Phys · cardiovascular
Cardiomyopathies
Also known as cardiomyopathy · hypertrophic cardiomyopathy · HOCM · dilated cardiomyopathy · arrhythmogenic right ventricular cardiomyopathy · ARVC · restrictive cardiomyopathy · cardiac amyloidosis · ATTR cardiomyopathy · AL amyloidosis · mavacamten · septal myectomy · alcohol septal ablation · Loeffler endocarditis
Consultant-physician-depth guide to the four cardiomyopathy phenotypes — hypertrophic (HCM), dilated (DCM), arrhythmogenic RV (ARVC), and restrictive cardiomyopathy — with genetics, cardiac MRI interpretation, sudden cardiac death risk stratification, and the guideline-driven management of each, structured for FRACP DWE and DCE preparation.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Cardiomyopathies

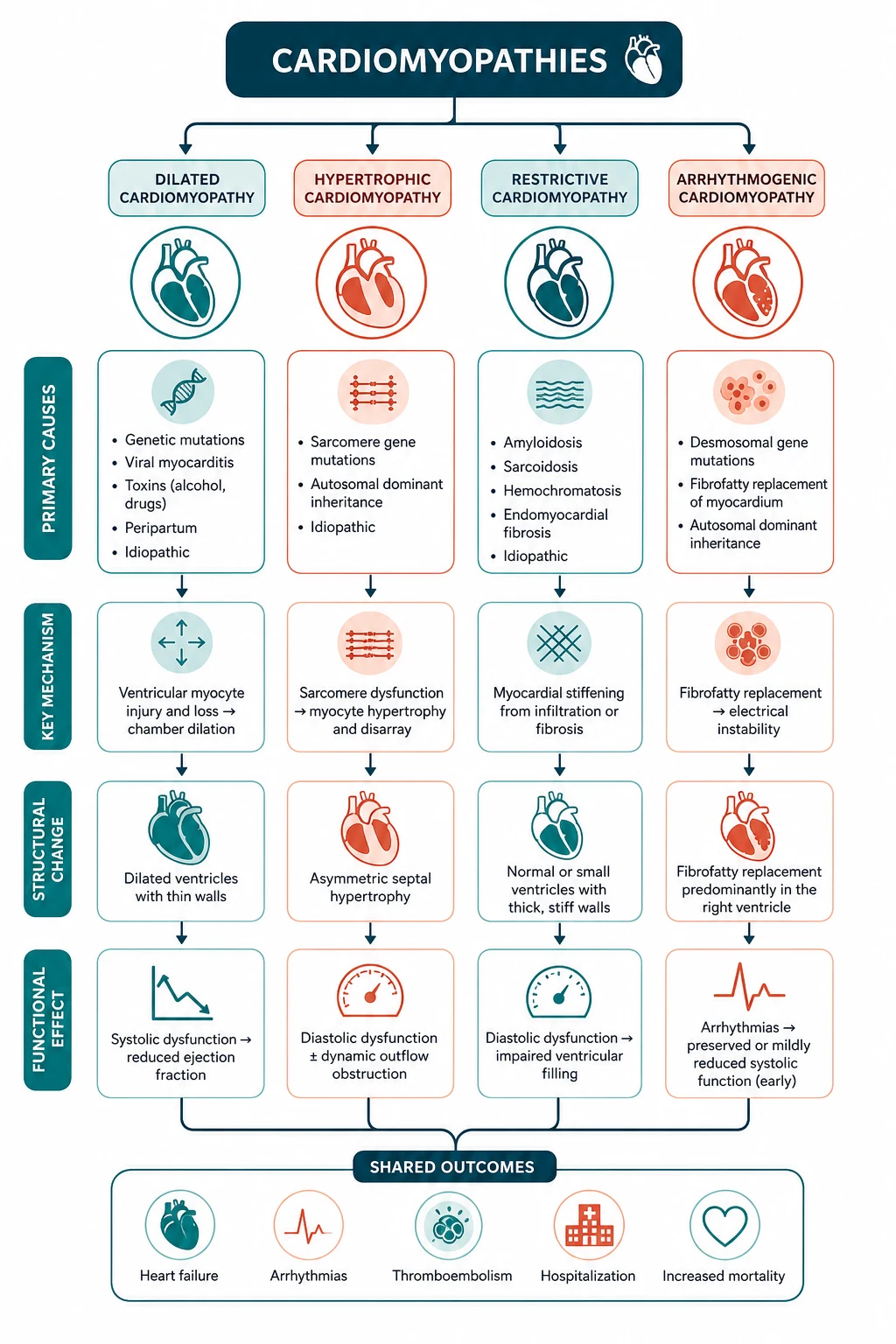

The answer first
A cardiomyopathy is a disease of the heart muscle. The two pivotal questions that govern everything below: [1]
- What is the phenotype? Define the morphology — hypertrophic (thick walls, small cavity), dilated (thin walls, large cavity, systolic dysfunction), arrhythmogenic RV (fibrofatty replacement of the RV, arrhythmia dominant), or restrictive (stiff, non-compliant ventricles, bi-atrial enlargement). The phenotype tells you the management logic before you even name the cause.
