Paeds · rheumatology-musculoskeletal-and-sports
Primary immunodeficiency with rheumatic manifestations
Also known as Rheumatic disease in inborn errors of immunity · Autoimmunity in primary immunodeficiency · CVID arthritis · Immune dysregulation syndromes with rheumatic features
A fellowship approach to the child in whom a primary immunodeficiency declares itself through rheumatic disease: recognise the overlaps that earn an immune work-up (CVID with symmetric polyarthritis and autoimmune cytopenia, chronic granulomatous disease with Crohn-like colitis, classical complement deficiency with childhood-onset lupus, and immune dysregulation syndromes), investigate the immune defect alongside the rheumatic phenotype, and treat both arms in parallel without letting the immunosuppression for the joints blind you to the infection behind it.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
Picture a six-year-old girl labelled with juvenile idiopathic arthritis because of a symmetric polyarthritis that has swollen both wrists and knees for three months. She has also had two pneumonias this year, chronic diarrhoea, and a recent bruising episode from a low platelet count. Her immunoglobulins come back low. Two diagnoses live inside that one child. Through one window she has ordinary juvenile idiopathic arthritis heading for methotrexate and a biologic. Through the other she has common variable immunodeficiency, where the arthritis and the autoimmune cytopenia are rheumatic fingerprints of a failing B-cell system, and the methotrexate will suppress an immune system that is already broken. Distinguishing those two children — and the CGD colitis from Crohn disease, and the complement-deficient lupus from ordinary lupus — is the whole skill this topic tests. [7] [8] [3]
R.H.E.U.M.A.T.I.C.
Overview & Definition
A primary immunodeficiency is an inborn error of immunity, and the rheumatic manifestations are the autoimmune, inflammatory, or granulomatous disease that some of these errors produce alongside their infection susceptibility. The two halves are joined at the root: the immune system exists to defend against pathogens and to tolerate the self, and a defect that weakens one arm often disturbs the other. Köstel Bal and colleagues framed the field precisely this way, describing rheumatological manifestations as a core feature of inborn errors of immunity rather than a coincidence. [3]
The reason this matters at the bedside is pattern recognition. A general paediatrician or rheumatology trainee who meets arthritis, lupus, vasculitis, or autoimmune cytopenia will reach for the standard rheumatic work-up, and in most children that is correct. The trap is the child in whom the rheumatic disease sits beside recurrent, persistent, severe, or unusual infection, because that child has a failing immune system and the standard immunosuppression may do harm. The defining task is to recognise the overlap and to quantify the immune defect before committing to a purely rheumatic label. [1] [2]
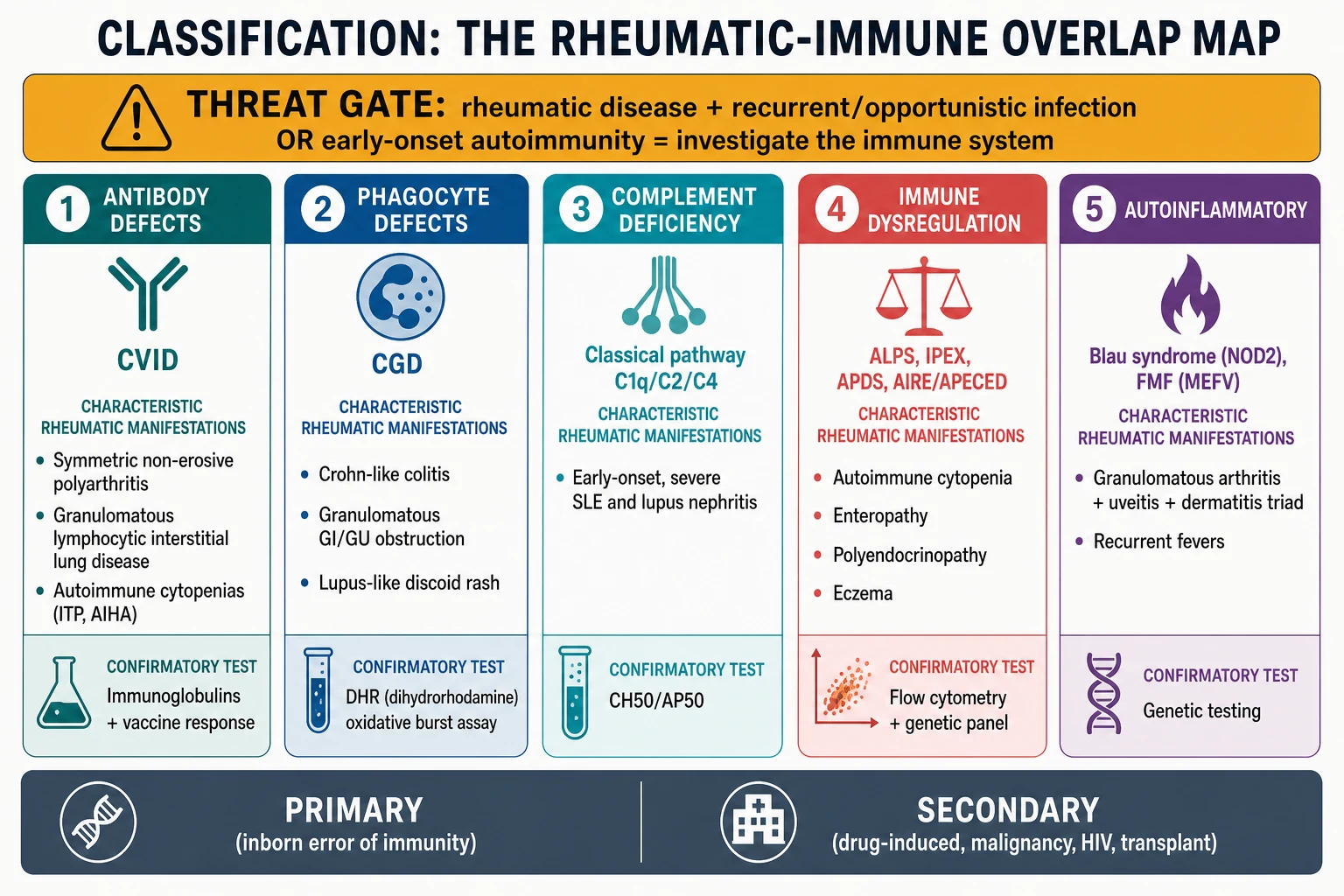
The IUIS 2022 phenotypic classification organises these conditions across the immune arms, and the rheumatic-Immune overlaps sit in several of them: predominantly antibody deficiencies, combined immunodeficiencies with syndromic features, diseases of immune dysregulation, congenital defects of phagocyte function, and complement deficiencies. A fellowship answer names the arm, the prototype disease, its rheumatic fingerprint, and the test that confirms it. [1]
Classification
Sort the rheumatic-immune overlaps by the immune arm at fault, because that single axis predicts the rheumatic phenotype, the discriminating test, and the management. The 2022 IUIS classification supplies the framework for naming each group. [1]
Antibody defects lead the list. Common variable immunodeficiency is the commonest symptomatic primary antibody deficiency, and a substantial minority of patients develop autoimmune or inflammatory disease — immune thrombocytopenia, autoimmune haemolytic anaemia, a symmetric non-erosive polyarthritis that mimics juvenile idiopathic arthritis, and granulomatous disease of the lung, lymph node, or spleen. The ESID registry burden study documented how often these non-infectious complications complicate CVID. [7] [8]
Phagocyte defects are dominated by chronic granulomatous disease. Here the rheumatic fingerprint is granulomatous rather than classical autoimmune: a Crohn-like colitis, gastric-outlet obstruction, genitourinary granulomatous obstruction, and occasionally a lupus-like discoid dermatitis. Because the failing NADPH oxidase produces dysregulated granulomatous inflammation on top of infection susceptibility, CGD sits naturally in a rheumatic-immune overlap topic. [4] [5]
Complement deficiencies of the early classical pathway produce lupus. Loss of C1q, C2, or C4 impairs clearance of apoptotic debris and immune complexes, and the un-cleared nuclear material drives antinuclear antibodies and childhood-onset systemic lupus erythematosus, often severe and early. C1q deficiency carries the highest risk, but C2 and C4 loss also feature, and the screen is the total haemolytic complement CH50 rather than a lone C3 or C4. [9] [10]
