Paeds · rheumatology-musculoskeletal-and-sports
Scleroderma, mixed connective-tissue disease and overlap syndromes
Also known as Juvenile localised scleroderma · Morphea · Juvenile systemic sclerosis · Linear scleroderma · En coup de sabre · Mixed connective tissue disease · Anti-U1-RNP overlap syndrome
Fellowship guide to paediatric scleroderma, mixed connective-tissue disease and the overlap syndromes. Distinguishes the localised scleroderma (morphea), confined to skin and subcutaneous tissue with the linear subtype commonest in children, from the rare juvenile systemic sclerosis defined by the proximal skin sclerosis and the internal organ disease, and frames the mixed connective-tissue disease as the high-titre anti-U1-RNP overlap of lupus, scleroderma and myositis. Covers the Zulian 2006 morphea classification of circumscribed, linear, generalised, pansclerotic and mixed subtypes, the PReS and ACR provisional criteria for juvenile systemic sclerosis, the scleroderma pathophysiology triad of microvascular injury, immune activation and fibroblast-driven fibrosis, the autoantibody map of anti-Scl-70, anti-centromere, anti-RNA polymerase III and anti-U1-RNP, the methotrexate first-line therapy for the active localised disease, the vascular and organ-specific therapy for the systemic disease, and the pulmonary arterial hypertension and interstitial lung disease surveillance that drives the prognosis.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
Overview & Definition
A mother notices that her eight-year-old daughter's forearm has grown a hard, shiny, ivory-coloured patch that she first put down to a bruise that never quite faded. On the same ward, a twelve-year-old boy arrives with cold blue fingers that turn white in the winter, hands that have grown stiff and tight, and a cough the doctor has begun to take seriously. These two children sit at opposite ends of a single spectrum called paediatric scleroderma, and the single decision that separates a good outcome from a harmed one is whether the clinician can tell the localised disease from the systemic one, name the antibody that predicts the organ damage, and start the methotrexate before the tissue is lost. [6][1]
The word scleroderma means hard skin, and the hardening is the visible signature of a disease in which collagen is laid down in excess wherever the small blood vessels have been injured. The great divide is between the localised scleroderma, also called morphea, which stays in the skin and the tissue just beneath it and never reaches the internal organs, and the juvenile systemic sclerosis, a rare multisystem disease in which the same fibrosis settles in the lungs, the heart, the gut and the kidneys. The localised form is far the commoner of the two in childhood, and it carries the better prognosis; the systemic form is uncommon but dangerous, and it is the one that shortens life. [6][8]
A third pattern sits across the boundary, and the fellow must hold it separately. The mixed connective-tissue disease is an overlap syndrome in which a child carries a high-titre anti-U1-RNP antibody and shows features of several connective-tissue diseases at once, the swollen puffy hands and the Raynaud phenomenon of scleroderma, the arthritis and the rash of lupus, and the muscle weakness of dermatomyositis. It is not scleroderma and it is not lupus, and the reason the fellow must name it is that its prognosis is driven by the one complication that does not declare itself early, the pulmonary hypertension. [10][11]
The first task at the bedside is therefore not to reach for a drug but to place the child correctly on the spectrum. Is this hard skin confined to one patch or band, which makes it morphea? Does it come with Raynaud phenomenon and internal organ involvement, which makes it systemic sclerosis? Or does it come with the arthritis and the myositis of an overlap, which makes it the mixed connective-tissue disease? The answer reshapes everything that follows, from the methotrexate for the localised disease, through the vascular and the pulmonary therapy for the systemic disease, to the pulmonary hypertension surveillance for the overlap syndrome. [1][9]
Classification
The most useful way to classify the paediatric scleroderma at the bedside is to ask a single question: does the disease stay in the skin, or does it reach the organs? The answer sorts the children into two families with entirely different prognoses and entirely different management. The localised scleroderma is the skin-confined family, and the juvenile systemic sclerosis is the organ-reaching family. The mixed connective-tissue disease and the other overlap syndromes sit as a third group, defined not by the skin alone but by the antibody and the constellation of the features. [6][8]
The localised scleroderma is itself a family of five subtypes, and the subtype changes the management because it changes the risk of the functional loss. The Zulian classification, drawn from the international study of seven hundred and fifty children, sorts the localised disease into the circumscribed morphea (the plaque), the linear scleroderma (the band), the generalised morphea (four or more plaques becoming confluent), the pansclerotic morphea (the circumferential involvement that is the most severe) and the mixed morphea. The single fact the fellow must carry is that the linear subtype is the commonest in children, accounting for roughly two-thirds of the juvenile localised scleroderma, which is the reverse of the adult pattern where the circumscribed plaque dominates. [6][8]
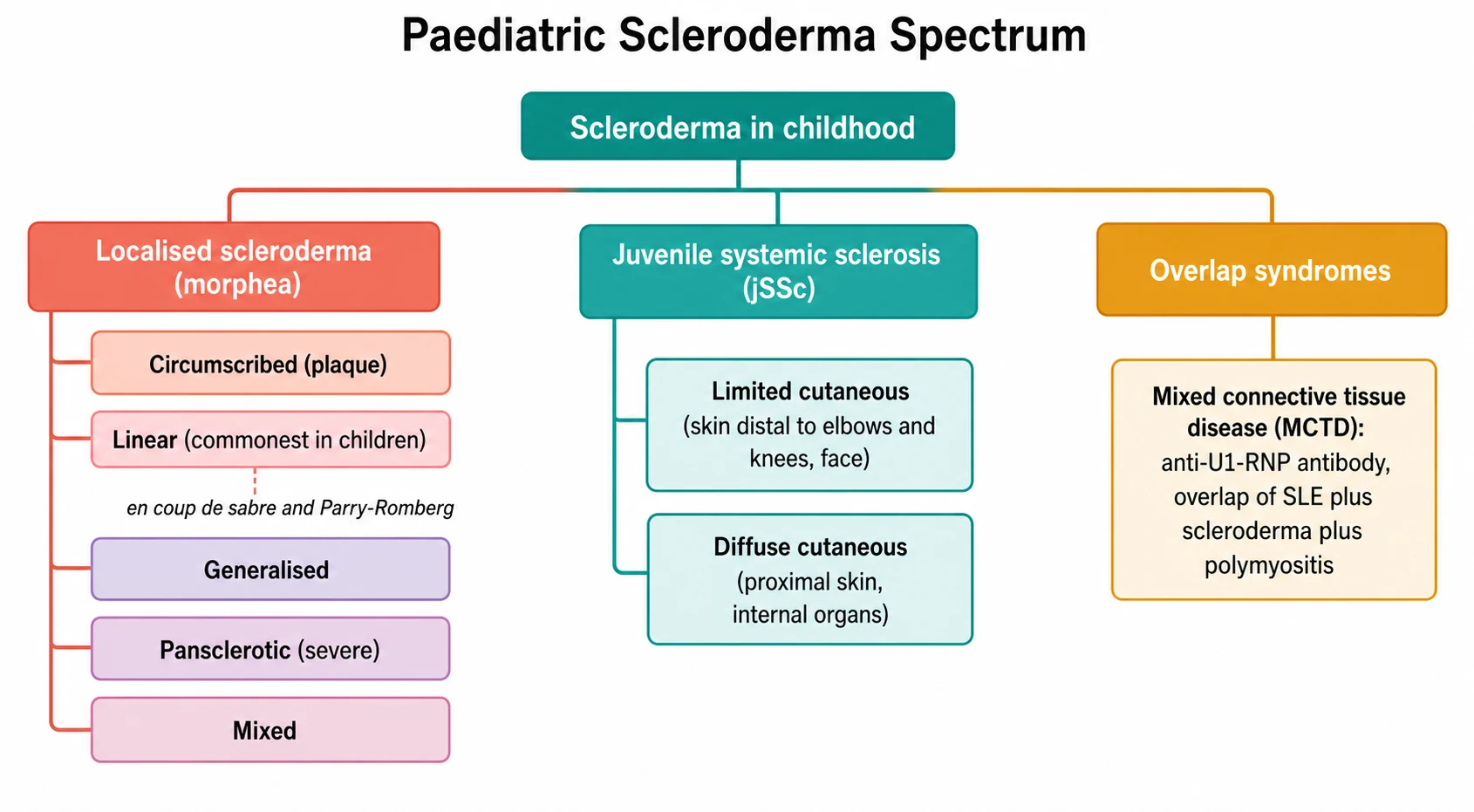