- What is the cause, and is the cause treatable? Once the phenotype is set, hunt for the cause — genetic, ischaemic, toxic, infiltrative, inflammatory. Some causes are reversible (alcohol, tachycardia-induced, thyroid, iron overload, anthracycline); some are not but are disease-modifying (tafamidis for ATTR amyloid, GDMT for DCM); and nearly all carry a need for family screening, because the inherited cardiomyopathies are autosomal dominant with 50 per cent transmission. [1]
The 2008 ESC (Elliott) and 2020 AHA/ACC frameworks classify cardiomyopathies morphologically, then sub-classify each phenotype into familial/genetic and non-familial [10]. This phenotype-first approach is what a FRACP examiner expects.
DWE high-yield: If you remember one sentence, remember this — a newly diagnosed cardiomyopathy demands a three-generation family history, a cardiac MRI with late gadolinium enhancement, and a referral to an inherited cardiac conditions service. The phenotype sets the management; the family history sets the surveillance. [1]
The four phenotypes at a glance
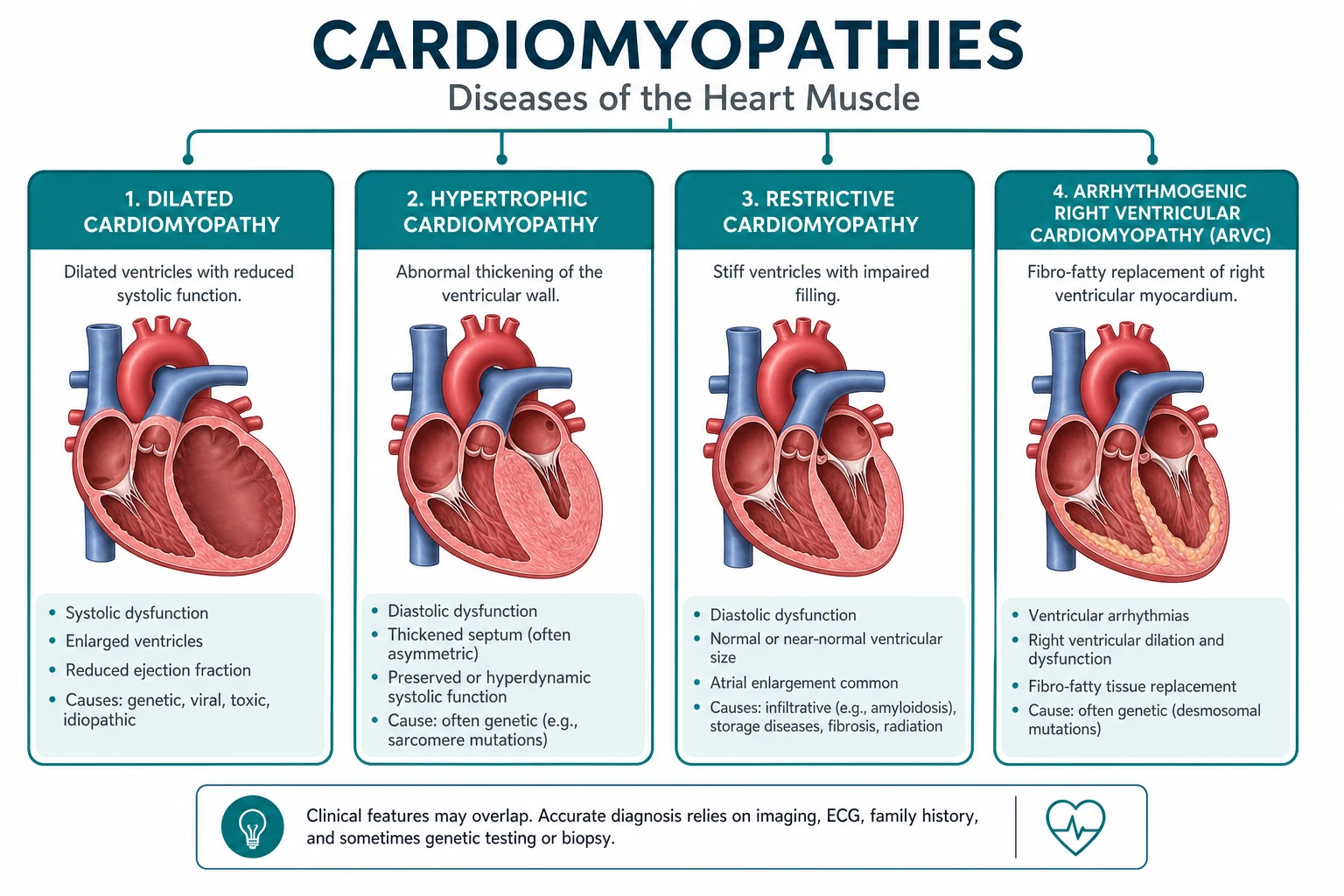

| Feature | Hypertrophic (HCM) | Dilated (DCM) | Arrhythmogenic RV (ARVC) | Restrictive |
|---|---|---|---|---|
| Morphology | Thick walls, small cavity; asymmetrical septal | Dilated LV (and often RV), thin walls, low EF | Fibrofatty RV replacement; LV can be involved | Stiff ventricles, bi-atrial enlargement, preserved EF |
| EF | Hyperdynamic or preserved | Reduced (below 45 per cent) | Often preserved early; may decline late | Preserved or mildly reduced |
