Paeds · rheumatology-musculoskeletal-and-sports
Systemic lupus erythematosus
Also known as Systemic lupus erythematosus · Childhood-onset SLE · Juvenile systemic lupus erythematosus · Lupus · cSLE
Fellowship guide to childhood-onset systemic lupus erythematosus: the SLICC 2012 and EULAR/ACR 2019 weighted classification criteria, the type I interferon and immune-complex pathophysiology behind low complement and anti-dsDNA, the multisystem clinical presentation dominated by adolescent girls, the hydroxychloroquine backbone for every patient with retinopathy screening, and the stepwise escalation from glucocorticoids through mycophenolate and azathioprine to belimumab and anifrolumab for refractory disease.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
Overview & Definition
A 13-year-old girl presents with three months of fatigue, joint pains in her wrists and fingers, painful mouth ulcers, and a red rash across her cheeks that worsens in sunlight. Her haemoglobin is low, her platelets are borderline, and bloods reveal a strongly positive antinuclear antibody, a high-titre anti-dsDNA, and low complement C3 and C4. This is childhood-onset systemic lupus erythematosus, a chronic multisystem autoimmune disease in which loss of tolerance to nuclear antigens generates pathogenic autoantibodies and immune complexes that deposit in tissues and drive inflammation across virtually every organ. In children the disease is more aggressive than in adults, with a higher burden of organ involvement, particularly renal, haematological, and neuropsychiatric disease. [4]
Systemic lupus erythematosus is fundamentally a disease of disordered immune regulation. The body loses self-tolerance, produces autoantibodies directed against nuclear material (most characteristically anti-double-stranded DNA and anti-Smith), and forms immune complexes that circulate and deposit in skin, joints, glomeruli, serosa, and the central nervous system. Complement is consumed in the process, which is why low C3 and C4 are the serological hallmark of active disease. A persistent type I interferon signature amplifies the immune response and sustains the loss of tolerance, linking the genetic susceptibility, the environmental triggers of ultraviolet light and infection, and the clinical phenotype. [5]
The distinction between childhood-onset and adult-onset disease matters for the exam. Children more often present with organ-threatening disease at or soon after diagnosis, accumulate damage faster, and require more aggressive and sustained immunosuppression. Lupus nephritis affects roughly 50 to 80 percent of children compared with 30 to 40 percent of adults, and haematological and neuropsychiatric involvement are likewise over-represented. Because the kidney, brain, and blood can be the presenting organ, any child with an unexplained multisystem illness, cytopenia, or nephritis needs lupus excluded with a focused autoantibody and complement screen. [4]
Classification
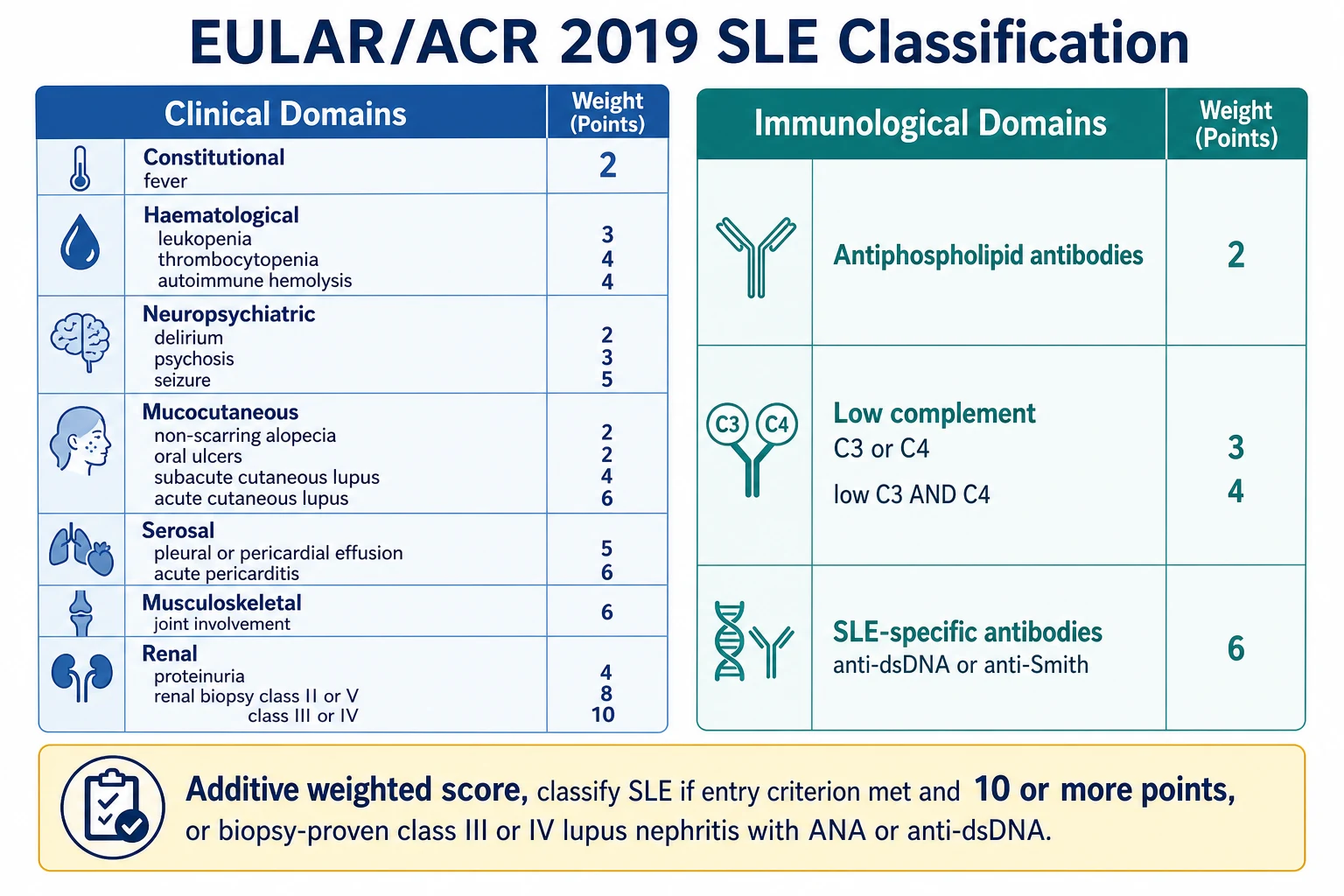
Classification criteria exist to standardise research and to give the clinician a defensible diagnostic scaffold; they are not meant to replace clinical judgement, and a child can have lupus that warrants treatment before the formal threshold is met. Three sets of criteria have shaped practice, and the exam expects familiarity with all three and an understanding of how they differ. The oldest still in use is the 1997 update of the 1982 American College of Rheumatology criteria, which require at least four of eleven criteria. The Systemic Lupus International Collaborating Clinics 2012 criteria, derived by Petri and colleagues, require at least four of seventeen criteria with at least one clinical and one immunological criterion, or biopsy-proven lupus nephritis with positive antinuclear antibody or anti-dsDNA. [3]
The current standard, and the one a candidate should lead with, is the 2019 European League Against Rheumatism and American College of Rheumatism classification, developed by Aringer and colleagues. It is structured as a weighted, additive system. The entry criterion is a positive antinuclear antibody at any time, on at least one occasion, using an immunofluorescence or equivalent assay. If the entry criterion is met, the candidate then sums weighted points from clinical and immunological domains, and classification requires a cumulative score of 10 or more points. Importantly, each criterion counts only once, the highest-weighting within a domain is taken, and a criterion is only counted if no more likely explanation exists. [1]
The clinical domains carry the weight of organ involvement. Constitutional disease (fever) scores 2 points. Haematological disease is split into leukopenia at 3 points, thrombocytopenia at 4 points, and autoimmune haemolysis at 4 points. Neuropsychiatric disease ranges from delirium at 2 points to psychosis at 3 points and seizure at 5 points. Mucocutaneous disease runs from non-scarring alopecia and oral ulcers at 2 points through subacute cutaneous lupus at 4 points to acute cutaneous lupus (the malar rash) at 6 points. Serosal disease gives 5 points for a pleural or pericardial effusion and 6 points for acute pericarditis. Musculoskeletal disease (joint involvement) scores 6 points, and renal disease gives 4 points for proteinuria, 8 points for biopsy class II or V lupus nephritis, and 10 points for biopsy class III or IV lupus nephritis. [1]
The immunological domains are where the laboratory anchors the diagnosis. Antiphospholipid antibodies score 2 points. Low complement gives 3 points for low C3 or C4, and 4 points for low C3 and C4 together. The SLE-specific antibodies (anti-dsDNA or anti-Smith) carry 6 points, reflecting their high specificity for lupus. A crucial shortcut is the biopsy clause: a patient with biopsy-proven class III or IV lupus nephritis in the presence of a positive antinuclear antibody or anti-dsDNA is classified as having systemic lupus erythematosus regardless of the point total, underscoring how nephritis dominates the disease. [1]
[2]Epidemiology & Risk Factors