The linear subtype deserves its own attention because it is the one that harms the growing child. A band of indurated skin that crosses a joint limits its movement and, in a child whose bones are still growing, produces a permanent flexion contracture and a shortened limb. A band that crosses the face produces the en coup de sabre, the sword-strike of the forehead, and its deeper variant the Parry-Romberg syndrome, the progressive hemifacial atrophy that shrinks the skin, the fat and the bone beneath the linear lesion. These facial variants are the ones that carry the extracutaneous complications, the seizures and the uveitis, because the inflammation can extend to the brain and the eye beneath the band. [7][8]
The juvenile systemic sclerosis is classified along a different axis, the extent of the skin involvement, because the extent maps to the organ risk. The diffuse cutaneous systemic sclerosis is the disease in which the skin thickening reaches proximal to the elbows and the knees and involves the trunk, and it carries the early and the aggressive internal organ disease, the interstitial lung disease and the renal crisis. The limited cutaneous systemic sclerosis is the disease in which the skin thickening stays distal to the elbows and the knees and involves the face, and it carries the later and the more indolent organ disease, the pulmonary arterial hypertension and the calcinosis. In children, the diffuse and the limited forms both occur, and the diffuse form tends to carry the heavier early burden. [3][1]
Localised scleroderma (morphea)
skin-confined
- Skin and subcutaneous tissue only, no internal organ disease
- Linear subtype commonest in children, around two-thirds
- En coup de sabre and Parry-Romberg for the facial variant
- Methotrexate first-line for the active disease
- Better prognosis, the cosmetic and the functional outcome the concern
Juvenile systemic sclerosis
multisystem
- Proximal skin sclerosis with the internal organ involvement
- Rare in childhood, around three to five percent of childhood rheumatic disease
- Diffuse cutaneous and limited cutaneous forms
- Pulmonary disease the leading cause of the mortality
- PReS and ACR criteria of the proximal sclerosis plus at least two minor
Mixed connective-tissue disease
overlap
- High-titre anti-U1-RNP antibody is the defining serology
- Overlap of lupus, scleroderma and myositis
- Raynaud phenomenon nearly universal
- Pulmonary hypertension the feared and the lethal complication
- Treated by the dominant feature while screening for evolution
The classification of the juvenile systemic sclerosis was formalised for the child specifically, because the adult criteria under-diagnose the early paediatric disease. The Pediatric Rheumatology European Society, the American College of Rheumatology and the European League Against Rheumatism provisional classification criteria, published by Zulian and colleagues in 2007, set a single major criterion, the proximal skin sclerosis or the induration of the skin proximal to the metacarpophalangeal joints, and they require it to be accompanied by at least two minor criteria drawn from the nine organ systems. The minor criteria span the cutaneous (the sclerodactyly), the vascular (the Raynaud phenomenon, the nailfold capillary abnormalities), the pulmonary (the pulmonary arterial hypertension, the reduced diffusion capacity, the fibrosis), the cardiac, the renal, the gastrointestinal, the neurologic, the musculoskeletal and the serologic domains. The criteria carry a high sensitivity and specificity in the definitive disease, and they are the ones the fellow reproduces at the viva. [2]
Epidemiology & Risk Factors
The epidemiology of the paediatric scleroderma is the story of a rare disease in which the localised form dominates and the systemic form is the exception. The juvenile localised scleroderma is the commoner of the two, with an estimated incidence of around one to three per one hundred thousand children, and it accounts for roughly three to four percent of all the paediatric rheumatic disease. The juvenile systemic sclerosis, by contrast, is rare, with an incidence of only a few per million children and a prevalence that places it among the rarest of the childhood rheumatic diseases. The peak age of the localised disease sits between five and ten years, and the peak of the systemic disease sits a little later, around ten to twelve years, with a female predominance of roughly two to four to one. [6][4]
The risk factors are weak and largely genetic and autoimmune rather than environmental, and the fellow should not overstate them. A family history of the autoimmune disease and the presence of other autoimmune conditions in the child are the consistent associations, and the HLA and the cytokine gene polymorphisms are the molecular layer that explains the clustering. An infective trigger has been sought, particularly the Borrelia burgdorferi in the European cases of the localised disease, but the evidence is inconsistent and the antibiotic treatment does not alter the course in the seropositive child. The lesson for the exam is that the paediatric scleroderma is an autoimmune and the genetic disease, and the environmental triggers are the ones to question rather than to assert. [8]
The epidemiology of the mixed connective-tissue disease follows a different curve, and the fellow must hold it separately because it presents in the older child and the adolescent. The juvenile-onset disease accounts for a small fraction of the total mixed connective-tissue disease, with the onset typically in the adolescence, a strong female predominance, and a clinical profile that differs from the adult by the higher frequency of the arthritis, the fever and the serositis and the relative sparing of the renal and the central nervous system disease. The recent multicentre cohort has refined the paediatric phenotype and confirmed the pulmonary hypertension as the decisive complication. [10][11]
Pathophysiology