| Hallmark | LVOT obstruction, SAM, dynamic murmur | Systolic heart failure phenotype | Ventricular arrhythmia dominant; epsilon waves | High filling pressures; the constrictive mimic |
| Genetics | AD sarcomeric (MYH7, MYBPC3 — ~70 per cent) | AD (titin TTN, LMNA, BAG3); also ischaemic, toxic, viral, peripartum | AD desmosomal (PKP2, DSG2, DSP) | TTR (ATTR hereditary); AL is acquired |
| SCD risk | High — the leading cause of SCD in young athletes | Moderate — related to EF and cause | High — arrhythmia is the presentation | Lower arrhythmia risk; death from HF |
| First-line therapy | Beta-blocker (+ disopyramide); septal reduction | GDMT four pillars (ARNI, BB, MRA, SGLT2i) | Beta-blocker; sport restriction; ICD if high-risk | Treat the cause (tafamidis for ATTR; chemo for AL) |
DCE trap — 'burned-out' HCM. HCM can evolve in late stage into a DCM-like phenotype (wall thinning, cavity dilatation, systolic dysfunction). This is end-stage HCM; the management switches from beta-blocker/disopyramide to GDMT for HFrEF, and transplant evaluation should begin. [1]
Hypertrophic cardiomyopathy (HCM)
Definition and genetics
HCM is the disease of the sarcomere. It is defined by unexplained left ventricular hypertrophy (maximal wall thickness at least 15 mm in adults, or at least 13 mm with a positive family history or pathogenic genotype), in the absence of another cardiac, systemic, or syndromic cause that would explain the hypertrophy [1].
The inheritance is autosomal dominant. A pathogenic sarcomeric variant is found in 30 to 60 per cent of probands. The two dominant genes: [1]
- MYH7 (beta-myosin heavy chain) — historically the first identified, often a missense mutation, associated with earlier onset and more severe hypertrophy.