Systemic lupus erythematosus is uncommon in children but far from rare in adolescence. The estimated annual incidence of childhood-onset disease is roughly 0.3 to 0.9 per 100,000 children, with a prevalence of about 3 to 9 per 100,000. The peak age of onset in childhood is early adolescence, between 11 and 15 years, and the disease is rare before the age of five. When lupus does occur in childhood it carries a greater burden of organ involvement and cumulative damage than adult-onset disease, which is why childhood-onset lupus is treated as a distinct and more aggressive phenotype. [4]
Sex and ethnicity are the two dominant risk factors. The female-to-male ratio is roughly 5 to 1 before puberty but rises to 9 to 1 or higher after puberty, reflecting the permissive effect of oestrogen on the immune system and the loss of the protective androgenic milieu. Children of African, Asian, Hispanic, and South Asian ancestry have a higher incidence, an earlier age of onset, and a more severe phenotype with a greater likelihood of proliferative lupus nephritis and neuropsychiatric disease. This ethnic gradient reflects a combination of genetic susceptibility, particularly variants in complement and interferon pathway genes, and socio-environmental determinants of health. [4]
Genetic and environmental factors interact at the level of immune regulation. Monozygotic twin concordance is around 25 to 50 percent, far higher than the dizygotic rate, and inherited complement deficiencies, particularly of the early classical pathway components such as C1q, C1r, C1s, C2, and C4, confer a very high lifetime risk of lupus. Ultraviolet light is the best-defined environmental trigger, both precipitating the initial disease and driving flares through keratinocyte apoptosis and antigen exposure. Infections, smoking, and certain drugs can also trigger disease or flares, and the type I interferon pathway sits at the centre of how these environmental insults translate into sustained autoimmunity. [5]
Pathophysiology
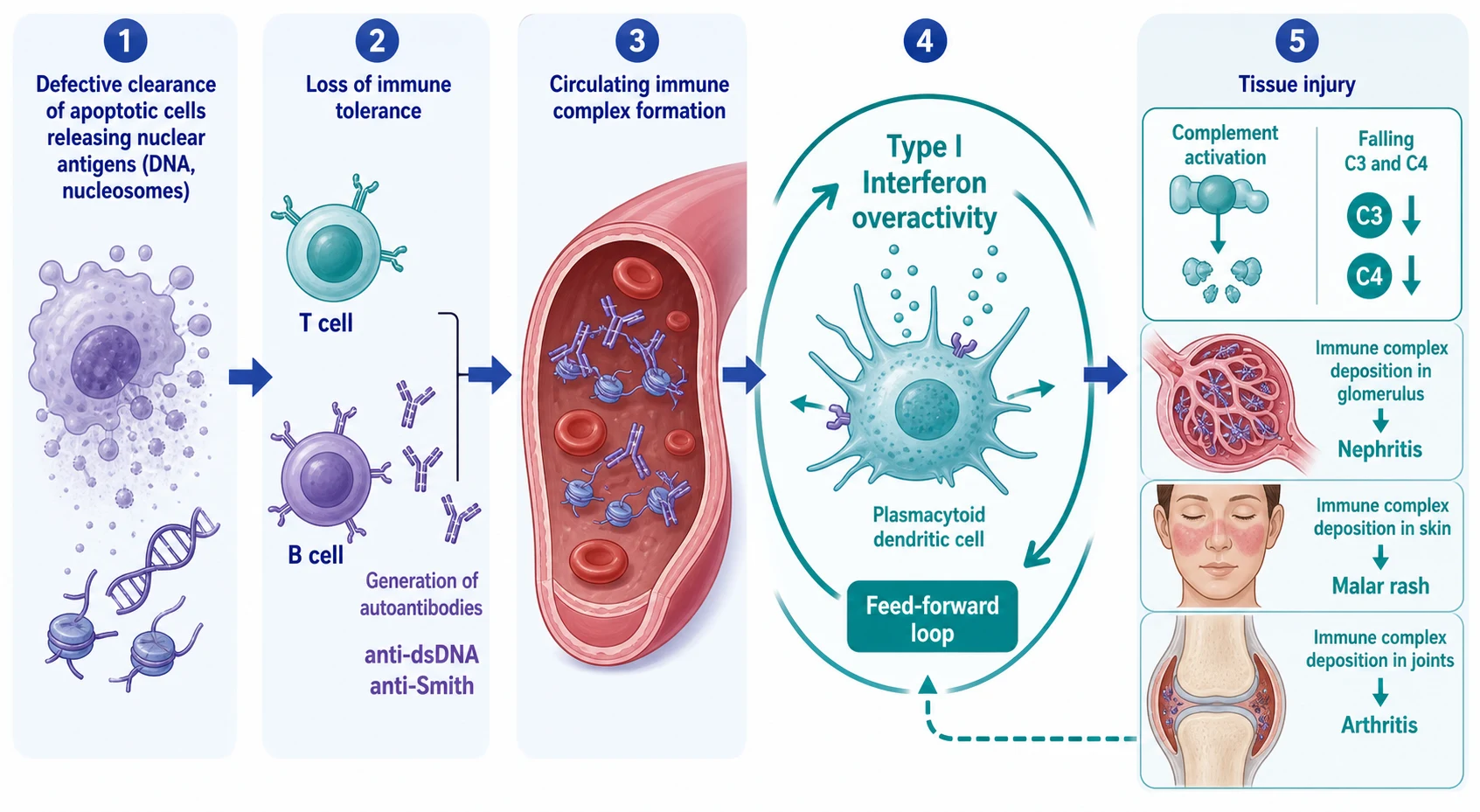
Lupus begins with a failure to clear the body's own dying cells. Apoptotic cells normally package and remove their nuclear debris cleanly, but in lupus this clearance is defective, and double-stranded DNA, nucleosomes, and ribonucleoproteins are released into the circulation and displayed to the immune system. The persistent exposure of these normally sequestered nuclear antigens is the first step in breaking tolerance. Children with inherited deficiencies of early complement components, whose job is to clear apoptotic debris and immune complexes, are disproportionately affected, which links the clearance defect directly to disease susceptibility. [5]
Once tolerance is broken, the immune system generates high-affinity autoantibodies against the exposed nuclear antigens. The two most important are anti-double-stranded DNA, which is highly specific for lupus and correlates with disease activity and nephritis, and anti-Smith, which is even more specific but less sensitive and marks the disease rather than its activity. Other autoantibodies include anti-Ro and anti-La, anti-ribosomal P, and the antiphospholipid antibodies. These autoantibodies are not mere markers; they form the circulating immune complexes that deposit in tissue and cause injury, and anti-dsDNA titre is a clinically useful barometer of disease activity. [5]
The type I interferon pathway is the amplifier that sustains the autoimmune loop. Plasmacytoid dendritic cells, activated by immune complexes containing nucleic acids, produce large quantities of type I interferons, which in turn activate antigen-presenting cells, promote B-cell maturation and autoantibody production, and upregulate the very pathways that generate more nuclear antigen. This feed-forward loop explains why lupus is chronic and relapsing, and it is the therapeutic rationale for anifrolumab, the type I interferon receptor antagonist. The interferon signature is detectable in the blood of most lupus patients and is a unifying feature of the disease across its many clinical faces. [12]
Immune complexes formed from autoantibody and nuclear antigen circulate and deposit in tissues, and it is this deposition that produces the organ damage. In the skin, deposition at the dermoepidermal junction with complement activation produces the malar rash, photosensitive eruption, and discoid lesions. In the joints, immune-complex deposition drives a non-erosive symmetrical arthritis. In the glomerulus, subendothelial, subepithelial, and mesangial deposits produce the proliferative, membranous, and mesangial patterns of lupus nephritis. In the serosa, deposition causes pleuritis and pericarditis, and in the central nervous system it contributes to the neuropsychiatric manifestations. Complement is consumed as the classical pathway is activated, which is why low C3 and C4 are the serological signature of active disease. [5]
S L E FLARE
Clinical Presentation
Lupus is the great imitator, and its presentation ranges from an insidious years-long prodrome of fatigue, arthralgia, and low-grade fever to a fulminant multisystem illness with renal failure, seizures, or severe cytopenias. The classic adolescent presentation is a girl with a constellation of mucocutaneous, musculoskeletal, and constitutional features: the malar butterfly rash across the cheeks and bridge of the nose that spares the nasolabial folds, oral or nasal ulcers that are typically painless, photosensitivity, non-scarring alopecia, Raynaud phenomenon, and a symmetrical non-erosive arthritis of the small joints of the hands, wrists, and knees. Constitutional symptoms of fever, fatigue, weight loss, and lymphadenopathy are common and may precede the specific organ features by months. [4]
The mucocutaneous manifestations are the most visible entry point to the diagnosis. The malar rash, or acute cutaneous lupus, is a confluent erythema over the cheeks and bridge of the nose that classically spares the nasolabial folds because those areas are shaded from ultraviolet light. Subacute cutaneous lupus produces a photosensitive, annular, or papulosquamous rash on the upper trunk and extensor arms, and is strongly associated with anti-Ro antibodies. Discoid lupus produces scarring, coin-shaped lesions that destroy hair follicles and leave permanent alopecia and pigment change. The distinction between scarring and non-scarring, and between sun-exposed and protected skin, is a recurring exam theme. [5]