The pathophysiology of the scleroderma is best understood as three injuries that run in parallel and feed one another, and the fellow who holds the three together can explain every clinical feature from the single mechanism. The first injury is the microvascular, the damage to the tiny blood vessels that begins, invisibly, long before the skin hardens. The second is the immune activation, the autoantibodies and the T-cells that sustain the inflammation and that produce the defining serology. The third is the fibroblast activation, the excessive production of the collagen that turns the injured tissue into the hard, scarred tissue that gives the disease its name. The three injuries converge on the same outcome, the fibrosis of the skin and the organs. [1][8]
The microvascular injury is the first event and the one that produces the Raynaud phenomenon, the sentinel sign of the disease. The endothelium of the small vessels is damaged, the vasodilator and the vasoconstrictor balance tips towards the vasoconstriction, and the structural capillaries are lost. The child experiences the cold-induced colour change of the Raynaud, the white then the blue then the red, and over time the capillary dropout produces the digital ischaemia and the ulcers. The nailfold capillaroscopy shows the dilated and the tortuous giant capillaries and the avascular areas that confirm the structural microangiopathy, and it is the bridge from the Raynaud to the diagnosis. [1][5]
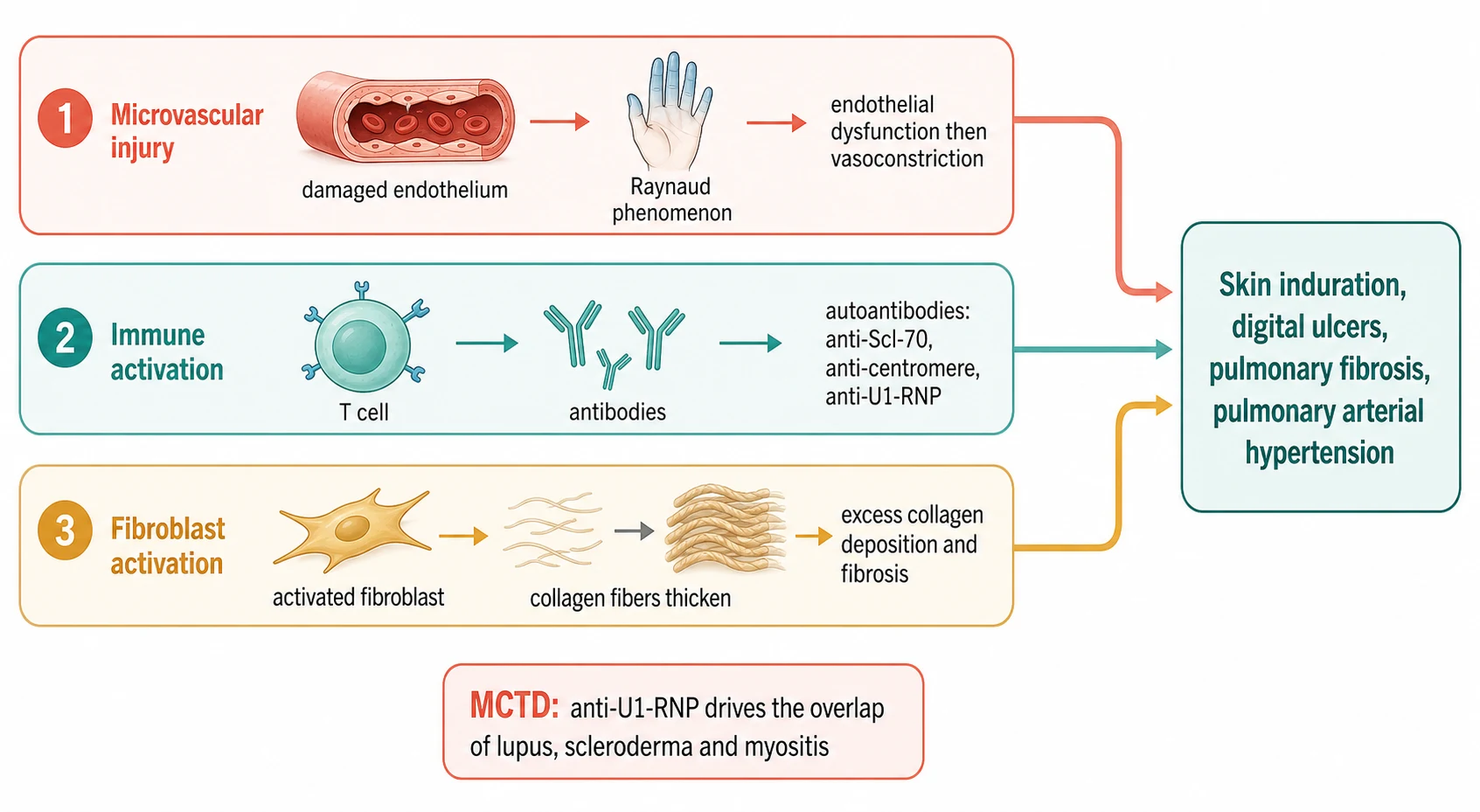
The immune activation produces the autoantibodies, and each antibody is a marker that predicts the organ risk, which is why the antibody panel is not a checkbox but a map of the disease. The anti-Scl-70, the antibody against the topoisomerase one, marks the diffuse cutaneous disease and the interstitial lung disease. The anti-centromere marks the limited cutaneous disease and the pulmonary arterial hypertension. The anti-RNA polymerase three marks the renal crisis. The anti-U3-RNP, the fibrillarin, marks the pulmonary hypertension and the gastrointestinal disease. In the child, the anti-Scl-70 is more common than the anti-centromere, which is the reverse of the adult pattern, and it is the antibody the fellow expects in the diffuse paediatric disease. [1][4]
The fibroblast activation is the third injury and the one that produces the visible disease. The activated fibroblasts, driven by the transforming growth factor beta and the other profibrotic signals, lay down the excess collagen and the extracellular matrix, and the tissue hardens. The skin thickens and loses its suppleness, the joints stiffen, and the same fibrosis settles in the lungs to produce the interstitial disease and in the vessels to produce the pulmonary arterial remodelling. The modified Rodnan skin score, which grades the thickness of the skin across seventeen body areas on a scale of zero to three, is the clinical measure of this fibrosis, and it is the one the fellow follows to judge the disease activity and the response to the therapy. [1][9]
The pathophysiology of the mixed connective-tissue disease sits on the same mechanism but with a different antibody at the centre. The high-titre anti-U1-RNP antibody is the defining serology, and it is directed against the U1 small nuclear ribonucleoprotein. The antibody is the marker, not in itself the cause of the organ damage, and the clinical phenotype is produced by the same triad of the microvascular injury, the immune activation and the fibrosis, expressed across the lupus, the scleroderma and the myositis features. The pulmonary hypertension that decides the prognosis is the vascular and the fibrotic injury expressed in the pulmonary arteries, and it is the reason the diffusion capacity and the echocardiography are followed for the life of the disease. [11][10]
Clinical Presentation
The localised scleroderma presents as a change in the skin that the parent or the child notices first, and the tempo and the pattern are the clues to the subtype. The circumscribed morphea begins as an area of the inflammation, a red or the violaceous patch, that evolves over weeks into the hard, the ivory-coloured plaque with a waxy surface and a lilac ring at the border. The linear scleroderma presents as a band of the indurated skin that follows a limb or crosses the face, and it is the band that the growing child cannot outgrow, because it tethers the joint and the bone beneath it. The generalised morphea presents as the multiple and the confluent plaques, and the pansclerotic as the circumferential involvement of a limb or the trunk that is the most disabling of the localised forms. [6][7]
The facial linear scleroderma deserves its own description because it carries the extracutaneous disease. The en coup de sabre presents as a linear depression of the forehead, often following the line of the fifth cranial nerve, that deepens over months as the skin, the fat and the bone beneath it atrophy. The Parry-Romberg syndrome, the progressive hemifacial atrophy, is the deeper variant in which one side of the face shrinks and the asymmetry becomes pronounced. These facial variants are the ones the fellow investigates for the central nervous system and the ocular involvement, because the inflammation beneath the band can produce the seizures, the headache and the uveitis, and the brain imaging is part of the workup of the symptomatic child. [7][8]
The juvenile systemic sclerosis presents differently, and the Raynaud phenomenon is almost always the first sign, often preceding the skin change by months or years. The child describes the cold-induced colour change of the fingers, the white then the blue then the red, and the parent may describe the puffy swelling of the hands that is the earliest skin change. The skin thickening begins in the fingers, the sclerodactyly, and extends proximally in the diffuse disease or stays distal in the limited disease. The internal organ disease may be silent at the presentation, which is the trap, and the dyspnoea, the reflux and the palpitations declare only as the lung, the gut and the heart are reached. [1][3]
