMID 10393598
- [8]De Benedetti F, Gattorno M, Anton J, et al Canakinumab for the Treatment of Autoinflammatory Recurrent Fever Syndromes N Engl J Med, 2018.PMID 29768139
- [9]Ter Haar N, Lachmann H, Ozen S, et al Treatment of autoinflammatory diseases: results from the Eurofever Registry and a literature review Ann Rheum Dis, 2013.PMID 22753383
- [10]Dingulu G, Georgin-Lavialle S, Kone-Paut I, et al Validation of the new classification criteria for hereditary recurrent fever in an independent cohort Rheumatology (Oxford), 2020.PMID 32125423
- [11]van der Hilst JCH, Bodar EJ, Barron KS, et al Long-term follow-up, clinical features, and quality of life in a series of 103 patients with hyperimmunoglobulinemia D syndrome Medicine (Baltimore), 2008.PMID 19011501