Organ-threatening presentations define the severity of childhood disease. Renal involvement presents with oedema, hypertension, haematuria, and proteinuria, and is the single strongest predictor of long-term morbidity and mortality. Haematological disease presents with autoimmune haemolytic anaemia, thrombocytopenia, leukopenia, or lymphopenia, and may be the first clue to lupus in a child presenting with easy bruising or pallor. Neuropsychiatric lupus spans headache, cognitive dysfunction, mood disturbance, seizures, psychosis, chorea, and cerebrovascular events, and is one of the hardest manifestations to diagnose with confidence. Serositis causes pleuritic chest pain, pleural effusion, and pericarditis with a rub, and a pericardial effusion occasionally progresses to tamponade. [4]
A serological flare is the pattern the exam rewards recognising. During a flare the anti-dsDNA titre rises and the complement C3 and C4 fall, often preceding the clinical relapse by days to weeks. The combination of a rising anti-dsDNA with a falling complement is therefore both diagnostic of activity and prognostic of impending organ inflammation, particularly nephritis. Conversely, a normal complement with a falling anti-dsDNA supports disease control. Children should be monitored with serial complement and anti-dsDNA alongside clinical assessment and urinalysis, because the kidney can flare silently before symptoms emerge. [5]
Differential Diagnosis
Because lupus touches every organ, its differential is correspondingly broad and depends on the presenting feature. A child with fever, arthralgia, and a rash can look like a systemic infection, a malignancy, or another rheumatological disease. The disciplined approach is to anchor on the autoantibody and complement profile, the pattern of organ involvement, and the temporal evolution, and to systematically exclude the mimics that share individual features but not the full picture. [4]
Infection is the first and most urgent exclusion. A child with fever, cytopenias, and a rash may have septicaemia, infective endocarditis, or a viral exanthem, and these must be excluded before committing to immunosuppression, because treating an infection with high-dose steroids is catastrophic. Blood cultures, inflammatory markers, and a careful exposure and travel history resolve most cases, and the persistent low complement with positive anti-dsDNA points away from simple infection toward lupus. Parvovirus B19 infection can mimic lupus with arthralgia, cytopenias, and a positive antinuclear antibody, but it is self-limiting and the complement is usually normal. [5]
Juvenile idiopathic arthritis is the principal rheumatological differential. Systemic juvenile idiopathic arthritis shares fever, rash, arthritis, and serositis with lupus, but its rash is evanescent and salmon-pink rather than fixed and photosensitive, its arthritis is more pronounced and persistent, and its antinuclear antibody and anti-dsDNA are negative with a normal or high complement rather than a low one. Acute rheumatic fever, now uncommon in many settings but still important, follows streptococcal infection and produces a migratory arthritis, carditis, and chorea, with raised antistreptolysin titres and no antinuclear antibody. Other connective tissue diseases such as juvenile dermatomyositis and mixed connective tissue disease overlap with lupus and are distinguished by their specific antibody and clinical patterns. [4]
[4]Malignancy, particularly leukaemia and lymphoma, must be excluded in any child with persistent cytopenias, fever, and lymphadenopathy, and a blood film and bone marrow examination settle the question when the counts do not recover. Macrophage activation syndrome, a secondary haemophagocytic syndrome, can complicate both lupus and systemic juvenile idiopathic arthritis and presents with fever, pancytopenia, high ferritin, low fibrinogen, and hepatosplenomegaly; it is a rheumatological emergency requiring urgent recognition and treatment. Drug-induced lupus, triggered by drugs such as hydralazine, procainamide, isoniazid, and certain anti-TNF agents, produces lupus-like symptoms with positive antihistone antibodies but typically spares the kidney and central nervous system and resolves on drug withdrawal. [5]
Clinical & Bedside Assessment
The history and examination in suspected lupus have two goals: to map the extent of organ involvement and to seek the triggers and complications that will shape management. The history probes each potentially affected system in turn, beginning with the constitutional and mucocutaneous features (fever, fatigue, weight loss, photosensitivity, rash, mouth ulcers, hair loss, Raynaud), then the musculoskeletal (joint pain, stiffness, swelling), cardiopulmonary (chest pain, breathlessness, palpitations), renal (urine colour, frothing, swelling, reduced output), neurological (headache, mood, cognition, seizures), and haematological (bruising, bleeding, pallor) symptoms. A careful drug, menstrual, sexual, and family history completes the picture. [5]
Examination looks for the cutaneous stigmata and the consequences of organ involvement. The malar rash sparing the nasolabial folds, discoid lesions, livedo reticularis suggesting antiphospholipid syndrome, painless oral and nasal ulcers, and non-scarring alopecia are the key skin and mucosal findings. The hands may show synovitis, nailfold capillary changes, and Raynaud. Cardiovascular examination checks the blood pressure, listens for a pericardial rub or murmur, and looks for signs of fluid overload. Respiratory examination seeks pleural effusion or pleural rub, and abdominal examination may reveal hepatosplenomegaly or ascites. A thorough neurological and mental state examination screens for the cognitive, psychiatric, and focal deficits of neuropsychiatric lupus. [5]