The mixed connective-tissue disease presents with the constellation rather than the single sign, and the Raynaud phenomenon and the swollen hands are the common gateway. The child describes the cold-induced colour change, the arthritis and the muscle weakness, and the examination shows the puffy swollen fingers, the inflammatory rash and the proximal muscle weakness. The serositis, the pleuritis and the pericarditis, is common in the paediatric disease, and the fever and the lymphadenopathy may accompany the onset. The pulmonary hypertension is usually silent until it is advanced, which is the reason the screening is built into the follow-up, and the dyspnoea on the exertion is the late and the dangerous sign. [10][11]
The atypical presentations are the ones that delay the diagnosis, and the fellow must hold them. The child who presents with the isolated Raynaud phenomenon for years before any skin change may have the evolving systemic sclerosis, and the nailfold capillaroscopy and the antibody panel are the tests that reveal it early. The child with the localised plaque that is mistaken for the eczema or the post-inflammatory hyperpigmentation may have the circumscribed morphea, and the induration on the palpation is the feature that distinguishes it. The child with the overlap features that do not yet fulfil any single criterion may have the undifferentiated connective-tissue disease that will declare itself over the months, and the serial review is the safeguard. [8][1]
Differential Diagnosis
The differential of the indurated skin in the child is built around whether the hardening is the true scleroderma or one of its mimics, and the biopsy and the antibody panel are the tests that resolve it. The lichen sclerosus presents as the white atrophic plaques, often in the genital region, and it lacks the deep induration of the morphea. The eosinophilic fasciitis presents as the painful induration of the limbs with the peripheral eosinophilia and the groove sign, and it spares the hands and the feet, which the systemic sclerosis does not. The chronic graft-versus-host disease presents in the child after the transplant, and the history is the discriminator. The deep morphea and the early systemic sclerosis are the ones that overlap, and the internal organ screen separates them. [8][9]
The differential of the Raynaud phenomenon is the question of the primary versus the secondary, and the nailfold capillaroscopy is the test that separates them. The primary Raynaud is the common, the benign and the familial phenomenon of the young woman, with the normal capillaroscopy, the normal antibody panel and the absence of the tissue damage. The secondary Raynaud is the phenomenon that accompanies the connective-tissue disease, with the abnormal capillaroscopy, the positive antibody and the digital ulcers or the pitting scars. The child who presents with the Raynaud phenomenon is screened for the secondary cause, because the Raynaud is the gateway sign of the systemic sclerosis and the mixed connective-tissue disease. [1][5]
The differential of the mixed connective-tissue disease is the question of which connective-tissue disease it is, and the anti-U1-RNP antibody is the discriminator. The systemic lupus erythematosus shares the arthritis, the rash and the serositis, but it carries the anti-dsDNA and the anti-Smith antibodies and the renal and the central nervous system disease that the mixed connective-tissue disease relatively spares. The juvenile dermatomyositis shares the proximal muscle weakness, but it carries the characteristic rash and the myositis-specific antibodies. The undifferentiated connective-tissue disease is the overlap that does not yet fulfil any single criterion, and it is the diagnosis that the mixed connective-tissue disease must be distinguished from by the high-titre anti-U1-RNP. [11][10]
Systemic lupus erythematosus
mimic
- Anti-dsDNA and anti-Smith positive, anti-U1-RNP low or absent
- Renal and central nervous system disease common
- Malar rash, photosensitivity, oral ulcers
- Complement consumption in the active disease
- Distinguished from MCTD by the antibody profile and the renal involvement
Juvenile dermatomyositis
mimic
- Proximal muscle weakness and the characteristic rash
- Gottron papules and heliotrope rash
- Myositis-specific antibodies
- Creatine kinase elevation
- Distinguished from MCTD by the rash and the myositis panel
Undifferentiated CTD
mimic
- Overlap features that fulfil no single criterion
- Anti-U1-RNP absent or low titre
- May evolve into a defined disease over time
- Serial review the safeguard
- Distinguished from MCTD by the titre of the anti-U1-RNP
The mimics of the systemic sclerosis itself are the rare fibrosing conditions, and the fellow names them to exclude them. The nephrogenic systemic fibrosis occurs in the child with the severe renal failure after the gadolinium exposure, and it produces the induration of the skin that mimics the scleroderma. The eosinophilia-myalgia syndrome followed the contaminated tryptophan and is now historical. The porphyria cutanea tarda and the phenylketonuria can produce the scleroderma-like skin. The history and the context are the discriminators, and the fellow who names the mimics demonstrates the breadth that the viva rewards. [8]
Clinical & Bedside Assessment
The bedside assessment of the child with the suspected scleroderma is a search for the skin lesion, its extent and its activity, the signs of the internal organ involvement, and the stigmata of the overlap. The assessment begins with the history of the onset, the progression and the cold sensitivity, and it turns to the family history of the autoimmune disease. The child is examined in the warm room, because the cold provokes the Raynaud and obscures the skin signs, and the examination is systematic across the skin, the joints, the nails, the chest and the abdomen. [6][1]
