Paeds · rheumatology-musculoskeletal-and-sports
Systemic juvenile idiopathic arthritis and macrophage activation syndrome
Also known as Systemic juvenile idiopathic arthritis · Still disease · Macrophage activation syndrome · MAS · Secondary hemophagocytic lymphohistiocytosis · Cytokine storm syndrome · Still disease spectrum
Fellowship guide to systemic juvenile idiopathic arthritis and its life-threatening complication, macrophage activation syndrome. Covers the ILAR classification of sJIA with its quotidian fever and evanescent rash, the reframing of sJIA as an autoinflammatory disease driven by the innate-immune IL-1 and IL-6 axis, the pathophysiology of the macrophage activation syndrome as an uncontrolled IFN-gamma cytokine storm, the 2016 EULAR ACR PRINTO classification criteria with their ferritin threshold and the platelet, AST, triglyceride and fibrinogen cut-offs, the HScore and HLH-2004 framework, the IL-1 blockade with anakinra and canakinumab, the IL-6 blockade with tocilizumab, the management of MAS with high-dose glucocorticoids, ciclosporin and anakinra, and the escalation to etoposide and emapalumab for the refractory case.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
Overview & Definition
A child with systemic juvenile idiopathic arthritis sits on a different kind of edge from the other JIA subtypes, because the disease is not primarily a disease of joints. It is an autoinflammatory disease of the innate immune system, driven by a storm of interleukin-one and interleukin-six, and the joints are sometimes the last thing to declare themselves. The fever comes first, spiking once or twice every day and returning to normal between spikes, and a salmon-pink rash appears with the fever and fades when it settles. The danger that makes this topic a fellowship must-know is the one complication that can kill a child within days: macrophage activation syndrome, the uncontrolled cytokine storm in which the T-cells and macrophages spiral out of control and the ferritin climbs to tens of thousands while the platelets and the fibrinogen collapse. [2][9]
The whole point of understanding sJIA is to stand between the child and the macrophage activation syndrome. The modern treatment is built on the recognition that the disease is driven by interleukin-one and interleukin-six, and the interleukin-one blockade with anakinra or canakinumab does not just control the arthritis and the fever but also protects against the macrophage activation syndrome by damping the very cytokine axis that drives it. The 2024 EULAR and PReS recommendations now formally reframe sJIA as Still disease, uniting it with adult-onset Still disease across a single spectrum of innate-immune dysregulation. [9]
Classification
Begin with the ILAR classification, because it is the framework every paediatric examination expects, and the systemic subtype stands apart from the other six for a reason that matters clinically. The ILAR criteria, published in their second revision in 2004, divide juvenile idiopathic arthritis into seven categories, all sharing the requirement of an onset before sixteen years and a duration of at least six weeks. The systemic subtype is defined not by the pattern of the arthritis but by the systemic fever that accompanies or precedes it. The child must have arthritis in one or more joints, with or preceded by a daily quotidian fever for at least two weeks, documented for at least three days of each week, and at least one of four extra-articular features: an evanescent and non-fixed erythematous rash, a generalised lymphadenopathy, a hepatomegaly or splenomegaly, and a serositis that most often involves the pericardium. [2]
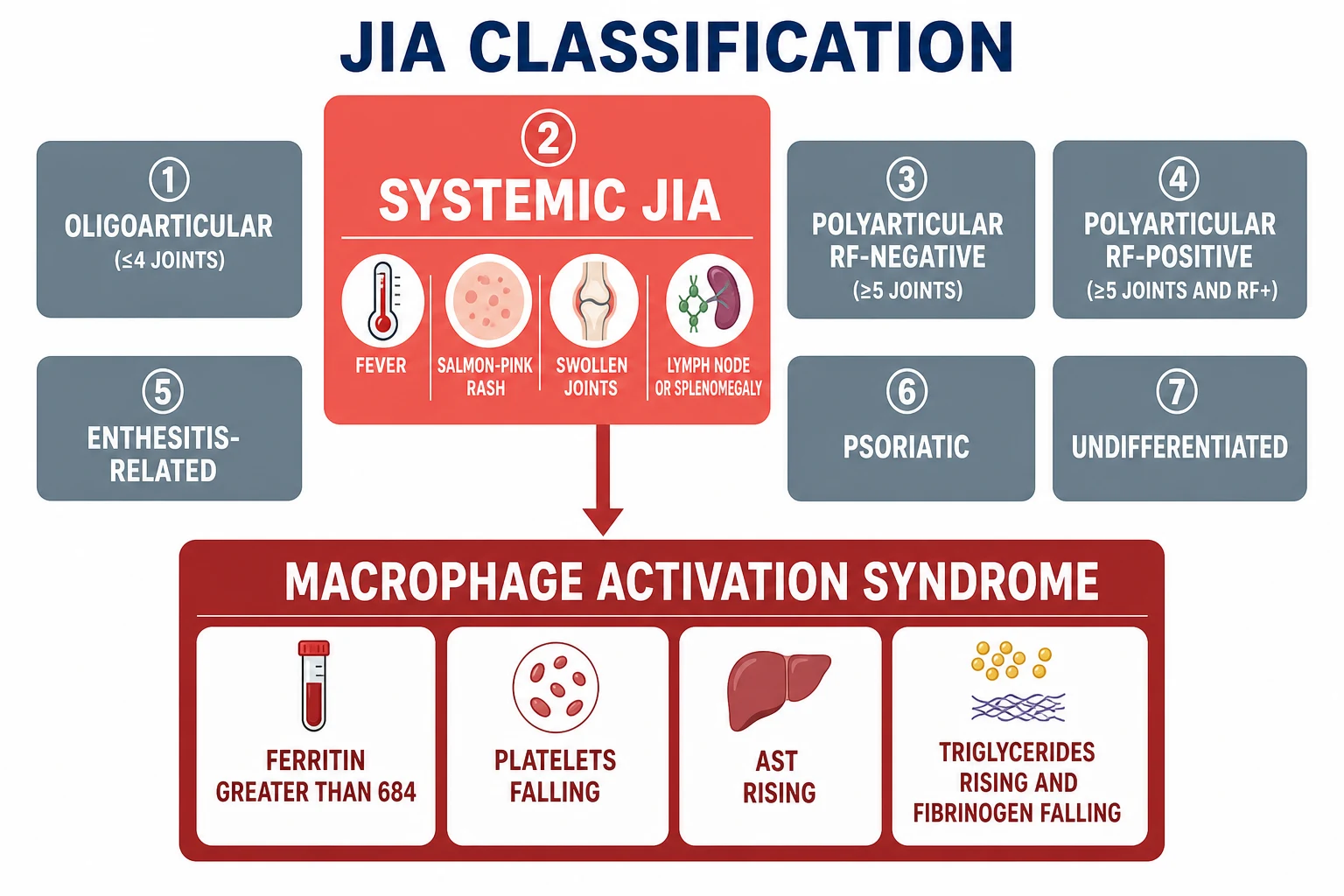
The other six ILAR categories exist to be distinguished from the systemic subtype, and the candidate should carry them as a short list. The oligoarthritis category affects four or fewer joints in the first six months and carries the highest risk of chronic anterior uveitis. The rheumatoid-factor-negative and the rheumatoid-factor-positive polyarthritis categories affect five or more joints. The enthesitis-related arthritis category overlaps with the spondyloarthropathies and is associated with HLA-B27. The psoriatic arthritis category pairs the arthritis with psoriasis or a psoriasis diathesis. The undifferentiated category holds the children who fulfil no category, more than one, or have an exclusion. The systemic subtype carries the exclusion of a first-degree relative with sJIA, which is why the family history matters. [2]
The reframing of sJIA as Still disease under the 2024 EULAR and PReS recommendations is the development that the current fellowship candidate must carry, because it changes how the disease is understood and treated. Still disease is reframed as a single entity across the age spectrum, driven by the innate-immune interleukin-one and interleukin-six axis rather than the adaptive-autoimmune pathways that drive the other JIA subtypes. This is why the interleukin-one and interleukin-six blockade is so effective in sJIA and not in the others, and it is why the macrophage activation syndrome complicates sJIA far more than any other subtype. [9]
Epidemiology & Risk Factors
Systemic juvenile idiopathic arthritis accounts for around ten to fifteen percent of all juvenile idiopathic arthritis, with an incidence of roughly zero point three to zero point nine per one hundred thousand children per year. It has a bimodal age distribution with peaks at one to four years and around nine to twelve years, and it affects boys and girls equally, unlike the female predominance of the oligoarticular and polyarticular subtypes. The disease occurs worldwide, with some studies suggesting a higher frequency and severity in populations of Asian ancestry, though the genetics are polygenic and not fully characterised. [9]