The single most important bedside manoeuvre in any child with known or suspected lupus is the urinalysis and blood pressure measurement, because renal involvement can be clinically silent. Dipstick proteinuria and haematuria, confirmed by microscopy showing dysmorphic red cells and casts, are the earliest renal signs and demand quantification of the protein-to-creatinine ratio and renal function. A child with active urinary sediment, hypertension, or a falling complement needs same-day nephrology involvement and consideration of renal biopsy, because the histological class of lupus nephritis determines the induction strategy. Recognising that an adolescent girl with swollen joints and a facial rash also has blood and protein in her urine is the bedside observation that triggers the pathway from suspicion to biopsy to treatment. [4]
Investigations
The investigation strategy confirms the diagnosis, gauges disease activity, screens for organ involvement, and excludes the mimics. Serology is the cornerstone. The antinuclear antibody is positive in over 95 percent of patients with lupus and is the screening test; indeed a positive antinuclear antibody at any time is the entry criterion for the 2019 classification criteria. A repeatedly negative antinuclear antibody makes lupus very unlikely, though not impossible. Anti-dsDNA antibodies are highly specific, correlate with disease activity and nephritis, and form the nephritogenic immune complexes; anti-Smith antibodies are the most specific marker of the disease but are less sensitive. [1]
Complement levels gauge activity. C3 and C4 are low during active disease because the classical pathway is being consumed by the deposited immune complexes, and a rising anti-dsDNA with a falling complement is the serological signature of a flare. The full blood count may reveal leukopenia, lymphopenia, autoimmune haemolytic anaemia with a positive direct antiglobulin test, or thrombocytopenia, all of which count toward the classification score. The erythrocyte sedimentation rate is typically high while the C-reactive protein is often normal or only mildly raised, and a disproportionately high C-reactive protein in a lupus patient should raise the suspicion of infection or serositis rather than pure lupus activity. [5]
Organ-specific screening is mandatory at diagnosis and at each review. Every child needs a urinalysis and protein-to-creatinine ratio, serum creatinine and estimated glomerular filtration rate, and blood pressure measurement to detect nephritis, because renal involvement is common, serious, and often silent. A renal biopsy is indicated whenever nephritis is suspected, because the ISN/RPS histological class (I minimal mesangial through VI advanced sclerosing) determines treatment. Antiphospholipid antibodies (lupus anticoagulant, anticardiolipin, anti-beta-2-glycoprotein I) define thrombotic risk and count toward the classification score, and they should be checked at baseline. An electrocardiogram, echocardiogram, and chest imaging screen for cardiac and pulmonary involvement when clinically indicated. [8]
Investigations must also exclude the mimics before immunosuppression begins. Blood cultures and infection screens rule out sepsis, a blood film and bone marrow examination rule out malignancy when cytopenias persist, and viral serology including parvovirus and hepatitis screens address infectious triggers. Before starting any immunosuppression, the child needs screening for latent tuberculosis, hepatitis B and C, and human immunodeficiency virus, because these will be reactivated or worsened by the therapy. Live vaccines, notably measles-mumps-rubella and varicella, should be given before immunosuppression begins or deferred until it is reduced, while pneumococcal, influenza, hepatitis B, and human papillomavirus vaccines are safe and recommended. [6]
Management — Resuscitation
Resuscitation addresses the immediate, organ-threatening or life-threatening presentations before the definitive long-term immunosuppression is established. A child presenting in a lupus flare with a severe cytopenia, acute nephritic syndrome, neuropsychiatric crisis, serosal tamponade, or macrophage activation syndrome needs prompt stabilisation. Severe autoimmune haemolysis or thrombocytopenia is treated with intravenous methylprednisolone pulses (30 mg per kg per dose, maximum 1 g, daily for 3 days) followed by oral prednisolone (0.5 to 1 mg per kg per day, maximum 60 mg per day), and transfusion is reserved for cardiovascularly unstable anaemia or active bleeding because transfused cells are themselves targeted. [6]
Acute lupus nephritis with hypertension, oedema, and renal impairment is managed in partnership with nephrology. Severe hypertension needs prompt control to prevent hypertensive encephalopathy and further renal injury, using a calcium channel blocker such as amlodipine (0.1 to 0.2 mg per kg per dose once daily, maximum 10 mg per day) or, in a hypertensive emergency, an intravenous infusion of labetalol (0.25 to 1 mg per kg per hour) or nicardipine (0.5 to 1 microgram per kg per minute). Fluid overload from oliguric renal failure is treated with salt restriction and a loop diuretic such as furosemide (1 to 2 mg per kg per dose), and dialysis is reserved for refractory hyperkalaemia, severe acidosis, or diuretic-unresponsive fluid overload. [8]
Neuropsychiatric and serosal emergencies and macrophage activation syndrome complete the resuscitation picture. A lupus seizure or acute psychosis is treated with anticonvulsants or antipsychotics alongside high-dose glucocorticoids, and a pericardial effusion causing tamponade needs urgent drainage. Macrophage activation syndrome, recognised by fever, pancytopenia, a ferritin typically above 10000 micrograms per litre, low fibrinogen, and hepatosplenomegaly, is treated as a rheumatological emergency with high-dose glucocorticoids and, if refractory, ciclosporin or an interleukin-1 receptor antagonist such as anakinra. In every acute presentation, infection must be excluded or covered before escalating immunosuppression, because treating an occult infection with high-dose steroids is catastrophic. [5]
Management — Definitive & Stepwise
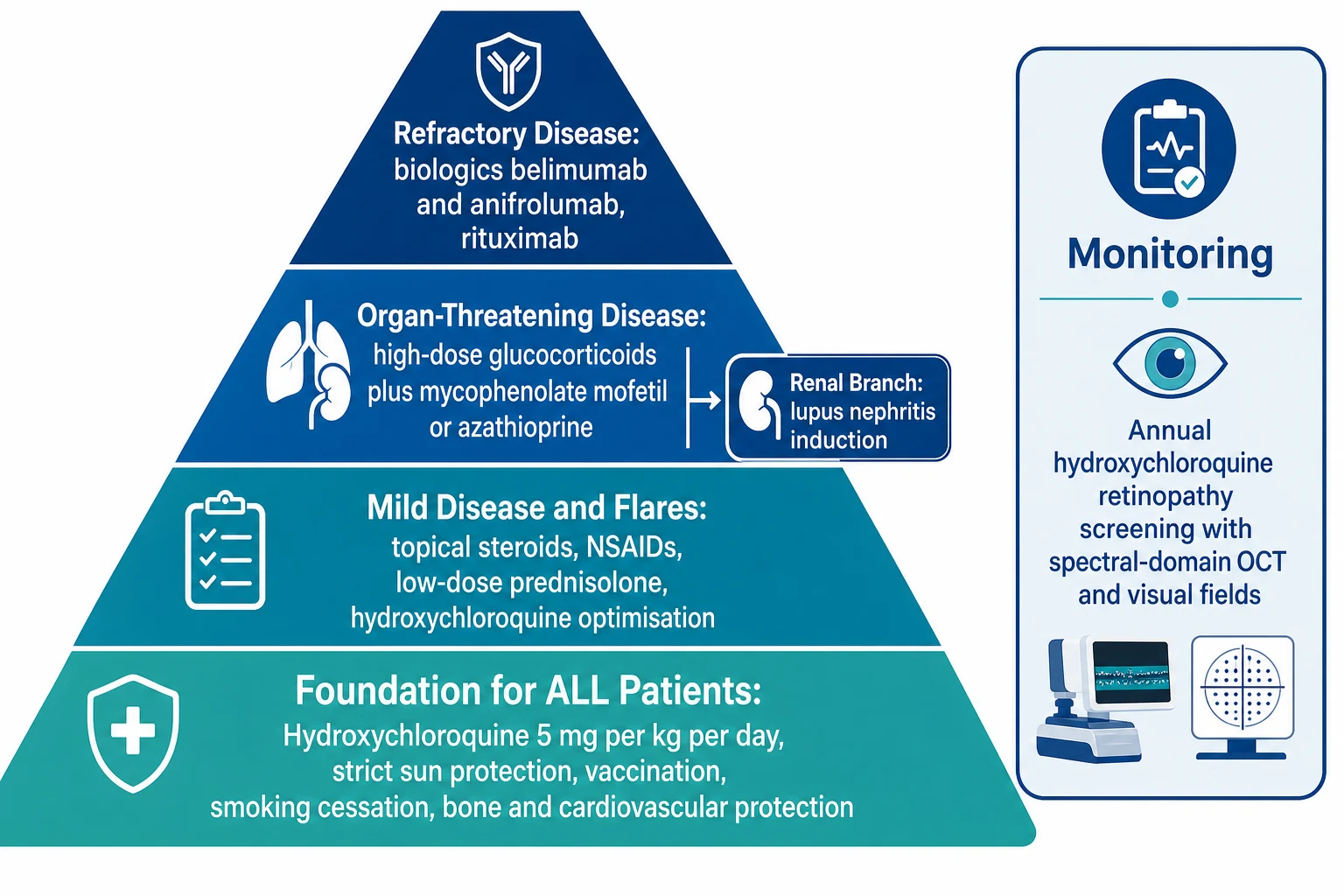