Immune dysregulation and autoinflammatory diseases form the final group. Autoimmune lymphoproliferative syndrome, IPEX (immunodysregulation polyendocrinopathy enteropathy X-linked), and the PI3K-dysregulation syndromes produce autoimmune cytopenia, enteropathy, eczema, and endocrinopathy. Blau syndrome, from a gain-of-function NOD2 variant, is the autoinflammatory archetype: a granulomatous arthritis with uveitis and dermatitis presenting before the age of five. [11] [12]
Epidemiology & Risk Factors
The rheumatic-immune overlaps are individually rare, but they cluster in patterns a candidate should be able to recite. CVID is the commonest symptomatic primary antibody deficiency, and registry data show that roughly a quarter to a third of patients develop autoimmune or inflammatory complications over their lifetime, with immune cytopenias the single most frequent. [7]
Chronic granulomatous disease has an incidence of about one in two hundred thousand live births, and roughly two-thirds of cases are X-linked, affecting boys. The autosomal recessive forms — most often p47phox — account for the rest, so consanguinity raises the rate. The Winkelstein registry of 368 patients documented that inflammatory and granulomatous complications are frequent and that the disease affects every system the phagocyte patrols. [4]
Classical complement deficiency is rare but carries a disproportionate lupus risk. C1q deficiency is uncommon, yet the great majority of affected individuals develop systemic lupus erythematosus, usually in childhood and often severe. C2 deficiency is more common in northern European populations and carries a lower but real lupus risk. The Lintner review set out the strength of these associations across the early classical components. [9]
Blau syndrome and the immune dysregulation syndromes are very rare individually. Blau syndrome is autosomal dominant from a gain-of-function NOD2 variant, so a family history of granulomatous arthritis, uveitis, and dermatitis is a strong clue. The immune dysregulation syndromes — ALPS, IPEX, and the PI3K-dysregulation disorders — are each monogenic and frequently present in infancy or early childhood with autoimmunity that can be mistaken for ordinary rheumatic disease. [11] [12]
The strongest risk factor across all of these is a family history that joins autoimmunity to immunodeficiency. Consanguinity, early male infant death, a known inborn error of immunity, or a relative with childhood-onset lupus or granulomatous arthritis should lower the threshold to send the immune screen whenever the clinical picture is suggestive. Indigenous, refugee, and socioeconomically disadvantaged families face added barriers of late presentation and reduced access to specialist immunology, which narrows the margin for delay when an overlap exists. [3]
Pathophysiology
The unifying idea is that the immune system defends against pathogens and tolerates the self through overlapping machinery, so a defect that weakens defence often disturbs tolerance. Each immune arm fails in its own way, and the mechanism predicts the rheumatic fingerprint. [3]
In common variable immunodeficiency, the terminal differentiation of B cells into antibody-secreting plasma cells and class-switched memory cells fails, but the failure is not confined to antibody production. Defective B-cell regulation lets autoreactive clones escape, and the dysregulated T-cell help that accompanies some CVID genotypes amplifies autoimmunity. The result is immune cytopenia, a symmetric non-erosive arthritis, and granulomatous inflammation of the lung and lymphoid tissue. A critical clinical point follows from this: immunoglobulin replacement fixes the infection susceptibility but does not reliably control the autoimmune or granulomatous features, which need separate management. [7] [6]
Chronic granulomatous disease is a failure of the respiratory burst. The phagocyte NADPH oxidase cannot generate the superoxide that fuels microbe killing, so catalase-positive organisms such as Staphylococcus aureus, Burkholderia cepacia complex, Serratia marcescens, Nocardia, and Aspergillus survive inside the cell. Because the organism is not killed, macrophages accumulate and wall it off with granulomatous inflammation — and that inflammation becomes dysregulated, obstructing the gut and urinary tract and producing a Crohn-like colitis. CGD is therefore a dual disease of infection and inflammation, which is why it belongs in a rheumatic-immune overlap. [4] [5]
Classical complement deficiency produces lupus through failed clearance. C1q, C2, and C4 belong to the classical activation pathway, and C1q in particular opsonises apoptotic cells so that phagocytes can clear them quietly. When C1q is absent, apoptotic debris accumulates, exposes nuclear antigens, and drives a break in B-cell tolerance followed by antinuclear antibodies and lupus. This is why complement-deficient lupus is severe and early: the failure is upstream, at the point where the immune system normally tidies up the body's own dying cells. [9] [10]