The macrophage activation syndrome is the complication that every candidate must anticipate. It develops in roughly ten to twenty percent of children with sJIA across the disease course, though the reported rates vary widely and the true incidence is probably higher because the mild and the subclinical cases go unrecognised. The risk is highest at the disease onset and during a flare, and it is heightened by a change in the medication, a new infection, or the paradoxical effect of the interleukin-six blockade that can mask the syndrome. The mortality of the untreated macrophage activation syndrome is high, historically reported at twenty to thirty percent and now reduced to below ten percent in the biologic era with the early recognition and the interleukin-one blockade. [8][10]
The triggers of the macrophage activation syndrome in the child with sJIA include the viral and bacterial infections, especially the Epstein-Barr virus, the cytomegalovirus and the parvovirus, a change in the immunosuppression including the tapering of the glucocorticoids or the biologics, and the disease flare itself. The child with a very high baseline interleukin-eighteen level is at a particular risk, because the interleukin-eighteen primes the T-cells and the macrophages for the uncontrolled activation when the second signal arrives. [10]
Pathophysiology
The key to understanding sJIA is to abandon the adaptive-autoimmune framework and adopt the autoinflammatory one. The innate immune cells, the monocytes and the macrophages and the neutrophils, are the primary drivers. They produce large amounts of interleukin-one-beta and interleukin-six in a dysregulated loop that is not held in check by the normal regulatory mechanisms. The interleukin-one-beta drives the quotidian fever by its effect on the hypothalamic prostaglandin pathway, and it activates the endothelial cells that produce the evanescent rash. The interleukin-six drives the acute-phase response, the thrombocytosis, the anaemia of chronic disease and the growth failure. The arthritis is a downstream effect of the systemic cytokine spillover into the synovium, which is why the arthritis may not appear for weeks or months after the fever begins. [9]
The interleukin-eighteen is the third cytokine that completes the pathogenic picture, and it is the bridge to the macrophage activation syndrome. The sJIA macrophages produce vast amounts of interleukin-eighteen, and the high interleukin-eighteen level primes the natural-killer cells and the T-cells for an exaggerated response. The interleukin-eighteen, together with the interleukin-one and the interleukin-six, keeps the innate immune system in a state of constant readiness, and when a second hit arrives, whether an infection or a flare, the system tips over into the uncontrolled cytokine storm. [10]
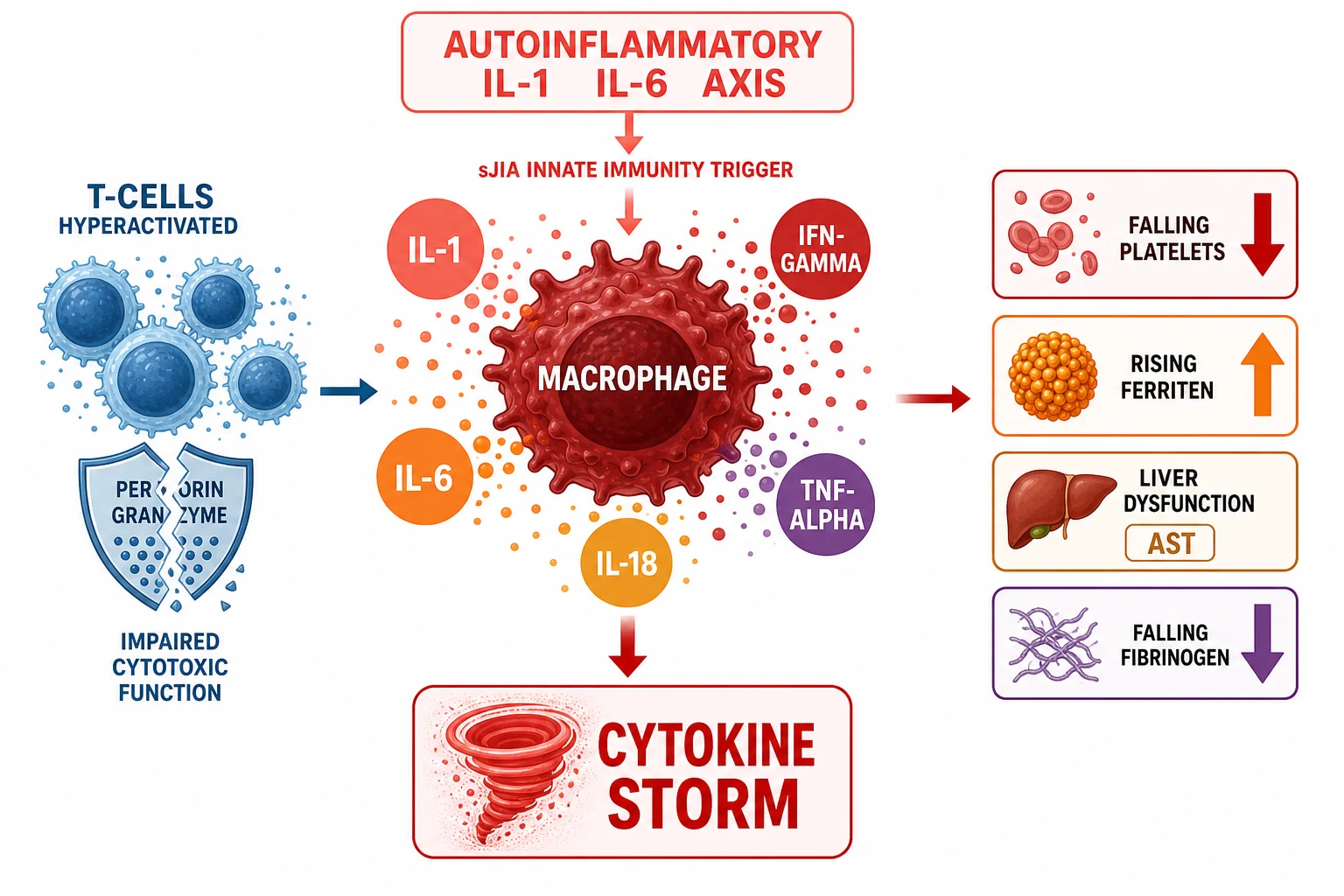
The macrophage activation syndrome is the endpoint of this uncontrolled activation, and it is driven primarily by interferon-gamma. The T-cells, activated beyond their normal regulatory threshold, produce enormous amounts of interferon-gamma, and the macrophages, activated by the interferon-gamma, release their entire arsenal of cytokines and consume themselves in a haemophagocytic frenzy. The ferritin is secreted by the activated macrophages and climbs to tens of thousands of nanograms per millilitre. The platelets and the fibrinogen are consumed by the combination of the haemophagocytosis and the disseminated intravascular coagulation. The liver is damaged by the direct cytokine toxicity and the Kupffer-cell activation, driving the AST and the bilirubin up. The triglycerides rise because the interferon-gamma upregulates the lipoprotein lipase suppression. [7][10]
The impaired cytotoxic function is the deeper mechanism that explains why the storm is not terminated. In the primary haemophagocytic lymphohistiocytosis, a genetic defect in the perforin or the degranulation pathway (the PRF1, the UNC13D, the STX11 or the STXBP2 genes) prevents the cytotoxic T-cells and the natural-killer cells from killing the infected or the activated cells, so the immune activation cannot be switched off. In the sJIA-associated macrophage activation syndrome, the cytotoxic function is impaired functionally rather than genetically, through the downregulation of the natural-killer-cell activity, but the consequence is the same: the immune response spirals on without a brake. [3][7]
Clinical Presentation
The presentation of sJIA begins with the fever, and the fever is the key to the diagnosis. The quotidian pattern means the fever spikes once or twice each day, usually in the late afternoon or the evening, and it returns to normal or below normal between the spikes. This return to baseline is what distinguishes the sJIA fever from the persistent fever of an infection or a malignancy, and it is the feature the examination rewards. The fever is typically high, over thirty-nine degrees Celsius, and it persists for at least two weeks before the diagnosis is made. [2]
The evanescent rash is the second hallmark, and it is the feature that the short-case examiner looks for. The rash is salmon-pink or erythematous, macular or slightly raised, and it appears on the trunk and the proximal limbs in concert with the fever spikes. It is non-pruritic and migratory, and it may be brought out by a warm bath or by rubbing the skin, the Koebner phenomenon. The rash fades when the fever settles, which is why it may be missed if the child is examined only when afebrile. [2]