The long-term goal of treatment is to control disease activity, prevent organ damage from both the disease and its treatment, and preserve growth, development, and quality of life through childhood and into adulthood. The 2019 European League Against Rheumatism management recommendations, updated by Fanouriakis and colleagues, frame a stepwise approach anchored on a foundation of hydroxychloroquine for every patient, strict photoprotection, vaccination, and cardiovascular and bone health. [6]
Hydroxychloroquine
Dose
5 mg per kg per day orally (use the lesser of 5 mg per kg per day or 400 mg per day)
Hydroxychloroquine is the single most important drug in lupus and is recommended for every patient unless contraindicated. It reduces flares, improves survival, protects against renal and cutaneous relapse, reduces thrombosis, and improves lipid profiles, and it is safe in pregnancy. The dose is 5 mg per kg per day, using the lesser of the weight-based dose or 400 mg per day to minimise retinopathy risk. Because hydroxychloroquine can cause a bull's-eye maculopathy, screening for retinopathy is mandatory: a baseline ophthalmology assessment including colour vision, visual fields, and optical coherence tomography within the first year of treatment, then annual screening after five years of therapy, or sooner in higher-risk patients such as those on higher doses, with renal impairment, or with concurrent tamoxifen use. The 2016 American Academy of Ophthalmology recommendations codified this weight-based dosing and screening schedule. [7]
Glucocorticoids remain indispensable for controlling acute organ-threatening disease but are the largest source of treatment-related damage, so the strategy is to use the lowest dose for the shortest time. Mild flares may respond to a short course of oral prednisolone at 0.5 mg per kg per day, tapered as quickly as the disease allows. Organ-threatening disease (nephritis, central nervous system disease, severe cytopenias, serositis) is treated with intravenous methylprednisolone pulses (30 mg per kg per dose, maximum 1 g, daily for 3 days) followed by oral prednisolone (0.5 to 1 mg per kg per day, maximum 60 mg per day) tapered to a maintenance dose below 5 mg per day within months where possible. Bone health is protected with calcium, vitamin D, and consideration of a bisphosphonate for prolonged courses, and growth, weight, blood pressure, glucose, and mood are monitored. [6]
[6]Steroid-sparing immunosuppressants are added for organ-threatening or relapsing disease and to permit glucocorticoid taper. Mycophenolate mofetil (target 2 to 3 g per day, or 600 mg per square metre twice daily in children) and azathioprine (1 to 3 mg per kg per day) are the workhorses for nephritis, haematological disease, and general disease control, and mycophenolate is preferred over cyclophosphamide first-line in children because cyclophosphamide is gonadotoxic. Methotrexate (10 to 15 mg per square metre per week) is useful for refractory arthritis and has a role in skin disease, while calcineurin inhibitors such as tacrolimus and ciclosporin are options for nephritis and cytopenias. For proliferative lupus nephritis, induction uses mycophenolate mofetil or low-dose Euro-Lupus cyclophosphamide (500 mg every 2 weeks for 6 doses) with glucocorticoids, followed by long-term maintenance with mycophenolate or azathioprine. [8]
Biologic therapy is reserved for refractory disease that has not responded to conventional immunosuppression. Belimumab, a B-cell-activating factor (BAFF) inhibitor licensed for adults and now with paediatric pharmacokinetic and safety data, is given as 10 mg per kg per dose intravenously at weeks 0, 2, and 4 then every 4 weeks, and reduces disease activity and glucocorticoid exposure. Anifrolumab, a type I interferon receptor antagonist demonstrated in the TULIP-2 trial by Morand and colleagues, addresses the interferon amplification loop at the heart of the disease and is an emerging option for refractory disease. Rituximab, a B-cell-depleting anti-CD20 antibody, is used off-label for refractory nephritis, cytopenias, and central nervous system disease. The choice of biologic is tailored to the dominant organ involvement and the patient profile. [10]
TULIP-2 (Morand 2020)
Population: Adults with active systemic lupus erythematosus despite standard therapy
Key finding
Significantly higher response rates, greater glucocorticoid tapering, and fewer flares than placebo
Specific Subtypes & Scenarios
Childhood-onset lupus behaves as several overlapping clinical subtypes, and recognising the dominant pattern refines the treatment. The lupus nephritis subtype, dominated by proliferative class III and IV disease, is the most severe and the strongest predictor of long-term outcome; it presents with active urinary sediment, hypertension, and renal impairment, and it dictates the induction-maintenance paradigm described in the dedicated nephritis pathway. The haematological subtype presents with autoimmune haemolytic anaemia, thrombocytopenia, or leukopenia, sometimes as the sole manifestation, and responds to glucocorticoids with immunosuppression for relapsing disease. The neuropsychiatric subtype, the hardest to characterise, spans cognitive dysfunction, headache, seizures, psychosis, chorea, and cerebrovascular events, and requires high-dose glucocorticoids with immunosuppression for the inflammatory forms. [4]
Cutaneous lupus is a distinct phenotype in which the disease is confined to the skin. It includes acute cutaneous lupus (the malar rash), subacute cutaneous lupus (the photosensitive annular or papulosquamous rash associated with anti-Ro antibodies), and chronic cutaneous (discoid) lupus, which scars and destroys hair follicles. Isolated cutaneous lupus is treated with hydroxychloroquine, rigorous photoprotection, and topical or intralesional corticosteroids, and immunosuppressants such as methotrexate or mycophenolate are added for refractory disease. A child with cutaneous lupus needs ongoing systemic surveillance, because a minority progresses to systemic disease, and serological monitoring (complement, anti-dsDNA) and urinalysis detect that transition. [5]
Severity
Mild cutaneous or musculoskeletal lupus