- MYBPC3 (myosin-binding protein C) — the most common single gene; tends to present later (middle age) with a more favourable natural history. [1]
Other genes: TNNT2 (troponin T — often mild hypertrophy but high SCD risk), TNNI3, ACTC1, TPM1, MYL2, MYL3. A negative gene panel does not exclude HCM — 30 to 50 per cent of clinically definite HCM are genotype-negative. The phenocopies (Fabry disease, amyloidosis, mitochondrial myopathies, Noonan/LEOPARD syndromes) must be considered in atypical presentations and are a recognised exam trap. [1]
Pathophysiology — why the disease behaves the way it does
The histological hallmarks are myocyte disarray, interstitial fibrosis, and small vessel disease. Disarray is the chaotic architectural arrangement of hypertrophied myocytes — it is the substrate for ventricular arrhythmia and the reason HCM is the leading cause of SCD in young athletes. Fibrosis, visible on cardiac MRI as late gadolinium enhancement at the RV insertion points and within hypertrophied segments, predicts SCD and progression to end-stage. Microvascular ischaemia (from medial thickening of intramural coronary arteries) explains the angina that occurs without epicardial disease. [1]
Asymmetrical septal hypertrophy narrows the left ventricular outflow tract. In about two-thirds of patients this produces dynamic LVOT obstruction — the gradient varies with loading conditions and is provocable (exercise, Valsalva, standing). Systolic anterior motion (SAM) of the mitral valve is the mechanism: the hypertrophied septum and the narrow outflow tract create a Venturi effect and drag forces that pull the anterior mitral leaflet toward the septum during systole. SAM causes two things simultaneously — worsening LVOT obstruction and mitral regurgitation (the leaflet fails to coapt). This is why the HCM murmur has a mitral regurgitant component radiating to the axilla in addition to the outflow murmur at the lower left sternal edge. [1]
Diastolic dysfunction is near-universal — the thick, stiff ventricle relaxes poorly, raising filling pressures and producing exertional dyspnoea even when there is no obstruction at rest. [1]
Clinical presentation
The classic symptoms map onto four mechanisms:
- Exertional dyspnoea — diastolic dysfunction (high filling pressures) and/or LVOT obstruction limiting cardiac output on exertion. This is the most common symptom.
- Angina — microvascular ischaemia (demand ischaemia in the hypertrophied myocardium despite clean coronaries).
- Presyncope or syncope — LVOT obstruction (fixed cardiac output), arrhythmia (AF, VT), or abnormal vascular responses to exercise.
- Palpitations — AF (which is poorly tolerated and signals disease progression) or ventricular ectopy/VT. [1]
Examination:
- Bifid apex beat — a palpable forceful atrial contraction followed by the ventricular contraction; highly specific for HCM.
- Ejection systolic murmur at the lower left sternal edge and apex, radiating poorly (no carotid radiation — the obstruction is subvalvular and dynamic).
- The murmur changes with dynamic manoeuvres — this is the PACES discriminator (see below).
- A mitral regurgitant murmur may accompany the outflow murmur (from SAM).
- Jerky, bisferiens carotid pulse (a rapid upstroke then a second wave) — in contrast to the slow-rising pulse of aortic stenosis. [1]
DWE trap — HCM vs AS murmur. Both are ejection systolic. HCM is at the lower left sternal edge, radiates poorly, and increases with Valsalva/standing. AS is at the upper right sternal edge, radiates to the carotids, and decreases with Valsalva (less preload means less stroke volume but no dynamic obstruction in a fixed stenotic valve — the murmur is softer). The dynamic manoeuvre response is the single most reliable bedside discriminator. [1]
Dynamic manoeuvres — memorise this table
| Manoeuvre | Effect on HCM murmur | Mechanism |
|---|---|---|
| Valsalva (strain phase) / standing | Louder | Decreased preload → smaller LV → septum and mitral leaflet closer → more obstruction |
| Squatting to standing | Louder on standing (same as above) | Preload drops on standing |
| Squatting | Softer | Increased preload and afterload → larger LV → septum and leaflet further apart → less obstruction |
| Handgrip | Softer | Increased afterload → larger LV → less obstruction |
| Passive leg raise | Softer | Increased preload → larger LV → less obstruction |
| Post-PVC beat | Louder (Brockenbrough-Braunwald sign) | Post-extrasystolic potentiation increases contractility → more obstruction; pulse pressure narrows (opposite of fixed AS) |
DCE short-case trap: Mitral valve prolapse is the only other murmur that changes with Valsalva — but MVP gets earlier and longer (the click moves closer to S1 and the murmur lengthens), whereas HCM gets louder. The examiner distinguishes candidates by whether they perform and interpret the dynamic manoeuvres. [1]
Investigations
- ECG: LVH with strain pattern, deep symmetric T-wave inversions (especially lateral leads), abnormal deep Q waves (septal hypertrophy — 'septal Q waves' in inferior/lateral leads), AF in advanced disease. A normal ECG in definite HCM is uncommon and should prompt reconsideration of the diagnosis (e.g., athlete's heart, phenocopy).