The arthritis is the paradox of sJIA, because it may not be present at the onset and it may never become the dominant feature. When it does appear, it can affect any number of joints, from a single large joint to a symmetrical polyarthritis of the small and the large joints. The child may also have a generalised lymphadenopathy, a hepatosplenomegaly, and a serositis that most often presents as a pericarditis or a pleuritis. The pericarditis can be silent or it can present with chest pain, and a pericardial effusion large enough to cause a tamponade is the rare but the recognised emergency. [2][9]
The presentation of the macrophage activation syndrome is the presentation that the candidate must not miss, and it is the one that changes everything. The fever pattern changes from the quotidian spikes to a persistent, unremitting fever that does not return to normal. The child becomes more ill, not less, despite the ongoing treatment. The platelet count, which was previously high or normal in the active sJIA, begins to fall, and this falling platelet trend is the earliest and the most reliable single sign. The ferritin rises sharply, often exceeding ten thousand nanograms per millilitre. The fibrinogen falls, which is why the ESR paradoxically drops even though the disease is worsening. The child may develop a coagulopathy with bleeding, an encephalopathy with confusion or seizures, a hepatic dysfunction, and a renal dysfunction. [1][8]
Differential Diagnosis
The differential of the child presenting with fever, rash and arthritis is wide, and the candidate must hold the dangerous mimics and the treatable look-alikes. The first mimic is the infection, because the quotidian fever and the rash of sJIA can resemble a viral exanthem, a Kawasaki disease, or a reactive arthritis. The sepsis and the osteomyelitis must be excluded, and the blood cultures, the echocardiogram and the joint aspirate are the tools. The subacute bacterial endocarditis can produce a fever and an arthritis through an immune-complex mechanism, and the blood cultures and the echocardiogram are the keys. [9]
The second group of mimics is the malignancy, and this is the one that must never be missed. The acute leukaemia can present with a fever, a bone pain and an arthritis, and the clue is the cytopenia, the blast on the blood film, and the infiltration on the marrow. The lymphoma, and especially the T-cell lymphoma with an anterior mediastinal mass, can present with the systemic features. The full blood count with the differential, the blood film, and the lactate dehydrogenase are the screening tools, and the bone marrow aspirate is the definitive test when the suspicion is high. [9][10]
sJIA
Leukaemia
Kawasaki disease
Autoinflammatory fever syndrome
The differential of the macrophage activation syndrome itself is narrower but no less critical. The syndrome must be distinguished from the sepsis, because the two share the fever, the multiorgan failure and the rising inflammatory markers, and the child with MAS may also have a triggering infection. The key is the ferritin and the fibrinogen: the MAS produces the very high ferritin with the falling fibrinogen, while the uncomplicated sepsis produces the high ferritin with the normal or rising fibrinogen. The other secondary haemophagocytic syndromes, from the lymphoma-associated HLH, the infection-associated HLH and the autoimmune lymphoproliferative syndrome, present in the same way and require the same framework. [3][4]
The drug reaction with eosinophilia and systemic symptoms, the DReSS syndrome, is the mimic that can be missed because it develops weeks after the exposure to a new drug and it produces the fever, the rash, the eosinophilia and the multiorgan failure. The history of the recent drug exposure, the eosinophilia, and the timing are the clues, and the distinction matters because the management is the withdrawal of the offending drug and the glucocorticoid rather than the interleukin-one blockade. [10]
Clinical & Bedside Assessment
The focused assessment of the child with suspected sJIA begins with the fever pattern, because the quotidian return to baseline is the feature that sets sJIA apart. Ask the parents to record the temperature at regular intervals, because the pattern is more informative than any single reading. Examine the skin during the fever spike, because the rash is evanescent and may be absent when the child is afebrile. Examine all the joints, because the arthritis may be subtle and the child may guard the affected joint. Palpate the lymph nodes, the liver and the spleen, because the organomegaly is part of the definition. Auscultate the heart and the lungs, because the pericarditis and the pleuritis may be silent. [2]
The assessment of the child with the suspected macrophage activation syndrome is the assessment that must be fast and focused, because the syndrome progresses within hours to days. The level of consciousness is the first thing, because the encephalopathy is the sign that the brain is involved and the indicator that the syndrome is severe. The blood pressure and the perfusion are the next, because the haemodynamic instability and the shock may develop. The skin is examined for the petechiae and the bruising of the coagulopathy. The liver is palpated for the enlargement and the tenderness. The urine output is monitored, because the oliguria is the sign of the renal involvement. [1][8]