Malar rash, oral ulcers, or non-erosive arthritis controlled with hydroxychloroquine, sun protection, and low-dose steroids. Excellent prognosis with adherence and surveillance.
Severity
Haematological or serosal lupus
Autoimmune cytopenias or serositis needing glucocorticoids and a steroid-sparing agent such as azathioprine or mycophenolate. Good prognosis with monitoring for relapse.
Severity
Nephritis, neuropsychiatric, or refractory lupus
Proliferative nephritis, central nervous system disease, or disease refractory to conventional therapy needing induction, biologics, and lifelong surveillance. High risk of cumulative damage.
The antiphospholipid syndrome, which can accompany lupus, adds a thrombotic dimension that changes management. Children with persistent lupus anticoagulant, anticardiolipin, or anti-beta-2-glycoprotein I antibodies are at risk of venous and arterial thrombosis, and those who have had a thrombotic event need lifelong anticoagulation. The combination of nephrotic-range proteinuria and antiphospholipid antibodies is particularly thrombogenic and warrants prophylactic anticoagulation when the serum albumin falls below 25 g per litre. Catastrophic antiphospholipid syndrome, with multi-vessel thrombosis and organ failure, is a rare but life-threatening emergency treated with anticoagulation, glucocorticoids, plasma exchange, and intravenous immunoglobulin. [5]
Drug-induced lupus and neonatal lupus are related but distinct scenarios that the exam expects candidates to separate from idiopathic disease. Drug-induced lupus, triggered by hydralazine, procainamide, isoniazid, minocycline, and anti-tumour necrosis factor agents, produces lupus-like symptoms with antihistone antibodies, typically spares the kidney and central nervous system, and resolves on withdrawing the offending drug. Neonatal lupus is a passively acquired autoimmune disease in which maternal anti-Ro and anti-La antibodies cross the placenta and cause the newborn a transient photosensitive rash and, more seriously, congenital heart block, which is permanent and often requires pacing; these mothers need specialist antenatal surveillance and the antibodies identify future lupus risk. [5]
Complications & Pitfalls
The complications of lupus arise from both the disease and its treatment, and the exam rewards a candidate who can distinguish damage from active inflammation. Disease-driven complications include progression to end-stage kidney disease from proliferative nephritis, accelerated atherosclerosis and premature cardiovascular disease from chronic inflammation, avascular necrosis of bone (particularly the femoral head), osteoporosis, neuropsychiatric sequelae, pulmonary hypertension and interstitial lung disease, and the thrombosis of antiphospholipid syndrome. Cumulative damage is measured by validated indices and accrues over years, driven by both persistent activity and glucocorticoid exposure. [4]
Treatment-driven complications are dominated by glucocorticoid toxicity, which is the largest single contributor to damage. Chronic glucocorticoids cause growth suppression, obesity, hypertension, diabetes, osteoporosis and avascular necrosis, cataracts and glaucoma, adrenal suppression, mood disturbance, and increased infection risk. Mycophenolate and azathioprine cause leukopenia, infection, and are teratogenic, demanding reliable contraception and pregnancy planning. Cyclophosphamide causes haemorrhagic cystitis, secondary malignancy, and irreversible gonadotoxicity that threatens future fertility, the central reason mycophenolate is preferred first-line in children. Calcineurin inhibitors cause nephrotoxicity, hypertension, hyperkalaemia, and glucose intolerance, and biologics increase infection and infusion-reaction risk. [6]
Infection is the leading cause of early mortality and a constant pitfall, because the disease and its immunosuppressive treatment both impair host defence. Every febrile child on immunosuppression needs a prompt infection screen, and opportunistic infections such as pneumocystis, candidiasis, and reactivated cytomegalovirus must be considered. Prophylaxis against pneumocystis with co-trimoxazole is often given to children on moderate-dose glucocorticoids plus another immunosuppressant. Vaccination against influenza, pneumococcus, hepatitis B, and human papillomavirus is safe and recommended, while live vaccines are avoided during significant immunosuppression. Macrophage activation syndrome, a secondary haemophagocytic lymphohistiocytosis, can complicate lupus and presents with fever, pancytopenia, high ferritin, and hepatosplenomegaly; it is a rheumatological emergency requiring urgent treatment. [5]
The most dangerous clinical pitfall is diagnostic and therapeutic delay. Delaying investigation in a child with a multisystem illness and positive autoantibodies delays the diagnosis and allows organ damage to accumulate. Under-treating severe organ-threatening disease by using inadequate induction or stopping maintenance prematurely invites relapse and cumulative damage. Conversely, over-treating mild disease with prolonged high-dose glucocorticoids inflicts needless toxicity. Failing to screen the urine in every lupus patient, dismissing a falling complement, and neglecting hydroxychloroquine adherence and retinopathy screening are all avoidable errors. Finally, mistaking an intercurrent infection for a lupus flare and escalating immunosuppression rather than treating the infection is a catastrophic error that a disciplined clinician avoids by culturing before escalating. [5]
Prognosis & Disposition
The prognosis of childhood-onset lupus has improved dramatically with hydroxychloroquine for all, structured immunosuppression, and treat-to-target monitoring, but it remains a serious chronic disease with significant morbidity and a measurable mortality. Five-year and ten-year survival now exceeds 90 percent in modern cohorts, but children accumulate damage over decades of disease, and the long-term burden of cardiovascular disease, renal failure, infection, and treatment toxicity defines the adult life of a patient diagnosed in adolescence. The strongest predictors of a poor outcome are organ-threatening disease at diagnosis (particularly nephritis and neuropsychiatric involvement), persistent disease activity, cumulative glucocorticoid exposure, low socioeconomic status, and poor adherence. [4]