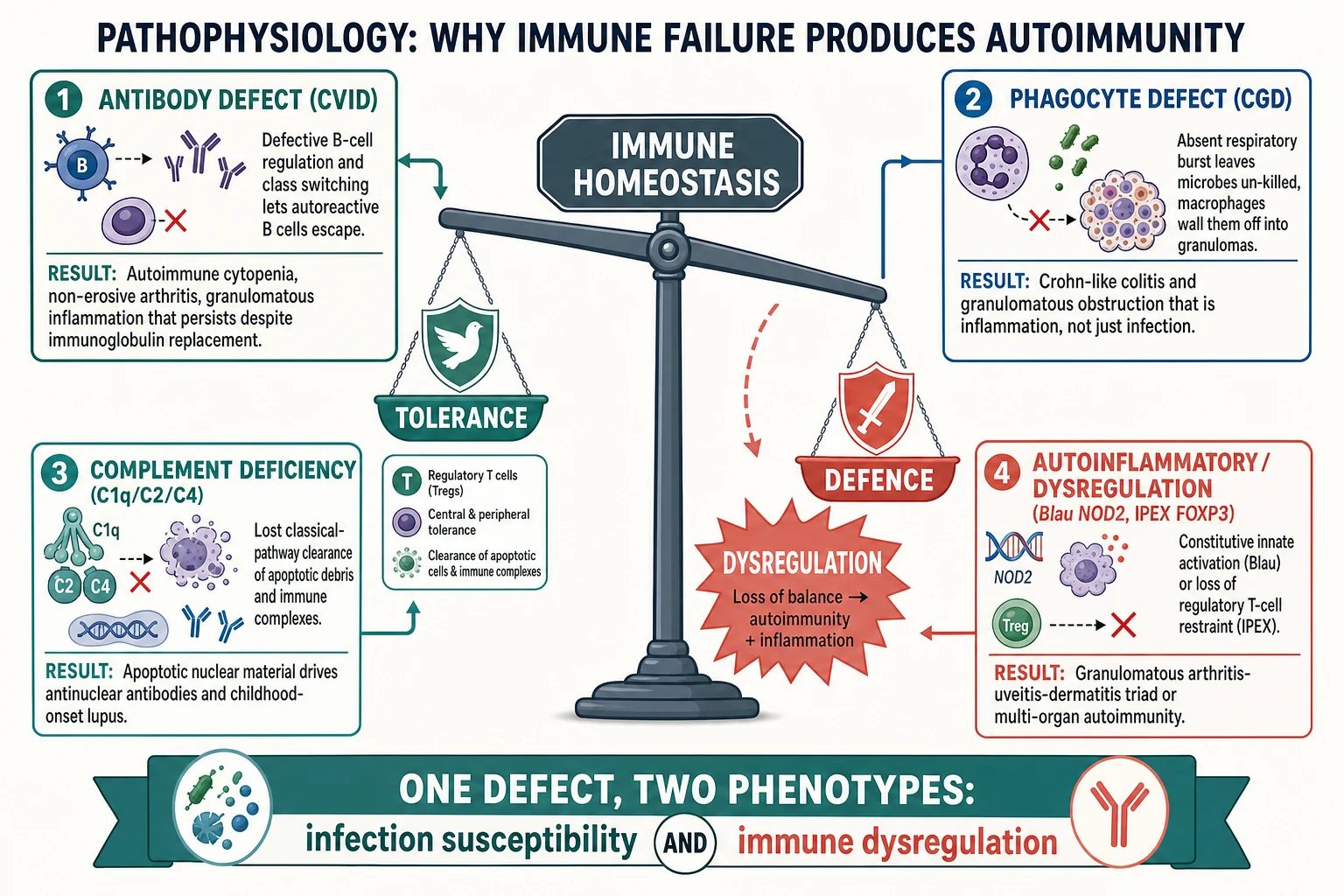
The immune dysregulation and autoinflammatory diseases fail tolerance directly. In IPEX, loss of the FOXP3 transcription factor abolishes regulatory T-cell function, so autoreactive effector cells run unchecked and produce autoimmune enteropathy, endocrinopathy, eczema, and cytopenia. In autoimmune lymphoproliferative syndrome, defective FAS-mediated apoptosis lets lymphocytes accumulate, producing chronic non-malignant lymphoproliferation and autoimmune cytopenia. In Blau syndrome, a gain-of-function NOD2 variant switches innate immune signalling on constitutively, driving granulomatous inflammation of the joints, eyes, and skin. These are the diseases in which autoimmunity is the presenting and dominant feature, with infection susceptibility secondary. [11] [12]
Clinical Presentation
The clinical task is to recognise the overlap pattern, and each arm has a signature. A candidate who hears the right combination of rheumatic disease and infection should reach for the immune screen rather than the rheumatic drug cupboard. [3]
CVID arthritis and autoimmunity presents as a symmetric, non-erosive polyarthritis that is clinically indistinguishable from juvenile idiopathic arthritis until the infection history and the immunoglobulins are examined. Alongside the arthritis, look for autoimmune cytopenia (bruising from immune thrombocytopenia, pallor from autoimmune haemolytic anaemia), granulomatous lymphadenopathy or splenomegaly, granulomatous lung disease, and chronic diarrhoea. The combination of an arthritis-like picture with recurrent sinopulmonary infection and cytopenia is the CVID fingerprint. [7] [8]
CGD colitis and granulomatous disease presents with a Crohn-like inflammatory bowel disease — diarrhoea, abdominal pain, weight loss, and perianal disease — alongside the signature abscess-forming infections. Granulomatous obstruction of the gastric outlet causes vomiting and failure to thrive, and genitourinary obstruction causes hydronephrosis. A lupus-like discoid rash occasionally appears. The child with recurrent abscesses plus a granulomatous colitis has CGD until the dihydrorhodamine assay proves otherwise. [4] [5]
Complement-deficient lupus presents as systemic lupus erythematosus, but with features that flag an immune defect: onset in early childhood, a male sex, a family history of lupus or meningococcal disease, or a persistently absent CH50. The lupus itself may be severe, with nephritis and neurological involvement, because the upstream clearance defect drives relentless autoantigen exposure. [9] [10]
Blau syndrome and the immune dysregulation syndromes present differently. Blau syndrome declares itself before the age of five with the triad of granulomatous arthritis, panuveitis, and a papular exanthema, often with a parent carrying the same NOD2 variant. The immune dysregulation syndromes present in infancy or early childhood with autoimmune cytopenia, severe eczema, enteropathy, or polyendocrinopathy, and a family history of early autoimmunity. These are the children in whom autoimmunity is the dominant and presenting feature. [11] [12]
Differential Diagnosis
The differential for a rheumatic-immune overlap is the differential of each rheumatic syndrome, and the discipline is to ask, in every case, whether the infection history and immune screen warrant an immune work-up. The common error is to anchor on the familiar rheumatic label and miss the immune defect. [3]
Juvenile idiopathic arthritis is the default label for a symmetric childhood polyarthritis, and it is far commoner than CVID arthritis. The discriminating features are the infection history, the presence of autoimmune cytopenia or granulomatous disease, and the immunoglobulin and vaccine-response results. Juvenile idiopathic arthritis alone does not carry recurrent pneumonia, chronic diarrhoea, or low immunoglobulins. [8]
Crohn disease is the default label for a granulomatous childhood colitis, and it overlaps closely with CGD colitis. Both produce transmural granulomatous inflammation and perianal disease, but the CGD colitis accompanies a history of abscess-forming infection with catalase-positive organisms and shows pigmented macrophages on biopsy. The dihydrorhodamine assay settles the question. Treating suspected Crohn disease with immunosuppression before excluding CGD can unmask serious infection. [4] [5]
Classic childhood-onset lupus is the default label for juvenile systemic lupus erythematosus, and the question is when to look for a complement deficiency. Early childhood onset, male sex, a family history of lupus or invasive meningococcal disease, and a persistently low or absent CH50 all warrant the complement work-up, because classical pathway deficiency is rare but carries a disproportionate burden of severe, early lupus. [9] [10]