The bedside synthesis that should trigger the concern for the MAS is the combination of a changing fever pattern, a falling platelet count, a rising AST, a falling fibrinogen, and a rapidly rising ferritin in the child with known or suspected sJIA. The candidate who is asked to present the findings at the bedside should lead with the trajectory, not the absolute numbers, because the trend is more informative than any single value, and the child whose platelets are falling from six hundred to four hundred to two hundred is heading for the MAS even if the absolute count is still in the normal range. [1][10]
Investigations
The first-line panel for the child with suspected sJIA is the full blood count, the inflammatory markers, the liver function, the ferritin, the fibrinogen, the triglycerides, and the infectious and the autoimmune screen. The active sJIA typically shows a leukocytosis with a neutrophilia, a thrombocytosis, an anaemia of chronic disease, a markedly elevated CRP and ESR, and a ferritin that is high but usually under ten thousand. The autoimmune screen, including the antinuclear antibody and the rheumatoid factor, is typically negative in sJIA, and this is part of what distinguishes it from the other subtypes. The infection screen excludes the mimics, and the blood film excludes the leukaemia. [2][9]
The monitoring of the ferritin trend is the cornerstone of the early MAS detection, because the ferritin rises sharply before the full-blown syndrome declares itself. The platelet count, the fibrinogen, the triglycerides and the AST are monitored alongside the ferritin, because the 2016 criteria require the ferritin plus two of these. The coagulation profile is checked for the disseminated intravascular coagulation, and the D-dimer is a sensitive marker of the activation. The lactate dehydrogenase is a marker of the tissue damage and the haemophagocytosis. [1]
The 2016 EULAR, ACR and PRINTO classification criteria for the macrophage activation syndrome complicating sJIA are the criteria the examination rewards, and they turn the clinical suspicion into an objective set of numbers. A febrile patient with known or suspected sJIA is classified as having MAS if the ferritin is over six hundred and eighty-four nanograms per millilitre and any two of the following are present: a platelet count under one hundred and eighty-one times ten to the nine per litre, an AST over forty-eight units per litre, triglycerides over one hundred and fifty-six milligrams per decilitre, and a fibrinogen under three hundred and sixty milligrams per decilitre. These criteria have a sensitivity of around seventy-three percent and a specificity of around ninety-nine percent, and they were derived and validated in a large international cohort. [1]
The HLH-2004 criteria, developed for the primary familial haemophagocytic lymphohistiocytosis, provide the broader diagnostic framework that extends beyond sJIA. The diagnosis is made by a molecular diagnosis consistent with HLH or by meeting five of eight criteria: a fever, a splenomegaly, a cytopenia of two or more lineages with a haemoglobin under ninety, a platelet count under one hundred and a neutrophil count under one, a hypertriglyceridaemia over three millimoles per litre and or a hypofibrinogenaemia under one point five grams per litre, a haemophagocytosis in the marrow or the spleen or the lymph node, a low or absent natural-killer-cell activity, a ferritin over five hundred micrograms per litre, and a soluble interleukin-two receptor over two thousand four hundred units per millilitre. These criteria are used for the MAS that does not meet the 2016 sJIA-specific criteria or for the child where the primary HLH is in the differential. [3]
The HScore, developed and validated by Fardet and colleagues in 2014, is the tool for the reactive haemophagocytic syndrome that combines the clinical and the laboratory variables into a single probability. It incorporates the known immunosuppression, the known malignancy, the temperature, the organomegaly, the number of cytopenias, the ferritin, the triglycerides, the fibrinogen, the AST and the serum albumin, and a score over around one hundred and sixty-nine carries a high probability of the reactive haemophagocytic syndrome. The HScore is useful when the 2016 criteria do not apply, because the patient does not have sJIA or the presentation is atypical. [4]
The bone marrow aspirate is not required to make the diagnosis of the MAS, because the haemophagocytosis is neither sensitive nor specific, and its absence does not exclude the syndrome. The aspirate is done to exclude the malignancy, and the flow cytometry for the perforin and the degranulation assays (the CD107a surface exposure) screens for the primary HLH when the family history or the early onset raises the suspicion. The soluble interleukin-two receptor, the natural-killer-cell function, and the genetic panel are the tools for the confirmed or the refractory case where the primary HLH is in the differential. [3][7]
Management — Resuscitation
The child with the established macrophage activation syndrome is a medical emergency, and the resuscitation follows the structured approach. The airway and the breathing are secured, because the encephalopathy may compromise the airway. The circulation is supported with the fluid boluses and the vasopressors, because the shock may be distributive from the cytokine storm and cardiogenic from the myocardial depression. The coagulopathy is corrected with the fresh frozen plasma and the platelets, and the bleeding is treated aggressively. The child is transferred to the paediatric intensive care early, because the syndrome can deteriorate within hours. [7]