Achieving and sustaining low disease activity is the central prognostic goal. A lupus low disease activity state, defined by controlled disease on tolerated medication with no new activity, is associated with less damage accrual and better survival, and it is the target the treatment ladder is built to reach. For nephritis, the strongest predictor of long-term renal survival is a complete renal response (proteinuria below 500 mg per day with creatinine at baseline) within the first year, and roughly 10 to 20 percent of children with severe proliferative disease progress to end-stage kidney disease long-term despite optimal therapy. Children who reach end-stage kidney disease are managed with dialysis and transplantation, generally deferred until six to twelve months of remission. [8]
Disposition depends on the severity and phase of the disease. Acute severe presentations (rapidly progressive nephritis, severe cytopenias, neuropsychiatric crisis, serosal tamponade, macrophage activation syndrome) are admitted for induction, organ support, and complication management, often in a high-dependency or intensive care setting. Stabilised children on maintenance therapy are managed as outpatients with regular monitoring of disease activity (complement, anti-dsDNA, full blood count, urinalysis and protein-to-creatinine ratio, renal function, blood pressure) and drug toxicity (glucose, lipids, bone density, ophthalmology). Every child needs a structured transition to adult rheumatology and nephrology services in late adolescence, because loss to follow-up in the transition period is a major cause of relapse, damage accrual, and progression. [5]
Special Populations
Adolescent girls are the largest paediatric group with lupus, and their management is shaped by two reproductive considerations that every candidate must articulate. First, mycophenolate, cyclophosphamide, methotrexate, and the newer biologics are teratogenic or have unknown fetal effects, so reliable contraception and pregnancy planning are non-negotiable, and treatment must be switched to pregnancy-compatible agents (hydroxychloroquine, azathioprine, low-dose glucocorticoids, and where appropriate calcineurin inhibitors) before conception is attempted. Second, cyclophosphamide threatens future ovarian reserve, which is the rationale for mycophenolate first-line and for gonadotropin-releasing hormone agonist protection when cyclophosphamide is unavoidable. Hydroxychloroquine is continued in pregnancy because it reduces flares and improves outcomes. [6]
Children of African, Asian, Hispanic, and South Asian ancestry, and Indigenous Australian, Maori, and Pacific children, have more severe disease with a greater burden of proliferative nephritis, neuropsychiatric involvement, and cardiovascular damage. This ethnic gradient reflects a combination of genetic susceptibility and socio-environmental determinants, and it means these children warrant a lower threshold for biopsy, close monitoring, and aggressive early treatment. Culturally competent care, attention to the social determinants of health, and equitable access to biologics and tertiary services are essential to closing the outcome gap, and shared decision-making with the young person and family improves adherence and outcomes. [4]
In Australia and New Zealand, childhood-onset lupus is managed collaboratively between paediatric rheumatology, nephrology, and immunology in tertiary centres, with all children started on hydroxychloroquine and treated according to EULAR, American College of Rheumatology, and SHARE-aligned protocols. Access to mycophenolate, calcineurin inhibitors, belimumab, and anifrolumab is available through tertiary paediatric services. Indigenous Australian and Maori and Pacific children are over-represented among the more severe phenotypes, reflecting the global pattern of ethnic predisposition and the local burden of social determinants of health. Structured transition to adult care is coordinated through young-adult rheumatology services. [5]
The rare prepubertal child with lupus (onset before ten years, and rarely before five) tends to have more aggressive disease with a higher frequency of organ involvement, and deserves early aggressive therapy and close surveillance. Children with antiphospholipid syndrome complicating their lupus need lifelong anticoagulation after a thrombotic event. Neonates exposed to maternal anti-Ro and anti-La antibodies need monitoring for congenital heart block and cutaneous lupus, and their mothers benefit from specialist antenatal surveillance because the antibodies identify future lupus risk. Young people transitioning to adult care need a structured handover of the serological profile, the cumulative cyclophosphamide dose, the cardiovascular and bone risk, and the reproductive plan, because these define the adult management. [5]
Evidence, Guidelines & Regional Differences
Three sets of classification criteria define the disease, and the exam expects the candidate to know the lineage and the differences. The 1997 update of the American College of Rheumatology criteria, by Hochberg, require at least four of eleven criteria and remain a historical reference. The Systemic Lupus International Collaborating Clinics 2012 criteria, by Petri and colleagues, require at least four criteria including at least one clinical and one immunological, and they add the lupus nephritis biopsy shortcut. The 2019 European League Against Rheumatism and American College of Rheumatism criteria, by Aringer and colleagues, are the current standard: a positive antinuclear antibody entry criterion followed by a weighted additive score of 10 or more, with each criterion counted once and the biopsy-proven class III or IV lupus nephritis shortcut preserved. [2]