Secondary immune dysregulation must be distinguished from the primary inborn errors. Drug-induced autoimmunity, malignancy-related autoimmunity, and post-infectious phenomena can mimic the immune dysregulation syndromes. A thorough drug history, a screen for lymphoproliferative disease, and the targeted genetic panel separate the monogenic immune dysregulation disorders from their mimics. [12]
| Pattern | Likely alternative | Key discriminator |
|---|---|---|
| Symmetric polyarthritis in a child | Juvenile idiopathic arthritis | No recurrent infection, normal immunoglobulins and vaccine response |
| Childhood-onset lupus | Classic juvenile SLE | Normal CH50, older age, female sex, no family meningococcal history |
| Autoimmune cytopenia in an infant | Secondary or idiopathic ITP | No eczema, enteropathy, or polyendocrinopathy, normal genetic panel |
Clinical & Bedside Assessment
The bedside task is to decide whether the rheumatic picture crosses the threshold for an immune work-up, and to gather the discriminating evidence at the first consultation. Begin with the threat gate: any rheumatic disease that coexists with recurrent, persistent, severe, or unusual infection, or with a family history of immunodeficiency, earns the immune screen. [3] [1]
The history is the discriminating instrument. Quantify the infection burden — number, site, severity, and documented microbiology of infections — and ask specifically about pneumonia confirmed on imaging, deep-tissue abscesses, opportunistic organisms, and chronic diarrhoea. Establish the rheumatic symptom set: joint pattern and duration, rash, oral ulceration, photosensitivity, and cytopenia symptoms. Take a family history for early-onset autoimmunity, immunodeficiency, consanguinity, early male death, and meningococcal or lupus clusters. Plot growth, because failure to thrive alongside colitis or recurrent infection narrows the differential toward CGD or CVID. [7] [4]
The examination carries the physical clues. Perform a full joint examination for the pattern and symmetry of arthritis, and look for dactylitis, enthesitis, and axial involvement that refine the rheumatic label. Examine the skin for the malar rash and photosensitivity of lupus, the papular exanthema of Blau syndrome, the discoid lupus-like rash of CGD, and the eczema of immune dysregulation. Examine the abdomen for hepatosplenomegaly and lymphadenopathy that suggest granulomatous or lymphoproliferative disease. Look for the sequelae of chronic disease: clubbing, chest wall change, and growth faltering. An ophthalmology assessment for uveitis belongs in the Blau and lupus work-ups. [3] [9]
Assess the family's practical context at the same visit. A child in a remote community, a refugee family, or a household with socioeconomic disadvantage may present late and face barriers to specialist immunology and genetic testing. Documenting these factors shapes the referral, the retrieval plan, and the coordination of shared care between immunology and rheumatology. [2]
Investigations
The investigation ladder is built to answer a single question first: which immune arm is failing? Pair that with the rheumatic work-up, and the two together place the child on the classification map. [1]
First-line immune tests are a full blood count with differential, total immunoglobulins (IgG, IgA, IgM) interpreted against age-adjusted reference ranges, a functional vaccine response, and a total haemolytic complement CH50. The CH50 is the functional screen for the classical and terminal complement pathways; a lone C3 or C4 misses a classical pathway deficiency because C3 and C4 can be normal in component deficiencies between flares. [9] [10]
Second-line immune tests characterise the cellular and functional defect. For suspected CVID, lymphocyte subset enumeration by flow cytometry and memory-B-cell phenotyping refine the diagnosis. For suspected CGD, the dihydrorhodamine flow cytometry assay quantifies the oxidative burst and is the decisive test — an absent or markedly reduced burst confirms a phagocyte killing defect. For suspected immune dysregulation, flow cytometry identifies the characteristic profiles of autoimmune lymphoproliferative syndrome (elevated double-negative T cells) and regulatory T-cell deficiency. [4] [12]
Genetic testing confirms the monogenic causes and enables family counselling and cascade screening. A targeted inborn-errors-of-immunity gene panel is now the standard approach when a rheumatic-immune overlap is suspected, distinguishing the monogenic immune dysregulation syndromes, Blau syndrome (NOD2), classical complement deficiencies, and genetically resolved CVID from their mimics. Genetic confirmation is particularly important when haematopoietic stem cell transplantation is under consideration. [5] [11]