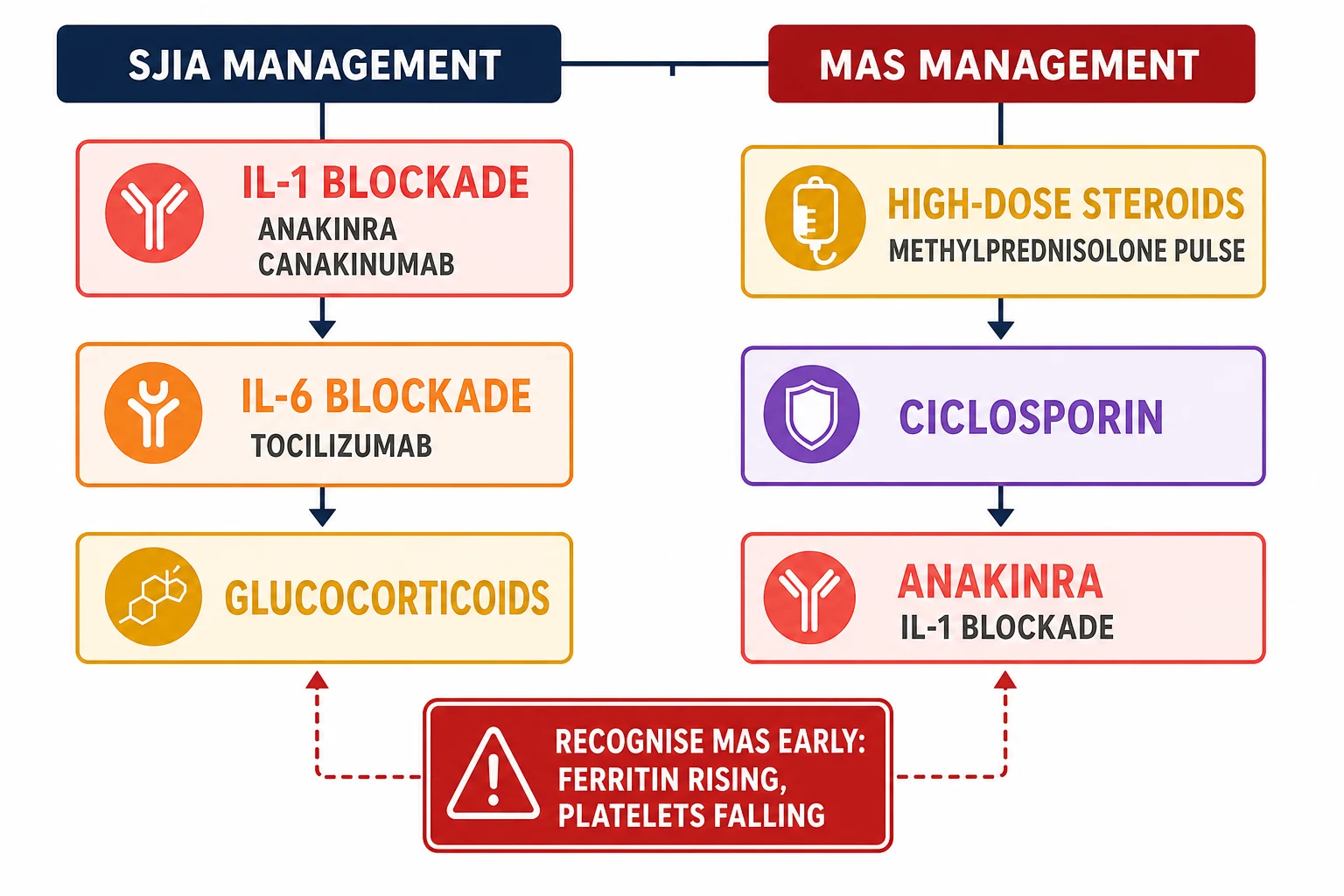
The high-dose glucocorticoid is the first specific therapy, and it is given as an intravenous methylprednisolone pulse at ten to thirty milligrams per kilogram per day, up to one gram per day, for three to five days, followed by a tapering oral or intravenous prednisolone. The glucocorticoid dampens the cytokine production and the macrophage activation, and it is the backbone of the MAS treatment alongside the ciclosporin and the anakinra. [7][10]
The ciclosporin, a calcineurin inhibitor that suppresses the T-cell activation, is added at two to seven milligrams per kilogram per day intravenously or orally, with the trough monitoring for the renal and the hepatic toxicity. The anakinra, the recombinant interleukin-one receptor antagonist, is given at two to four milligrams per kilogram per day subcutaneously and escalated to four to eight milligrams per kilogram per day in the refractory case. The anakinra is increasingly used as the first-line therapy for the sJIA-associated MAS, because the interleukin-one is central to the pathogenesis, and the early blockade can abort the syndrome before the full-blown multiorgan failure develops. [10]
The trigger is sought and treated concurrently, because the MAS is often precipitated by an infection that must be identified and managed. The broad-spectrum antibiotics are given empirically for the presumed sepsis until the cultures are negative, because the sepsis and the MAS are clinically indistinguishable at the onset and may coexist. The intravenous immunoglobulin is considered for the suspected viral trigger, and the underlying disease flare is controlled by the optimisation of the sJIA therapy. [7][10]
Management — Definitive & Stepwise
The definitive treatment of sJIA is built on the interleukin-one and the interleukin-six blockade, and the modern approach is the treat-to-target with the early biologic to achieve the clinically inactive disease and to spare the glucocorticoids. The interleukin-one blockade is the first-line biologic for the systemic features, because the interleukin-one-beta is the primary driver. The anakinra, a recombinant interleukin-one receptor antagonist, is given at one to two milligrams per kilogram per day subcutaneously and is titrated up; it has the advantage of the rapid onset and the short half-life that allows both the aggressive dosing and the rapid withdrawal if the infection is the concern. The canakinumab, a fully human monoclonal antibody against the interleukin-one-beta, is given at four milligrams per kilogram every four weeks subcutaneously, up to a maximum of three hundred milligrams per dose, and it is approved for the sJIA in children aged two years and older. [6][9]
The interleukin-six blockade is the second pillar of the sJIA treatment. The tocilizumab, a monoclonal antibody against the interleukin-six receptor, was proven in the 2012 phase-three trial by De Benedetti and colleagues to be effective in the systemic features and the arthritis. The dose is twelve milligrams per kilogram intravenously every two weeks for the child under thirty kilograms, and eight milligrams per kilogram every two weeks for the child of thirty kilograms or above, up to a maximum of eight hundred milligrams per dose. The subcutaneous formulation at one hundred and sixty-two milligrams every week for the child of thirty kilograms or above is an alternative. [5]
The glucocorticoids are the bridge, not the foundation, of the sJIA treatment in the biologic era. They are given as an intravenous methylprednisolone pulse for the severe presentation, followed by an oral prednisolone at one to two milligrams per kilogram per day that is tapered as the biologic takes effect. The aim is to spare the glucocorticoids, because the chronic exposure causes the growth failure, the osteoporosis, the Cushingoid appearance, and the adrenal suppression, and the modern treat-to-target with the early interleukin-one blockade reduces the cumulative dose significantly. The methotrexate and the other conventional disease-modifying drugs are used for the arthritis but they are less effective for the systemic features, because the systemic inflammation is driven by the innate-immune cytokines that the methotrexate does not target. [9]
The management of the refractory macrophage activation syndrome escalates to the etoposide and the emapalumab. The etoposide, the topoisomerase inhibitor that suppresses the activated T-cells and the macrophages, is given at one hundred and fifty milligrams per square metre per dose on the HLH-2004 protocol, and it is reserved for the case that has not responded to the glucocorticoids, the ciclosporin and the anakinra. The emapalumab, a monoclonal antibody against the interferon-gamma, is the newer agent that targets the key cytokine of the MAS directly, and it is approved for the refractory primary HLH and increasingly used for the refractory sJIA-associated MAS. The ruxolitinib, a JAK inhibitor that blocks the interferon-gamma signalling, is another emerging option. The haematopoietic stem cell transplantation is the curative option for the primary HLH and is used for the refractory sJIA with the lung disease in the specialist centre. [3][7]
Recognise the MAS: persistent fever with falling platelets and rising ferritin in the febrile child with sJIA; apply the 2016 criteria
Resuscitate: secure the airway and the circulation, correct the coagulopathy, transfer to the PICU early
First-line therapy: intravenous methylprednisolone pulse at ten to thirty mg per kg per day, ciclosporin at two to seven mg per kg per day, anakinra at two to four mg per kg per day