The skin examination is the heart of the assessment, and each finding carries weight. The localised lesion is examined for the induration, the lilac ring of the active inflammation, the atrophy of the established plaque, and the relationship to the underlying joint. The linear band is examined for its crossing of the joint line and its effect on the range of movement, because the band that crosses a joint is the one that produces the contracture. The modified Rodnan skin score is performed in the systemic disease, grading the thickness of the skin across the seventeen body areas on the scale of zero for the normal, one for the mild, two for the moderate and three for the severe, with the total score of zero to fifty-one tracking the disease burden. [1][9]
The focused examination of the child with the suspected scleroderma
Inspect the skin in the warm room for the indurated plaque, the linear band, the puffy fingers and the sclerodactyly, and plot the extent on a body map
Palpate the lesion for the induration, the atrophy and the lilac ring of the active inflammation, and photograph it for the serial tracking
Examine the joints for the range of movement, because the linear band that crosses a joint produces the contracture, and the growth of the limbs for the asymmetry
Perform the nailfold capillaroscopy for the dilated capillaries, the microhaemorrhages and the avascular areas that confirm the systemic sclerosis pattern
Measure the blood pressure and examine the chest for the fine crackles of the interstitial lung disease and the loud pulmonary component of the pulmonary hypertension
Examine for the overlap features: the malar rash and the oral ulcers of the lupus, the proximal weakness of the myositis, and the telangiectasia and the calcinosis of the limited disease
The nailfold capillaroscopy is the bedside test that separates the primary from the secondary Raynaud and that confirms the early systemic sclerosis, and the fellow performs it with the dermoscope and the immersion oil. The normal capillaroscopy shows the uniform hairpin capillaries in the neat rows. The systemic sclerosis pattern shows the dilated and the giant capillaries, the microhaemorrhages and the avascular areas that follow the capillary dropout, and it is the pattern that confirms the microangiopathy before the skin disease declares itself. The capillaroscopy is the bridge from the isolated Raynaud to the diagnosis, and it is the test the fellow names at the viva. [1][4]
The assessment of the activity is the question that drives the treatment, and it separates the active inflammation that warrants the immunosuppression from the established damage that does not. The active localised lesion shows the erythema and the lilac ring and the warmth, and it is the lesion that the methotrexate treats. The established lesion is the atrophic, the pigmentary and the indurated plaque without the inflammation, and it is the damage that the physiotherapy and the reconstructive surgery address. The disease activity is tracked by the clinical photography, the modified Rodnan skin score and the localised scleroderma severity index, and the fellow treats the activity and not the score. [9][7]
Investigations
The investigation of the suspected paediatric scleroderma moves in three steps: the antibody panel that names the subtype and predicts the organ risk, the nailfold capillaroscopy and the skin assessment that confirm the microangiopathy and the skin disease, and the organ screen that defines the internal involvement. The antinuclear antibody is near universal in the systemic disease and is the first test, and the specific autoantibody panel follows: the anti-Scl-70 for the diffuse disease and the interstitial lung disease, the anti-centromere for the limited disease and the pulmonary hypertension, the anti-RNA polymerase three for the renal crisis, and the anti-U1-RNP for the mixed connective-tissue disease. [1][11]
The organ screen is built around the lung, because the pulmonary disease is the leading cause of the mortality. The pulmonary function tests, with the forced vital capacity and the diffusion capacity for the carbon monoxide, are performed at the baseline and the regular intervals; the isolated fall of the diffusion capacity is the early sign of the pulmonary vascular disease and the pulmonary hypertension. The high-resolution computed tomography of the chest defines the interstitial lung disease, the ground-glass and the reticular changes and the honeycombing of the established fibrosis. The echocardiography estimates the pulmonary artery pressure from the tricuspid regurgitant jet velocity and the right ventricular size, and the pro-B-type natriuretic peptide is the serum marker that the fellow follows for the evolving pulmonary hypertension. [5][12]
The European Respiratory Society and the European Alliance of Associations for Rheumatology clinical practice guidelines for the connective-tissue-disease-associated interstitial lung disease, applied across Australia, Aotearoa New Zealand, the United Kingdom, Europe, the United States and Canada, set the screening at the baseline and the regular intervals with the pulmonary function tests including the diffusion capacity, the high-resolution computed tomography and the echocardiography. The mycophenolate mofetil and the cyclophosphamide are the first-line immunosuppression for the progressive interstitial lung disease, and the antifibrotic agents are the adjuncts for the progressive disease. The fellow follows the local protocol, but the screening and the first-line therapy are the international standard.