The rheumatic work-up runs in parallel. Antinuclear antibody, anti-double-stranded-DNA, extractable nuclear antigen, and complement C3 and C4 quantify the lupus phenotype; renal assessment with urinalysis, blood pressure, and creatinine screens for lupus nephritis; and joint imaging and an ophthalmology review complete the picture. Interpret the rheumatic serology in the light of the immune defect: a low C3 and C4 during a lupus flare reflects consumption, so the CH50 must be checked when the child is well to exclude a true deficiency. [9]
Management — Resuscitation
Resuscitation in a rheumatic-immune overlap means treating the acute presentation safely while avoiding the two harms specific to this group: immunosuppression in a child with an undiagnosed immune defect, and live vaccines in a possible combined immunodeficiency. [1]
A child presenting with acute severe infection receives the standard paediatric sepsis pathway: prompt cultures, empirical intravenous antibiotics, and fluid and oxygen as required. The microbiology is guided by the likely organisms — encapsulated bacteria in antibody deficiency, the catalase-positive CGD cluster in phagocyte defects, and opportunists in combined or dysregulation defects. Empirical cover should be broadened when an opportunistic organism is suspected, and any indwelling line or focus must be sought. [4] [5]
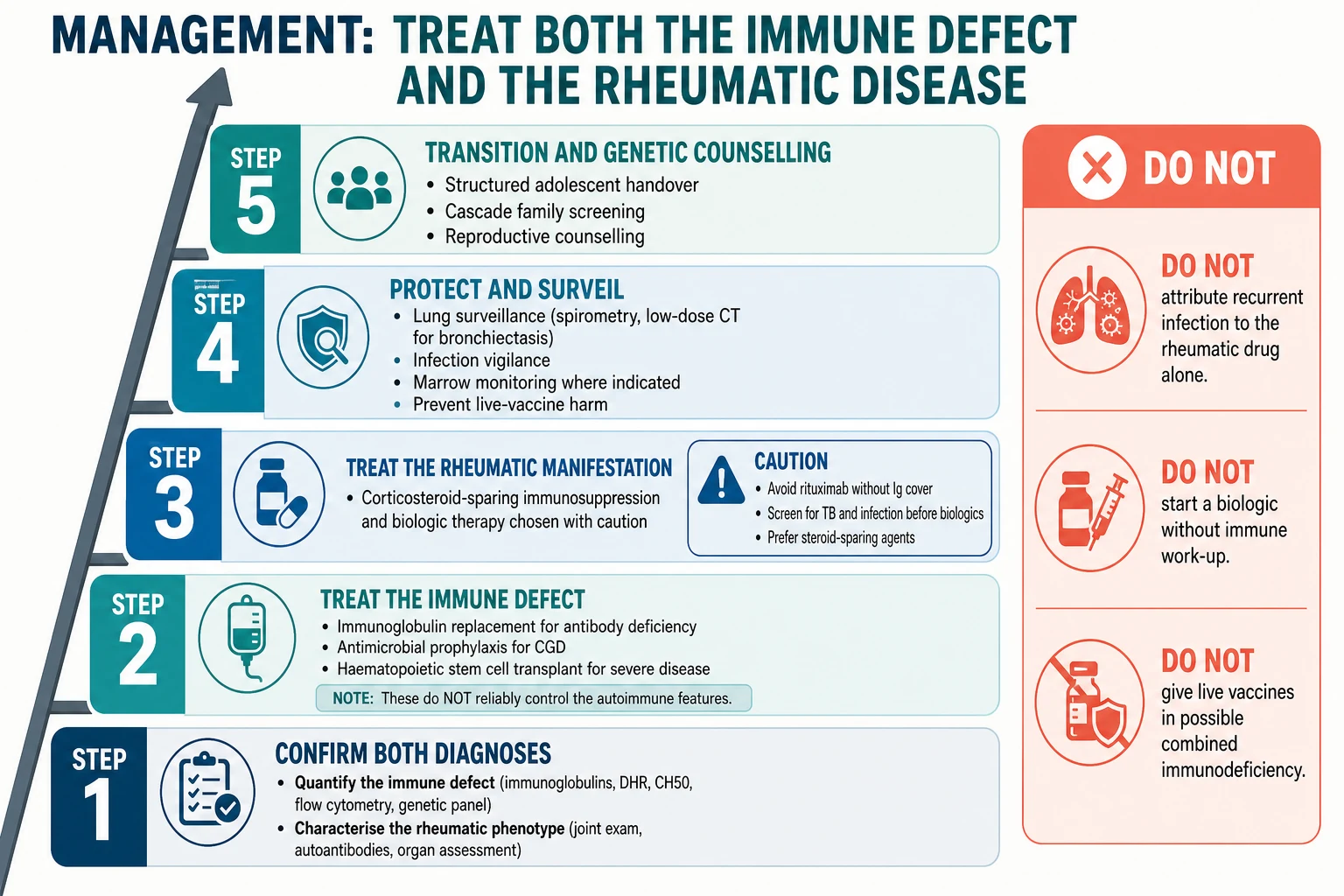
The first overlap-specific hazard is immunosuppression before the immune screen is back. A child presenting with a polyarthritis or a colitis may be started on corticosteroid, methotrexate, or a biologic before the immune work-up is complete. If an immune defect is even suspected, defer the immunosuppression until the immunoglobulins, the DHR assay, and the CH50 return, because suppressing a failing immune system can precipitate overwhelming infection. Where the rheumatic disease is severe and treatment cannot wait, involve immunology immediately and choose the least broadly immunosuppressive option that controls the flare. [3]
The second hazard is live vaccination in a possible combined or severe immunodeficiency. If there is any suspicion of a combined T- and B-cell defect or a severe phagocyte disorder, defer live vaccines such as measles-mumps-rubella, varicella, and BCG until the immune status is clarified, because live vaccines can cause disseminated disease in severely immunocompromised children. [1] [2]
Resuscitation of the family's understanding matters equally. A child newly diagnosed with a rheumatic-immune overlap will need a structured plan from two services, and parents benefit from an early, honest explanation of the dual diagnosis, the treatment plan, and the role of genetic testing, before the detail of long-term management is introduced. [3]
Management — Definitive & Stepwise
Definitive management follows a five-step ladder that treats the immune defect and the rheumatic manifestation in parallel. The discipline is to address both arms — treating only the joints in a child with a failing immune system fails both. [3] [1]
Step one is to confirm both diagnoses before treating. Quantify the immune defect with the matched test and characterise the rheumatic phenotype, because the long-term plan depends on the underlying cause rather than on the symptom alone. A child with CVID arthritis, CGD colitis, complement-deficient lupus, and Blau syndrome each need a different plan, and the label must be right before committing to lifelong therapy. [8] [9]
Step two is to treat the immune defect. Immunoglobulin replacement is the foundation for the antibody defects — intravenous or subcutaneous, dosed to keep the trough IgG in the normal range. Antimicrobial prophylaxis is the foundation for CGD, with co-trimoxazole covering bacterial infection and an anti-mould azole covering Aspergillus. Haematopoietic stem cell transplantation is curative for the severe forms, particularly CGD and the severe immune dysregulation syndromes, and survival after transplant for CGD is high in modern series. [5] [12]
Step three is to treat the rheumatic manifestation with corticosteroid-sparing intent. Choose immunosuppression and biologic therapy with caution: avoid rituximab in a child without immunoglobulin cover because it deepens the antibody defect, screen for tuberculosis and latent infection before any biologic, and prefer steroid-sparing agents to limit the infection and bone risks of long-term corticosteroid. In CVID, remember that immunoglobulin replacement does not reliably control the autoimmune or granulomatous features, so these need their own therapy. [7] [6]
Step four is to protect and surveil. Establish baseline and repeat spirometry and low-dose chest computed tomography when bronchiectasis is suspected in the antibody defects, monitor marrow and infection in CGD, screen the eyes for uveitis in Blau syndrome and lupus, and screen the kidneys and blood pressure in lupus. The aim is to detect the complications of both arms early, because established bronchiectasis and lupus nephritis are the dominant long-term morbidities. [7] [9]
Step five is to plan transition and genetic counselling. A structured adolescent handover to adult immunology and rheumatology preserves continuity of the immunoglobulin regimen, the prophylaxis, and the rheumatic therapy. Genetic counselling for the family — including carrier testing for the X-linked and autosomal recessive forms and cascade screening of relatives — belongs here too. [5] [11]