The management evidence base is built on the 2019 European League Against Rheumatism recommendations, the SHARE initiative recommendations for childhood-onset disease, and the American College of Rheumatology lupus nephritis guideline. These converge on hydroxychloroquine for every patient, rigorous photoprotection, glucocorticoids at the lowest effective dose for the shortest time, and steroid-sparing immunosuppression (mycophenolate, azathioprine, methotrexate, calcineurin inhibitors) for organ-threatening or relapsing disease. The pivotal belimumab BLISS-52 trial established B-cell-activating factor inhibition in adults, and paediatric pharmacokinetic and safety data now support its use in children, while the TULIP-2 trial established anifrolumab by blocking the type I interferon receptor at the core of the disease. [6]
The three classification systems at a glance
Regional differences are shaped by ethnicity, drug access, and care structure. The multi-target triple-therapy approach combining mycophenolate with a calcineurin inhibitor is favoured in East Asia for nephritis, and it is increasingly adopted elsewhere. Access to belimumab and anifrolumab is expanding but remains concentrated in higher-income health systems, while resource-limited settings lean more heavily on glucocorticoids and cyclophosphamide. In all settings the core principles hold: hydroxychloroquine for every patient, early recognition and treatment of organ-threatening disease, steroid-sparing immunosuppression, treat-to-target low disease activity, and structured transition to adult care. The SHARE initiative recommendations specifically address the diagnosis and treatment of childhood-onset disease and are the paediatric reference standard. [5]
Exam Pearls
The classification criteria are the highest-yield topic. The EULAR/ACR 2019 system has an entry criterion of a positive antinuclear antibody at any time, then a weighted additive score of 10 or more across clinical domains (constitutional, haematological, neuropsychiatric, mucocutaneous, serosal, musculoskeletal, renal) and immunological domains (antiphospholipid antibodies, complement, SLE-specific antibodies). The heaviest weights go to acute cutaneous lupus and joint involvement at 6 points each, the SLE-specific antibodies at 6 points, and biopsy class III or IV lupus nephritis at 10 points, with a biopsy shortcut that classifies lupus regardless of the point total. Each criterion counts only once, the highest within a domain is taken, and a more likely alternative explanation excludes the criterion. [1]
The serological pattern of a flare is high-yield and clinical. During a flare the anti-dsDNA titre rises and the complement C3 and C4 fall, often preceding the clinical relapse, and the combination is both diagnostic of activity and prognostic of impending organ inflammation, particularly nephritis. Anti-Smith is the most specific antibody and marks the disease rather than its activity. A normal complement with a falling anti-dsDNA supports control, and a high erythrocyte sedimentation rate with a normal C-reactive protein is characteristic of active lupus, while a high C-reactive protein should prompt a search for infection or serositis. [5]
The treatment ladder is the management backbone and should be rehearsed in order. First, hydroxychloroquine (5 mg per kg per day) for every patient, with strict sun protection, vaccination, and cardiovascular and bone protection. Second, glucocorticoids for flares, at the lowest effective dose for the shortest time, with methylprednisolone pulses (30 mg per kg, maximum 1 g) for organ-threatening disease followed by a prednisolone taper. Third, steroid-sparing immunosuppression (mycophenolate, azathioprine, methotrexate, calcineurin inhibitors) for organ-threatening or relapsing disease, with mycophenolate preferred over cyclophosphamide first-line in children to spare the gonads. Fourth, biologics (belimumab, anifrolumab, rituximab) for refractory disease. Cyclophosphamide is reserved for the most severe induction and uses the low-dose Euro-Lupus regimen (500 mg every 2 weeks for 6 doses). [6]
References
- [1]Aringer M, Costenbader K, Daikh D, et al 2019 European League Against Rheumatism/American College of Rheumatology Classification Criteria for Systemic Lupus Erythematosus. Arthritis Rheumatol, 2019.PMID 31385462
- [2]Petri M, Orbai AM, Alarcon GS, et al Derivation and validation of the Systemic Lupus International Collaborating Clinics classification criteria for systemic lupus erythematosus. Arthritis Rheum, 2012.PMID 22553077
- [3]Hochberg MC Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum, 1997.PMID 9324032
- [4]Kamphuis S, Silverman ED Prevalence and burden of pediatric-onset systemic lupus erythematosus. Nat Rev Rheumatol, 2010.PMID 20683438
- [5]Groot N, de Graeff N, Avcin T, et al European evidence-based recommendations for diagnosis and treatment of childhood-onset systemic lupus erythematosus: the SHARE initiative. Ann Rheum Dis, 2017.PMID 28877866
- [6]Fanouriakis A, Kostopoulou M, Alunno A, et al 2019 update of the EULAR recommendations for the management of systemic lupus erythematosus. Ann Rheum Dis, 2019.PMID 30926722
- [7]Marmor MF, Kellner U, Lai TY, et al Recommendations on Screening for Chloroquine and Hydroxychloroquine Retinopathy (2016 Revision). Ophthalmology, 2016.PMID 26992838
- [8]Fanouriakis A, Kostopoulou M, Cheema K, et al 2019 Update of the Joint European League Against Rheumatism and European Renal Association-European Dialysis and Transplant Association (EULAR/ERA-EDTA) recommendations for the management of lupus nephritis. Ann Rheum Dis, 2020.PMID 32220834
- [9]Sammaritano LR, Askanase A, Bermas BL, et al 2024 American College of Rheumatism (ACR) Guideline for the Screening, Treatment, and Management of Lupus Nephritis. Arthritis Rheumatol, 2025.PMID 40331662
- [10]Navarra SV, Guzman RM, Gallacher AE, et al Efficacy and safety of belimumab in patients with active systemic lupus erythematosus: a randomised, placebo-controlled, phase 3 trial (BLISS-52). Lancet, 2011.PMID 21296403
- [11]Brunner HI, Abud-Mendoza C, Viola DO, et al Pharmacokinetics, Pharmacodynamics, and Safety of Subcutaneous Belimumab in Pediatric Patients With Systemic Lupus Erythematosus. Arthritis Care Res (Hoboken), 2026.PMID 41261055
- [12]Morand EF, Furie R, Tanaka Y, et al Trial of Anifrolumab in Active Systemic Lupus Erythematosus. N Engl J Med, 2020.PMID 31851795