[12]The skin biopsy is performed when the diagnosis of the localised scleroderma is in doubt, because the clinical appearance is usually sufficient and the biopsy is reserved for the atypical lesion. The histology shows the thickening of the dermis with the collagen bundle hyalinisation and the perivascular lymphocytic infiltrate of the active lesion, and it confirms the morphea and excludes the mimics. The biopsy is avoided in the typical systemic sclerosis when the clinical and the serological picture is clear, because the diagnosis is made on the combined skin, the capillaroscopy and the antibody assessment. [8][9]
The baseline bloods and the urinalysis are sent to define the organ involvement and to screen for the overlap. The full blood count, the erythrocyte sedimentation rate and the C-reactive protein track the inflammation; the creatine kinase is sent for the myositis of the overlap; the renal function and the urinalysis are followed for the renal crisis. The echocardiography, the pulmonary function tests and the high-resolution computed tomography complete the baseline organ screen, and the right heart catheterisation is the test that confirms the pulmonary arterial hypertension when the echo raises the suspicion. [1][4]
Management — Resuscitation

The child with the paediatric scleroderma is rarely the dramatic resuscitation, but the three emergencies demand the immediate response. The scleroderma renal crisis is the first, and it presents as the new hypertension with the rising creatinine and the microangiopathic haemolysis. The child is admitted, the blood pressure is controlled, and the angiotensin-converting enzyme inhibitor is begun immediately and titrated, because the inhibition of the renin-angiotensin axis is the one intervention that reverses the crisis. The creatinine may rise in the first days of the treatment, and the fellow holds the inhibitor and titrates it to the renal function, because the long-term outcome depends on the early and the sustained inhibition. [1][4]
The acute digital ischaemia is the second emergency, and it presents as the painful blue or the black digit that is threatened by the critical ischaemia. The child is warmed, the vasodilator is escalated, and the intravenous prostacyclin, the iloprost, is the agent that the fellow uses for the severe ischaemia, titrated to the tolerance and the blood pressure. The pain control, the infection surveillance and the surgical review for the debridement complete the management, and the digit-sparing outcome depends on the early vasodilation. [1]
The pulmonary hypertensive crisis is the third emergency, and it presents as the severe dyspnoea, the syncope and the right heart failure. The child is oxygenated, the right heart catheterisation is performed to confirm the diagnosis and the severity, and the pulmonary arterial hypertension therapy is begun, with the phosphodiesterase five inhibitor, the endothelin receptor antagonist and the prostacyclin as the agents. The intensive care and the specialist pulmonary hypertension service are involved early, because the pulmonary hypertension is the complication that decides the prognosis of the systemic sclerosis and the mixed connective-tissue disease. [5][10]
Management — Definitive & Stepwise
The definitive management of the paediatric scleroderma is built around the subtype, because the localised disease, the systemic disease and the overlap syndrome are treated along different ladders. The overarching principle is that the localised disease is treated with the methotrexate to halt the active inflammation before the damage, the systemic disease is treated with the immunosuppression for the skin and the organ involvement and the vascular and the organ-specific therapy, and the overlap syndrome is treated by its dominant feature while the pulmonary hypertension is screened. [9][1]
[9] [1] [10]The methotrexate is the first-line systemic therapy for the active juvenile localised scleroderma, and it is the agent that halts the inflammation before the damage is fixed. The methotrexate is given at fifteen milligrams per square metre per week, by the subcutaneous or the oral route, with the folic acid supplementation, and it is the agent that the fellow reaches for the moment the active linear or the deep disease is recognised. The systemic corticosteroid is added for the active and the severe disease, as the intravenous methylprednisolone pulse of thirty milligrams per kilogram per day capped at one gram for three consecutive days repeated monthly for three months, or the oral prednisolone at one milligram per kilogram per day capped at fifty milligrams tapered over the months, as the bridging therapy that controls the inflammation while the methotrexate takes effect. [9][7]
Methotrexate for the active juvenile localised scleroderma
Dose
The methotrexate is the first-line systemic therapy for the active linear, the deep, the generalised and the pansclerotic disease, given at fifteen milligrams per square metre per week capped at around twenty-five milligrams per dose, by the subcutaneous or the oral route, with the folic acid. A systemic corticosteroid is added as the bridging for the active and the severe disease, as the intravenous methylprednisolone pulse of thirty milligrams per kilogram per day capped at one gram for three consecutive days repeated monthly for three months, or the oral prednisolone at one milligram per kilogram per day capped at fifty milligrams tapered over the months. The mycophenolate mofetil or the mycophenolic acid is the second-line agent for the refractory disease, and the phototherapy with the ultraviolet A one is the adjunct for the superficial plaque
The physiotherapy and the occupational therapy are the partners of the methotrexate, and they are the ones that prevent the contracture and the functional loss in the linear disease. The stretching and the splinting maintain the joint range, and the serial casting and the strengthening address the established limitation. The cosmetic and the reconstructive surgery, such as the tissue expansion and the fat transfer for the facial asymmetry, is deferred until the disease is quiescent, because the surgery in the active disease fails. The psychological support and the school liaison address the body image and the participation, because the facial and the limb deformity carry a heavy burden for the child and the adolescent. [7][9]
The skin-directed therapy for the juvenile systemic sclerosis is built around the immunosuppression, and the agent is chosen by the severity and the organ involvement. The methotrexate is the first-line for the early and the indolent skin disease, the mycophenolate mofetil for the skin disease with the interstitial lung disease, and the cyclophosphamide for the severe and the progressive organ disease, especially the interstitial lung disease. The vascular therapy is built around the calcium channel blocker, the nifedipine or the amlodipine, as the first-line for the Raynaud phenomenon, the phosphodiesterase five inhibitor, the sildenafil, for the severe Raynaud and the digital ulcers, and the endothelin receptor antagonist, the bosentan, for the prevention of the new digital ulcers and the pulmonary arterial hypertension. [1][5]
Specific Subtypes & Scenarios
The linear scleroderma and the facial variants
The linear scleroderma is the commonest subtype of the juvenile localised scleroderma and the one that carries the functional and the cosmetic burden, and it is the subtype the fellow must manage early. The band that crosses a joint produces the contracture, and the band that crosses the face produces the en coup de sabre and the Parry-Romberg. The methotrexate is begun at the recognition of the active linear disease, the physiotherapy is started to preserve the joint range, and the facial variant is investigated for the central nervous system and the ocular involvement when the symptoms suggest it. The reconstructive surgery is deferred until the disease is quiescent, and the psychological support runs alongside. [7][8]
The juvenile systemic sclerosis with the interstitial lung disease
The juvenile systemic sclerosis with the interstitial lung disease is the subtype that decides the prognosis, and the screening and the early therapy are the management. The pulmonary function tests with the diffusion capacity and the high-resolution computed tomography are performed at the baseline and the regular intervals, and the mycophenolate mofetil is the first-line immunosuppression for the progressive disease, with the cyclophosphamide for the severe and the refractory disease. The antifibrotic agent, the nintedanib, is the adjunct for the progressive fibrosis, and the oxygen and the pulmonary rehabilitation are the supportive measures. The fellow who screens and treats the interstitial lung disease early is the one who changes the outcome. [5][12]
The mixed connective-tissue disease
The mixed connective-tissue disease is managed by the dominant feature, and the high-titre anti-U1-RNP antibody is confirmed first to settle the diagnosis. The arthritis is treated with the methotrexate and the hydroxychloroquine, the myositis with the corticosteroid and the immunosuppression, and the Raynaud with the calcium channel blocker. The pulmonary hypertension is screened with the echocardiography and the pro-B-type natriuretic peptide at the baseline and the regular intervals, and the right heart catheterisation confirms it. The pulmonary arterial hypertension therapy, with the phosphodiesterase five inhibitor, the endothelin receptor antagonist and the prostacyclin, is begun early, because the pulmonary hypertension is the complication that decides the prognosis. [10][11]
SCLERO
The severe and the refractory systemic sclerosis