Escalate if refractory: increase the anakinra to four to eight mg per kg per day, consider the etoposide or the emapalumab
Treat the trigger: broad-spectrum antibiotics empirically, intravenous immunoglobulin for the suspected viral cause, optimise the sJIA biologic therapy
Plan the recovery: glucocorticoid taper, biologic optimisation, infection surveillance and the long-term follow-up
Specific Subtypes & Scenarios
The newly diagnosed child at the first presentation is the scenario where the MAS risk is highest, because the disease is at its most active and the treatment is not yet in place. The initial workup includes the exclusion of the infection and the malignancy, and the ferritin and the coagulation are checked at the baseline. The interleukin-one blockade is started early, because the early treatment with the anakinra reduces both the disease activity and the MAS risk, and the glucocorticoid is used as the bridge. [9][10]
The paradoxical MAS under the tocilizumab is the scenario that the examination loves, because it tests the understanding of the mechanism. The interleukin-six blockade suppresses the fever and the CRP, which are the usual warning signs of the MAS, so the syndrome can develop silently with only the falling platelets and the rising ferritin to betray it. The mechanism is that the tocilizumab blocks the interleukin-six but not the interferon-gamma, so the underlying interferon-gamma storm continues unabated beneath the masked exterior. The management is to stop or to reduce the tocilizumab and to add the anakinra and the glucocorticoid. This is why the regular monitoring of the ferritin and the platelet count is essential for the child on the tocilizumab, even when the CRP is normal. [5][10]
The sJIA-associated lung disease is the complication that has emerged as a major cause of the morbidity and the mortality in the biologic era, and it is the one that the candidate should mention to show the depth. It manifests as a chronic dry cough, an exertional dyspnoea, a pulmonary arterial hypertension, and an interstitial lung disease, and it is associated with the high interleukin-eighteen and the chronic macrophage activation. The high-resolution CT of the chest, the echocardiogram, and the pulmonary function testing are the tools, and the management is the aggressive immunosuppression with the biologics, the glucocorticoids and, in the severe case, the haematopoietic stem cell transplantation. [9]
The very young child and the adolescent present their own challenges. The infant and the toddler with sJIA require the weight-based dosing of the anakinra and the careful attention to the growth and the development, and the live vaccines are deferred while the biologics are given. The adolescent with sJIA needs the transition planning into the adult care, with the continuation of the biologic therapy, the reproductive counselling for the teratogenic drugs such as the methotrexate, and the attention to the adherence and the psychosocial adjustment to the chronic illness. [9]
Complications & Pitfalls
The complications of the macrophage activation syndrome itself are the multiorgan failure, the disseminated intravascular coagulation, the encephalopathy, the renal failure, the hepatic failure and the death. The child who survives may have a long intensive-care stay, a prolonged recovery, and the cognitive sequelae of the encephalopathy. The MAS can also trigger the sJIA lung disease, and the chronic macrophage activation is a risk for the pulmonary arterial hypertension. [7][10]
The complications of the biologic therapy are the infection and the paradoxical inflammation. The interleukin-one and the interleukin-six blockade increase the risk of the serious and the opportunistic infections, and the screening for the latent tuberculosis, the hepatitis and the other infections is done before the start. The tocilizumab causes the neutropenia and the transaminitis, which are monitored with the regular blood counts and the liver function. The paradoxical MAS under the tocilizumab is the complication described above, and the canakinumab may rarely trigger the MAS. The chronic glucocorticoid exposure causes the growth failure, the osteoporosis, the adrenal suppression and the metabolic syndrome. [5][6]
The classic pitfalls are the missing of the early MAS by the reliance on a single normal ferritin or a normal CRP at one time point, the reassurance by a falling ESR, and the delay in the anakinra and the intensive-care escalation. The MAS is a clinical and a laboratory trend diagnosis, and the single static value is misleading. The candidate who is asked about the pitfall at the viva should state that the trend matters more than any single value, and that the child whose platelets and fibrinogen are falling and whose ferritin is rising has the MAS regardless of the absolute CRP or the ESR. [1][10]
The iatrogenic harm is prevented by the judicious use of the immunosuppression, the screening for the infections before the biologic, the monitoring of the blood counts and the liver function, and the aggressive treatment of the infection when it occurs. The child on the biologics is counselled to present early with the fever, and the broad-spectrum antibiotic is given empirically if the serious infection is suspected, because the immunosuppressed child may not mount the usual inflammatory response. [9]
Prognosis & Disposition