Specific Subtypes & Scenarios
Each major rheumatic-immune overlap carries a distinctive decision point, and a fellowship answer earns depth by handling them individually rather than as a single block. [3]
CVID arthritis and autoimmunity is the commonest overlap in practice. The arthritis is symmetric and non-erosive, and it coexists with autoimmune cytopenia and granulomatous disease in a substantial minority. The management principle that earns marks is that immunoglobulin replacement controls the infection susceptibility but does not reliably control the autoimmune and granulomatous features, which need corticosteroid-sparing immunosuppression chosen alongside immunology. The ESID registry burden study quantified how often these complications occur. [7] [6]
CGD colitis and granulomatous disease is the overlap most easily mistaken for ordinary inflammatory bowel disease. The colitis is granulomatous and Crohn-like, but the accompanying abscess-forming infection with catalase-positive organisms, the perianal disease, and the pigmented macrophages on biopsy point to CGD. The dihydrorhodamine assay confirms the diagnosis, and management combines antibacterial and anti-mould prophylaxis with anti-inflammatory therapy for the granulomatous complications, reserving haematopoietic stem cell transplantation for severe disease. [4] [5]
Complement-deficient lupus is the overlap that changes the lupus prognosis. Classical pathway deficiency produces severe, early, often male lupus driven by failed apoptotic clearance, and the management adds infection prophylaxis — meningococcal vaccination and antibiotic prophylaxis for the terminal and alternative pathway components — to the standard lupus therapy. The CH50, checked when the child is well, is the screen that earns the diagnosis. [9] [10]
Why a biologic can be the wrong first move
A child presents with a symmetric polyarthritis and is labelled juvenile idiopathic arthritis; the next step is often methotrexate and a tumour-necrosis-factor inhibitor. If the underlying cause is common variable immunodeficiency or chronic granulomatous disease, the biologic suppresses an immune system that is already failing and can precipitate serious or overwhelming infection. The protection is to send the immunoglobulins, the vaccine response, and — when infection clusters around abscess-forming organisms — the DHR assay before the first dose. [7] [4]
Blau syndrome and the immune dysregulation syndromes are the rare but high-yield overlaps. Blau syndrome presents before five with granulomatous arthritis, panuveitis, and a papular rash, often with an affected parent, and a gain-of-function NOD2 variant confirms it; management is corticosteroid-sparing and biologic-based. The immune dysregulation syndromes — autoimmune lymphoproliferative syndrome, IPEX, and the PI3K-dysregulation disorders — present in infancy or early childhood with autoimmunity, and management combines immunosuppression with consideration of haematopoietic stem cell transplantation for the severe forms. [11] [12]
Complications & Pitfalls
The complications divide into the consequences of untreated immune failure, the consequences of untreated rheumatic disease, and the iatrogenic harm of treating one arm without the other. A fellowship answer handles all three. [7] [4]
Infection remains the dominant complication of the immune defect. Recurrent or undertreated sinopulmonary infection in antibody deficiency leads to bronchiectasis, the catalase-positive organisms of CGD cause deep abscesses and invasive fungal disease, and the broad susceptibility of immune dysregulation syndromes permits opportunistic infection. These are both complications and diagnostic clues, and surveillance for them is part of routine care. [5]
End-organ rheumatic damage drives long-term morbidity in the rheumatic arm. Lupus nephritis in complement-deficient lupus, uveitis and joint destruction in Blau syndrome, and chronic granulomatous lung and gastrointestinal disease in CVID and CGD each require structured surveillance. Established organ damage is the strongest predictor of poor long-term outcome. [9] [6]
The iatrogenic pitfalls are the heart of this topic. Starting a biologic or methotrexate before excluding an immune defect, giving rituximab without immunoglobulin cover in a child with an antibody defect, giving live vaccines in a possible combined immunodeficiency, and assuming that immunoglobulin replacement alone will control the autoimmune and granulomatous features of CVID are all recognised harms. The commonest pitfall, however, is anchoring on the familiar rheumatic label and never sending the immune screen at all. [3] [7]
Prognosis & Disposition
Prognosis is dictated by the underlying immune defect, the timeliness of diagnosis, and the quality of surveillance for both arms. With early diagnosis and consistent dual therapy, many children do well, but the rarer defects carry a heavier burden. [3]