The severe and the refractory systemic sclerosis, with the progressive skin and the organ disease despite the conventional therapy, is the scenario for the autologous haematopoietic stem cell transplant. The transplant is the high-intensity, the specialist-centre therapy that resets the immune system, and it is reserved for the selected child with the severe, the progressive and the organ-threatening diffuse disease after the multidisciplinary assessment. The transplant carries the treatment-related mortality and the risk of the infection, and the benefit is the halting of the skin and the lung progression in the responder. The fellow holds the transplant as the last resort and cites the trials rather than the enthusiasm. [1][4]
Complications & Pitfalls
The complications of the paediatric scleroderma divide into the disease-related and the treatment-related, and the fellow must hold both. The disease-related complications of the localised scleroderma are the joint contracture, the growth asymmetry, the facial deformity and the extracutaneous involvement of the central nervous system and the eye in the facial variant. The disease-related complications of the systemic sclerosis are the interstitial lung disease, the pulmonary arterial hypertension, the renal crisis, the cardiac disease and the gastrointestinal dysmotility. The disease-related complication of the mixed connective-tissue disease is the pulmonary hypertension, the one that decides the prognosis. [1][10]
The pitfall of the high-dose corticosteroid in the systemic sclerosis is the one the fellow must never forget, because the high-dose corticosteroid above fifteen milligrams per day of prednisolone equivalent precipitates the renal crisis in the diffuse disease. The child with the diffuse systemic sclerosis is managed with the steroid-sparing immunosuppression, and the corticosteroid is reserved for the low dose and the short course, and the renal function and the blood pressure are monitored closely when the corticosteroid is unavoidable. The fellow who avoids the high-dose corticosteroid in the diffuse disease avoids the renal crisis. [1][4]
The treatment-related complications are the costs of the therapy, and the fellow must anticipate them. The methotrexate produces the hepatotoxicity, the myelosuppression and the mucositis, and the regular monitoring of the full blood count and the liver function is the safeguard. The cyclophosphamide produces the cytopenia, the infection and the gonadal toxicity, and the fertility preservation is discussed before the gonadotoxic therapy. The prostacyclin produces the hypotension and the headache, and the pulmonary arterial hypertension therapy produces the oedema and the hepatic effects. The autologous stem cell transplant carries the treatment-related mortality and the long-term infection risk. [1][4]
Prognosis & Disposition
The prognosis of the paediatric scleroderma is determined by the subtype, and the localised disease and the systemic disease sit at the opposite ends of the spectrum. The juvenile localised scleroderma carries a good prognosis for the survival, with the disease course that runs over the years and the outcome determined by the functional and the cosmetic result. The linear scleroderma of the limb produces the contracture and the limb-length discrepancy, and the linear scleroderma of the face produces the asymmetry, and the early methotrexate and the physiotherapy are the ones that improve the outcome. The disease often enters the remission after the years, but the damage that is allowed to fix is the damage that remains. [7][9]
The prognosis of the juvenile systemic sclerosis is the more serious, and it is driven by the pulmonary disease. The paediatric disease carries a better survival than the adult, but the interstitial lung disease and the pulmonary arterial hypertension remain the leading causes of the mortality. The survival is reported around the eight to the nine in ten at the five years in the modern cohorts, with the diffuse cutaneous disease and the pulmonary and the renal involvement the predictors of the worse outcome. The early diagnosis and the early organ-specific therapy are the ones that change the prognosis, and the fellow who screens and treats early is the one who improves the outcome. [3][4]
In Australia and Aotearoa New Zealand, the child with the juvenile systemic sclerosis or the mixed connective-tissue disease is managed in the tertiary paediatric rheumatology centre, with the multidisciplinary team that includes the paediatric rheumatology, the cardiology, the respiratory, the nephrology, the dermatology and the physiotherapy. The long distances are the reason the early recognition and the safe transfer from the regional or the rural hospital are so heavily weighted in the exam, and the telehealth supports the shared care with the regional services. The family is supported by the social work and the educational liaison through the long and the complex treatment.
[1]The prognosis of the mixed connective-tissue disease in the child is the intermediate, and it is decided by the pulmonary hypertension. The overall survival is favourable in the modern therapy, but the child who develops the pulmonary arterial hypertension carries the worse prognosis, and the early screening and the early therapy are the ones that change it. The disease may evolve over the years into a defined connective-tissue disease, the lupus or the systemic sclerosis, and the serial review is the safeguard. The fellow who screens for the pulmonary hypertension and the evolution is the one who manages the overlap syndrome well. [10][11]
Special Populations
The young child with the linear scleroderma is the special population in whom the growth drives the complication, because the band that crosses the joint or the face tethers the growing bone and produces the asymmetry that the child cannot outgrow. The methotrexate is begun early in the young child with the active linear disease, and the physiotherapy and the occupational therapy run alongside, and the growth and the limb length are monitored for the asymmetry. The methotrexate is dosed by the body surface area, and the dose and the monitoring are adjusted to the weight and the age. [7][9]
The adolescent with the scleroderma is prepared for the transition to the adult rheumatology and the adult scleroderma service, and the transition is the clinical act as important as the diagnosis. The reproductive and the fertility counselling, the fertility preservation before the gonadotoxic therapy, the contraception and the pregnancy planning, and the late-effects surveillance are addressed before the handover. The psychological support is the particular need of the adolescent with the facial and the limb deformity, and the body image and the peer support are the priorities. The young person leaves the paediatric service with the transition plan and the named adult provider. [1][4]
The socioeconomic disadvantage and the remoteness shape the access to the diagnosis and the specialist care, and they are the reason the early recognition in the primary care and the regional hospital is so heavily emphasised. The child far from the specialist centre may first present to the clinician who sees few such cases, and the indurated skin lesion and the Raynaud phenomenon are the bridge to the retrieval and the specialist care. The language and the cultural barriers are addressed with the interpreter and the cultural support, and the telehealth supports the shared care across the distance. The migrant and the refugee child may present late, and the index of suspicion is the safeguard. [6][8]
Evidence, Guidelines & Regional Differences
The landmark evidence for the paediatric scleroderma is the product of the international collaborations that built the classification, the cohorts that defined the phenotype, and the consensus that standardised the treatment. The Zulian classification of the juvenile localised scleroderma, drawn from the seven hundred and fifty children, is the one that established the five subtypes and the dominance of the linear form in the child. The provisional classification criteria for the juvenile systemic sclerosis, built by the Pediatric Rheumatology European Society, the American College of Rheumatology and the European League Against Rheumatism, are the ones that standardised the diagnosis for the child. The recent multicentre cohort of the juvenile-onset mixed connective-tissue disease has refined the paediatric phenotype and confirmed the pulmonary hypertension as the decisive complication. [6][2][10]
The evidence for the methotrexate in the juvenile localised scleroderma is the product of the trials and the consensus, and the Childhood Arthritis and Rheumatology Research Alliance consensus treatment plans have standardised the methotrexate-based therapy for the active disease. The evidence supports the methotrexate with the systemic corticosteroid bridging for the linear, the deep and the generalised disease, and the mycophenolate for the refractory disease. The evidence for the systemic sclerosis therapy is drawn largely from the adult trials and extrapolated to the child, with the mycophenolate and the cyclophosphamide for the interstitial lung disease and the nintedanib for the progressive fibrosis. [9][12]
The classification of the mixed connective-tissue disease rests on the four criteria sets, the Sharp, the Alarcon-Segovia, the Kasukawa and the more recent Bennett criteria, and all require the high-titre anti-U1-RNP antibody alongside the overlapping clinical features. The criteria differ in the precise clinical and the serological combination, and the comparison studies show the concordance and the discordance across the sets. The fellow should know the common thread, the high-titre anti-U1-RNP, and the distinguishing feature of each criterion, and cite the comparison rather than the single criterion.