The prognosis of the sJIA in the biologic era has been transformed by the interleukin-one and the interleukin-six blockade. The disease that was once managed with the high-dose glucocorticoids and the methotrexate, with the high rates of the growth failure and the chronic disability, is now managed with the early biologic that achieves the clinically inactive disease in the majority of the children. The remission rates have risen, and the glucocorticoid exposure has fallen. The MAS, when recognised and treated early, has a mortality of under ten percent, down from the twenty to thirty percent of the pre-biologic era. [9][10]
The prognosis of the macrophage activation syndrome depends on the speed of the recognition and the early treatment. The child who is treated with the glucocorticoid, the ciclosporin and the anakinra within the first day or two of the syndrome has a good outcome, and the child who presents late with the established multiorgan failure has a guarded prognosis. The sJIA lung disease is the complication that carries the highest mortality, and it is the reason for the heightened vigilance in the child with the chronic or the recurrent MAS. [7]
The disposition of the child with sJIA is the specialist paediatric rheumatology centre, with the multidisciplinary team that includes the rheumatologist, the paediatrician, the nurse specialist, the physiotherapist, the occupational therapist, the psychologist and the social worker. The child with the established MAS is managed in the paediatric intensive care unit, with the paediatric rheumatology, the haematology and the infectious diseases in the team. The retrieval and the transfer to the specialist centre are arranged early, because the MAS can deteriorate within hours and the child needs the centre with the full range of the therapies and the intensive care. [9][10]
After the resolution of the MAS, the child is followed up for the recurrence, the infection surveillance, the biologic optimisation, and the long-term outcome. The glucocorticoid is tapered, the biologic is optimised, and the family is taught the warning signs of the recurrence. The school, the growth and the development are monitored, and the transition planning into the adult care is begun in the adolescence. [9]
Special Populations
The very young child with sJIA, the infant and the toddler, requires the careful weight-based dosing of the anakinra and the tocilizumab, and the tocilizumab is approved down to the age of two years. The anakinra is used off-label in the very young child, and the dose is titrated to the response. The live vaccines are deferred while the biologics are given, and the killed-vaccine schedule is maintained, though the response may be blunted. The growth and the development are closely monitored, because the chronic inflammation and the glucocorticoid both retard the growth, and the early biologic therapy may restore the growth trajectory. [9]
The adolescent with sJIA needs the transition planning into the adult care, with the formal transition programme that addresses the medical, the psychosocial and the developmental needs. The biologic therapy is continued, the reproductive counselling addresses the teratogenic drugs such as the methotrexate and the emerging data on the biologics in the pregnancy, and the adherence is supported through the education and the self-management. The mental health and the substance use are screened, because the chronic illness from the childhood carries a high burden of the anxiety and the depression. [9]
The socioeconomic disadvantage and the remoteness affect the access to the biologics, which are expensive and require the cold-chain and the specialist administration. The child in the remote or the rural area may have the delayed diagnosis and the delayed treatment, and the retrieval and the telehealth are the tools to bridge the gap. The indigenous child and the migrant-refugee child may have the additional barriers of the language, the culture and the health literacy, and the culturally safe care and the interpreter service are essential. [9]
The child with the primary immunodeficiency and the haemophagocytic lymphohistiocytosis is distinguished from the sJIA-associated MAS by the earlier age of onset, the family history, the consanguinity, the failure to thrive, and the very early and the recurrent episodes. The perforin and the degranulation flow cytometry and the genetic panel are the tools, and the management is the HLH-2004 protocol with the etoposide and the dexamethasone and the curative haematopoietic stem cell transplantation, which is not the pathway for the typical sJIA-associated MAS. [3][7]
Evidence, Guidelines & Regional Differences
The landmark evidence for the modern sJIA management is the pair of the 2012 phase-three trials published in the same issue of the New England Journal of Medicine. The trial by De Benedetti and colleagues proved the tocilizumab for the systemic features and the arthritis, and the two trials by Ruperto and colleagues proved the canakinumab. These trials established the interleukin-one and the interleukin-six blockade as the standard of care and transformed the sJIA from a glucocorticoid-dependent disease into a biologic-responsive one. The 2016 classification criteria for the MAS, developed by the EULAR, the ACR and the PRINTO, gave the field the objective and the validated tool for the recognition. [1][5][6]
Tocilizumab in sJIA (TENDER trial)
Population: Children aged two to seventeen years with active sJIA
Key finding
The interleukin-six blockade with tocilizumab at twelve or eight milligrams per kilogram every two weeks achieved the JIA American College of Rheumatology thirty response in the majority of the children with refractory sJIA, with the reduction of the glucocorticoid exposure.