CVID arthritis carries a prognosis shaped by the autoimmune, granulomatous, and lymphoproliferative complications of CVID rather than by the arthritis alone, which is why surveillance for cytopenia, granulomatous disease, and lymphoma is part of routine care. CGD colitis prognosis depends on infection control and on the decision and timing of haematopoietic stem cell transplantation. [7] [5]
Complement-deficient lupus carries a more guarded prognosis than classic childhood lupus because the upstream clearance defect drives severe, early disease, and the infection susceptibility of some complement defects adds a second threat. Blau syndrome and the immune dysregulation syndromes carry a variable prognosis that depends on the genotype and on the success of biologic therapy or transplantation. [9] [12]
Disposition for a general paediatrician is shared care between clinical immunology and rheumatology. Immunology owns the diagnostic characterisation, the immunoglobulin regimen, the prophylaxis, the genetic counselling, and the transplantation decision; rheumatology owns the joint, skin, eye, and renal surveillance and the corticosteroid-sparing immunosuppression. Early referral to both services at the point of suspicion — rather than after a confirmed label — is the disposition rule, because the work-up is specialised and the therapy is lifelong. [2] [1]
Special Populations
A rheumatic-immune overlap behaves differently across populations, and a fellowship answer recognises that access, adherence, and late presentation shape outcome as much as the genotype. [3]
Indigenous children, particularly in Australia and New Zealand, face a high background burden of recurrent respiratory and skin infection, crowded housing, and reduced access to specialist immunology. A confirmed immune defect must be managed intensively to prevent rapid progression to bronchiectasis and to control the rheumatic complications, and home-based subcutaneous immunoglobulin can improve adherence where feasible. [7]
Migrant, refugee, and asylum-seeking families may have incomplete vaccination records, uncertain family history, and barriers to genetic testing and specialist access. A careful reconstruction of the infection and autoimmune history, confirmation of vaccination status, and a plan that accounts for mobility and language are essential, with interpreter use and trauma-informed communication throughout. [2]
Socioeconomically disadvantaged families carry a disproportionate burden of late presentation and recurrent infection. Subcutaneous home immunoglobulin therapy, where feasible, improves adherence and reduces travel burden, and the aim is to fit the therapy to the family's reality rather than the reverse. [7]
Adolescents in transition are a population in their own right. Adherence to immunoglobulin therapy and immunosuppression declines in adolescence, the risk-taking that accompanies chronic illness rises, and the handover to adult care is a vulnerable point. A structured, documented transition that preserves continuity of both the immunoglobulin regimen and the rheumatic therapy is the safeguard. [3]
Evidence, Guidelines & Regional Differences
The evidence base rests on three pillars: the IUIS classification that defines the phenotypes, the registry and cohort data that describe the burden, and the consensus definitions that standardise diagnosis. [1]
The 2022 IUIS update of the phenotypic classification of inborn errors of immunity is the current reference framework for naming and grouping these conditions across the immune arms. It supersedes earlier editions and reorganises the categories with updated gene assignments, and a fellowship answer should reference the most recent classification. The ESID registry working definitions provide the standardised clinical diagnostic criteria that complement the classification. [1] [2]
Registry and cohort data quantify the burden of the rheumatic complications. The ESID registry burden study documented the frequency of autoimmune and inflammatory disease in CVID, the Winkelstein registry of 368 patients defined the CGD phenotype, and the Lintner review set out the strength of the classical complement deficiency-lupus associations. Köstel Bal and colleagues synthesised the rheumatological manifestations of inborn errors of immunity across the field. [7] [4] [9] [3]
Genetic and translational evidence underpins the immune dysregulation and autoinflammatory diseases. The Miceli-Richard report of CARD15 (NOD2) mutations in Blau syndrome established the molecular basis of the autoinflammatory archetype, and the Chandrakasan case-based review set out the spectrum of primary immune regulatory disorders. These sources anchor the rare but high-yield end of the topic. [11] [12]