[11]The controversies and the open questions are the live ones. The optimal duration of the methotrexate in the localised scleroderma, balanced between the relapse and the overtreatment, is one. The role of the biologic agents, the tocilizumab and the abatacept, in the refractory disease is being explored. The place of the autologous haematopoietic stem cell transplant in the severe paediatric systemic sclerosis, balanced against the treatment-related mortality, is the open question that the specialist centres are addressing. The fellow holds these as the open questions and cites the trials and the guidelines rather than the dogma. [1][4]
Exam Pearls
The high-yield facts for the exam are the ones that change a decision at the bedside, and they are worth carrying as the sharp statements. The localised scleroderma is the commoner paediatric form, the linear subtype dominates with around two-thirds, and the systemic features are absent. The juvenile systemic sclerosis is defined by the proximal skin sclerosis plus at least two minor criteria under the PReS and ACR provisional criteria, and the pulmonary disease is the leading cause of the mortality. The methotrexate is the first-line systemic therapy for the active localised disease, given at fifteen milligrams per square metre per week. [6][2][9]
The anti-U1-RNP antibody is the defining serology of the mixed connective-tissue disease, and the pulmonary hypertension is its feared complication. The scleroderma renal crisis is treated with the angiotensin-converting enzyme inhibitor even in the renal failure, and the high-dose corticosteroid above fifteen milligrams per day of prednisolone equivalent is avoided because it precipitates the crisis. The anti-Scl-70 marks the diffuse disease and the interstitial lung disease, and the anti-centromere marks the limited disease and the pulmonary hypertension; in the child, the anti-Scl-70 is more common than the anti-centromere. [1][10][11]
The final pearls are the ones that catch the candidate who has learned the headline and forgotten the corner. The en coup de sabre and the Parry-Romberg are the facial linear scleroderma, and they carry the seizures and the uveitis. The nailfold capillaroscopy is the bridge from the isolated Raynaud to the diagnosis, and the dilated capillaries and the dropout confirm the systemic sclerosis pattern. The CREST syndrome, the calcinosis, the Raynaud, the esophageal dysmotility, the sclerodactyly and the telangiectasia, is the limited cutaneous systemic sclerosis. The pulmonary hypertension is screened with the echocardiography and the pro-B-type natriuretic peptide, and the right heart catheterisation confirms it. The corners are where the marks are won. [1][7]
References
- [1]Foeldvari I, Li SC, Wu E, Ting TV, Stevens AM Juvenile systemic sclerosis Best Pract Res Clin Rheumatol, 2026.PMID 41638996
- [2]Zulian F, Woo P, Athreya BH, Laxer RM, Medsger TA Jr, Lehman TJ, Cerinic MM, Martini G, Ravelli A, Russo R, Cuttica R, de Oliveira SK, Denton CP, Cozzi F, Foeldvari I, Ruperto N The Pediatric Rheumatology European Society/American College of Rheumatology/European League against Rheumatism provisional classification criteria for juvenile systemic sclerosis Arthritis Rheum, 2007.PMID 17330294
- [3]Foeldvari I, Tarantino HA, Shah U, Li SC, Stevens A, Akgul O, Goldzweig O, Fuhlbrigge RC, Jerath R, Lasky A, O'Neil KM, Vora SS, Zeft A Are diffuse and limited juvenile systemic sclerosis different in clinical presentation? Clinical characteristics of a juvenile systemic sclerosis cohort J Scleroderma Relat Disord, 2019.PMID 35382144
- [4]Jindal AK, Pilkington CA, Vignesh P, Rawat A, Sharma M, Suri D, Gupta A, Saikia B, Singh S Clinical profile, long-term follow-up and outcome of juvenile systemic scleroderma: 25 years of clinical experience from North-West India Clin Exp Rheumatol, 2021.PMID 34251299
- [5]Di Pasquale G, Santilli F, Mosca M, Tonti C, D'Onghia S, Marrani E, Candelli M, Caffarelli C, Foeldvari I, Simonini G, Resti M Pulmonary manifestations of juvenile vs. adult systemic sclerosis: insights into pathophysiological and clinical features Pediatr Pulmonol, 2025.PMID 39545645
- [6]Zulian F, Athreya BH, Laxer R, Nelson AM, de Oliveira SK, Punaro MG, Cuttica R, Anton J, Rakov N, Medsger TA Jr, Garcia-Consuegra J, Ozen S, Laxer RM Juvenile localized scleroderma: clinical and epidemiological features in 750 children. An international study Rheumatology (Oxford), 2006.PMID 16368732
- [7]Martini G, Fadanelli G, Agazzi A, Vittadello F, Meneghel A, Zulian F Disease course and long-term outcome of juvenile localized scleroderma: Experience from a single pediatric rheumatology Centre and literature review Autoimmun Rev, 2018.PMID 29729451
- [8]Kaushik A, Gupta M, Singh S Paediatric morphoea: a holistic review. Part 1: epidemiology, aetiopathogenesis and clinical classification Clin Exp Dermatol, 2020.PMID 32472964
- [9]Kaushik A, Gupta M, Singh S Paediatric morphoea: a holistic review. Part 2: diagnosis, measures of disease activity, management and natural history Clin Exp Dermatol, 2020.PMID 32449205
- [10]Chevalier K, Bader-Meunier B, Kone-Paut I, Terrier B, Hachulla E, Mouthon L, Chaigne B, Costedoat-Chalumeau N Juvenile-onset mixed connective tissue disease: A multicenter retrospective cohort study Semin Arthritis Rheum, 2026.PMID 41412094
- [11]John KJ, Panizza AR, Antony T, Nadaraj C, Sridhar S, Mathew JL, Batra N, Vyankatesh T, Balamugesh T, Gupta R Clinical and Immunological Profile of Mixed Connective Tissue Disease and a Comparison of Four Diagnostic Criteria Int J Rheumatol, 2020.PMID 32411251
- [12]Antoniou KM, Margaritopoulos GA, Tzouvelekis A, Goh NS, Bonifazi M, Fui A, Arroyo MJ, Windisch W, Renzoni E, Solomon JJ, Boeynaems JM, Vassallo R, De Sadeleer L, Blanco-Dominguez R, Bargagli E, Gerosa M, De Lorenzis R, Kamińska J, Lasky SU, Wells AU ERS/EULAR clinical practice guidelines for connective tissue disease-associated interstitial lung disease developed by the European Task Force Ann Rheum Dis, 2026.PMID 40912974