Canakinumab in sJIA
Population: Children aged two to nineteen years with active sJIA
Key finding
The interleukin-one-beta blockade with canakinumab at four milligrams per kilogram every four weeks achieved the adapted JIA American College of Rheumatology thirty response in the majority of the children, with the reduction of the fever within days.
The 2024 EULAR and PReS recommendations for the Still disease are the current guideline framework, and they reframe the sJIA as Still disease across the age spectrum and recommend the early interleukin-one blockade as the first-line biologic. The CARRA consensus treatment plans from the United States and the SHARE recommendations from Europe provide the operational pathways for the sJIA and the MAS. The regional differences are in the access to the biologics, the first-line choice between the anakinra and the canakinumab, and the place of the etoposide and the emapalumab in the refractory MAS. [9][10]
The controversies in the field include the optimal dosing of the anakinra for the MAS, where the evidence is still maturing, the place of the etoposide in the sJIA-associated MAS as opposed to the primary HLH, the role of the JAK inhibitors and the emapalumab for the refractory disease, and the biomarker-guided approach to the early MAS detection using the ferritin, the soluble interleukin-two receptor and the interleukin-eighteen. The sJIA lung disease is the complication that needs the urgent research, because it is the leading cause of the mortality in the biologic era. [9][10]
The Australasian approach, guided by the Australian Paediatric Rheumatology Group, uses the anakinra as the first-line interleukin-one blockade for the systemic features and the MAS, with the canakinumab and the tocilizumab as the alternatives. The access to the biologics is through the Pharmaceutical Benefits Scheme for the canakinumab and the tocilizumab, and the anakinra is used off-label under the specialist guidance. The retrieval to the specialist centre is arranged early for the child with the established MAS.
[9]Exam Pearls
What is the single most reliable early sign of macrophage activation syndrome in the child with sJIA?
The falling platelet count. In the active sJIA, the platelets are typically high or normal from the interleukin-six-driven thrombocytosis. When the platelet trend reverses and begins to fall, the macrophage activation syndrome is developing, even if the absolute count is still in the normal range. The trend matters more than the absolute number.
[1]FEVER
The MAS classification criteria are reproduced verbatim by every candidate who passes the examination. The ferritin over six hundred and eighty-four, the platelet count under one hundred and eighty-one, the AST over forty-eight, the triglycerides over one hundred and fifty-six and the fibrinogen under three hundred and sixty are the numbers that separate the MAS from the active sJIA, and they are derived from the large international cohort by Ravelli and the PRINTO group. The candidate who can reproduce these five numbers and the criterion of the ferritin plus any two of the remaining four has the core of the examination. [1]
The reframing of sJIA as Still disease under the 2024 EULAR and PReS recommendations is the fact that the current candidate should know to show the currency. The sJIA is an autoinflammatory disease of the innate immune system, driven by the interleukin-one and the interleukin-six, and this is why the interleukin-one and the interleukin-six blockade is so effective. The adaptive-autoimmune framework that governs the other JIA subtypes does not apply, and the methotrexate and the tumour-necrosis-factor blockade that work for the polyarticular subtypes are less effective for the systemic features. [9]
The paradoxical MAS under the tocilizumab is the exam favourite, because it tests the understanding of the mechanism. The interleukin-six blockade suppresses the fever and the CRP, so the usual warning signs are masked, and the MAS develops silently with only the falling platelets and the rising ferritin to betray it. The management is to reduce or to stop the tocilizumab and to add the anakinra and the glucocorticoid. The lesson is that the ferritin and the platelet trend must be monitored regularly for the child on the tocilizumab, even when the disease appears controlled. [5][10]
References
- [1]Ravelli A, Minoia F, Davì S, et al 2016 Classification Criteria for Macrophage Activation Syndrome Complicating Systemic Juvenile Idiopathic Arthritis: A European League Against Rheumatism/American College of Rheumatology/Paediatric Rheumatology International Trials Organisation Collaborative Initiative Ann Rheum Dis, 2016.PMID 26865703
- [2]Petty RE, Southwood TR, Manners P, et al International League of Associations for Rheumatology classification of juvenile idiopathic arthritis: second revision, Edmonton, 2001 J Rheumatol, 2004.PMID 14760812
- [3]Henter JI, Horne A, Aricó M, et al HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis Pediatr Blood Cancer, 2007.PMID 16937360
- [4]Fardet L, Galicier L, Lambotte O, et al Development and validation of the HScore, a score for the diagnosis of reactive hemophagocytic syndrome Arthritis Rheumatol, 2014.PMID 24782338
- [5]De Benedetti F, Brunner HI, Ruperto N, et al Randomized trial of tocilizumab in systemic juvenile idiopathic arthritis N Engl J Med, 2012.PMID 23252525
- [6]Ruperto N, Brunner HI, Quartier P, et al Two randomized trials of canakinumab in systemic juvenile idiopathic arthritis N Engl J Med, 2012.PMID 23252526
- [7]Jordan MB, Allen CE, Weitzman S, Filipovich AH, McClain KL How I treat hemophagocytic lymphohistiocytosis Blood, 2011.PMID 21828139
- [8]Ravelli A, Magni-Manzoni S, Pistorio A, et al Preliminary diagnostic guidelines for macrophage activation syndrome complicating systemic juvenile idiopathic arthritis J Pediatr, 2005.PMID 15870661
- [9]Fautrel B, Mitrovic S, Gonzalez-Chiappe S, et al EULAR/PReS recommendations for the diagnosis and management of Still's disease, comprising systemic juvenile idiopathic arthritis and adult-onset Still's disease Ann Rheum Dis, 2024.PMID 39317417
- [10]Boom V, Anton J, Lahdenne P, et al Evidence-based diagnosis and treatment of macrophage activation syndrome in systemic juvenile idiopathic arthritis Pediatr Rheumatol Online J, 2015.PMID 26634252