In Australia and New Zealand, a suspected rheumatic-immune overlap is referred jointly to a specialist clinical immunology and a paediatric rheumatology service. The immunology service owns the diagnostic characterisation, the immunoglobulin product (funded through national blood arrangements), the prophylaxis, and the genetic and transplantation work-up; rheumatology owns the joint, eye, and renal surveillance and the immunosuppression. Subcutaneous home immunoglobulin therapy is increasingly used for stable patients to improve adherence, particularly in remote communities. Involve both services early — the work-up and lifelong dual therapy are specialist-led. [1] [2]
Regional differences are practical rather than scientific. Australia and New Zealand follow the IUIS and ESID frameworks, with local product availability and funding arrangements shaping the choice between intravenous and subcutaneous immunoglobulin. Indigenous-health and remote-access considerations intensify the need for early referral and home-based therapy, and the warning-sign overlaps apply universally, with clinical judgement as the final arbiter. [3]
Exam Pearls
A fellowship candidate answering on a rheumatic-immune overlap should land five anchor points and avoid four classic traps. The anchors are the framework examiners listen for; the traps are where candidates lose easy marks. [1] [3]
Anchor one: autoimmunity and immunodeficiency are two faces of one failing immune system. A child with rheumatic disease and recurrent or unusual infection earns an immune work-up as eagerly as a child with sepsis. The pattern, not the label, drives the investigation. [3]
Anchor two: classify by the immune arm and match the test. Immunoglobulins and vaccine response for antibody defects, the dihydrorhodamine oxidative burst assay for CGD, the CH50 for classical complement deficiency, and flow cytometry with a targeted gene panel for immune dysregulation. [4] [9]
Anchor three: treat both arms in parallel. Immunoglobulin replacement, prophylaxis, and transplantation manage the immune defect; corticosteroid-sparing immunosuppression manages the rheumatic manifestation. Treating only the joints fails both. [7] [5]
Anchor four: immunoglobulin replacement does not reliably control the autoimmune and granulomatous features of CVID. These need their own therapy, and assuming otherwise is a recognised error. [6]
Anchor five: defer immunosuppression and live vaccines until the immune screen is back. A biologic, methotrexate, or rituximab given before the immune defect is excluded can precipitate overwhelming infection. [1]
The four traps to avoid are labelling CVID arthritis as juvenile idiopathic arthritis without an immune screen, treating CGD colitis as Crohn disease before the DHR assay, sending a lone C3 and C4 instead of a CH50, and giving rituximab without immunoglobulin cover. Avoid these and the rest of the answer falls into place. [8] [4]
References
- [1]Bousfiha A, Moundir A, Tangye SG, Picard C, Ochs HD, Al-Herz W, et al. The 2022 Update of IUIS Phenotypical Classification for Human Inborn Errors of Immunity. J Clin Immunol, 2022.PMID 36198931
- [2]Seidel MG, Kindle G, Gathmann B, Quinti I, Buckland M, van Montfrans J, et al. The European Society for Immunodeficiencies (ESID) Registry Working Definitions for the Clinical Diagnosis of Inborn Errors of Immunity. J Allergy Clin Immunol Pract, 2019.PMID 30776527
- [3]Köstel Bal S, Pašmandi J, Bhatta B, Boztug K. Rheumatological manifestations in inborn errors of immunity. Pediatr Res, 2020.PMID 31581173
- [4]Winkelstein JA, Marino MC, Johnston RB Jr, Boyle J, Curnutte J, Gallin JI, et al. Chronic granulomatous disease. Report on a national registry of 368 patients. Medicine (Baltimore), 2000.PMID 10844935
- [5]Kang EM, Marciano BE, DeRavin S, Zarember KA, Holland SM, Malech HL. Chronic granulomatous disease: overview and hematopoietic stem cell transplantation. J Allergy Clin Immunol, 2011.PMID 21497887
- [6]Ardeniz O, Cunningham-Rundles C. Granulomatous disease in common variable immunodeficiency. Clin Immunol, 2009.PMID 19716342
- [7]Odnoletkova I, Kindle G, Quinti I, Grimbacher B, van Montfrans J, Gathmann B, et al. The burden of common variable immunodeficiency disorders: a retrospective analysis of the European Society for Immunodeficiency (ESID) registry data. Orphanet J Rare Dis, 2018.PMID 30419968
- [8]Ameratunga R, Gillis D, Steele R. Diagnostic criteria for common variable immunodeficiency disorders. J Allergy Clin Immunol Pract, 2016.PMID 27587325
- [9]Lintner KE, Wu YL, Yang Y, Zhou B, Bledsoe JR, Hebert C, et al. Early Components of the Complement Classical Activation Pathway in Human Systemic Autoimmune Diseases. Front Immunol, 2016.PMID 26913032
- [10]Walport MJ. Complement. First of two parts. N Engl J Med, 2001.PMID 11287977
- [11]Miceli-Richard C, Lesage S, Rybojad M, Prieur AM, Manouvrier-Hanu S, Häfner R, et al. CARD15 mutations in Blau syndrome. Nat Genet, 2001.PMID 11528384
- [12]Chandrakasan S, Chandra S, Davila Saldana BJ, Venkateswaran S, Goyal RK, Strouse C, et al. Primary immune regulatory disorders for the pediatric hematologist and oncologist: A case-based review. Pediatr Blood Cancer, 2019.PMID 30697957