- Transthoracic echo: the diagnostic test. Maximal wall thickness at least 15 mm (or at least 13 mm with family history/genotype), asymmetrical septal dominance common, LVOT gradient measured at rest and with Valsalva/provocable manoeuvre (obstructive if gradient at least 30 mmHg at rest or at least 50 mmHg with provocation), SAM of the mitral valve, diastolic dysfunction (raised E/e', reduced tissue Doppler velocities).
- Cardiac MRI with late gadolinium enhancement: essential for accurate wall thickness measurement (especially apical HCM, which echo underestimates), detection of apical aneurysms, and quantification of LGE (a prognostic marker — extensive LGE predicts SCD). Patchy LGE at the right ventricular insertion points and within hypertrophied segments is the HCM pattern.
- Holter monitoring: for non-sustained VT (NSVT) — a major SCD risk factor. Usually 24 to 48 hours; some centres use extended monitoring for better NSVT detection.
- Exercise testing: for the blood pressure response — a failure to rise by at least 20 mmHg systolic, or a fall in blood pressure, during exercise is a major SCD risk factor. Also useful to provoke an LVOT gradient in patients with latent obstruction.
- Genetic testing: a sarcomeric gene panel. If a pathogenic variant is identified in the proband, cascade genetic testing of first-degree relatives enables targeted surveillance of genotype-positive relatives and discharge of genotype-negative relatives. [1]
Sudden cardiac death risk stratification — the centrepiece of HCM care
HCM is the leading cause of sudden cardiac death in young athletes. The single most important decision in HCM is whether to implant an ICD for primary prevention. Two frameworks coexist: [1]
The HCM Risk-SCD calculator (ESC-aligned) — developed by O'Mahony and colleagues from a multinational cohort of 3675 patients [2]. It integrates seven routinely measured variables to estimate the 5-year risk of SCD:
- Age at evaluation
- Maximal LV wall thickness
- Left atrial diameter
- Maximal LVOT gradient (rest or provocable)
- Family history of SCD (at least one first-degree relative under 40, or SCD in a first-degree relative at any age with no alternative explanation)
- Non-sustained VT on Holter
- Unexplained syncope
The 5-year risk score determines the ICD recommendation:
- At least 6 per cent (high risk): ICD recommended.
- 4 to less than 6 per cent (intermediate): consider ICD — individualise.
- Below 4 per cent (low risk): ICD not indicated. [1]
The AHA/ACC 2020 guideline uses a simpler major-risk-factor approach. The presence of one or more of these major risk factors supports consideration of an ICD:
- Massive LVH (maximal wall thickness at least 30 mm)
- Unexplained syncope (especially within the preceding 6 months, or exertional/arrhythmic)
- Family history of SCD in a first-degree relative (at least one relative under 50, or appropriate sudden death at any age)
- NSVT on ambulatory ECG (especially in patients under 30, or runs that are frequent, prolonged, or fast)
- Abnormal blood pressure response to exercise (fall or failure to rise) [1]
When the decision is uncertain, shared decision-making and the patient's values are central. The Maron 2000 NEJM registry established the efficacy of the ICD for terminating life-threatening arrhythmias in high-risk HCM, with an appropriate shock rate of about 5 per cent per year in the primary prevention cohort [4].
DWE trap — secondary prevention is not a calculator question. A patient who has survived a cardiac arrest or spontaneous sustained VT gets an ICD regardless of the HCM Risk-SCD score. The calculator is for primary prevention only. [1]
Management of symptomatic obstructive HCM

Step 1 — First-line medical therapy:
- Beta-blocker (propranolol, metoprolol, bisoprolol, atenolol) is first-line. The mechanism is reduced heart rate and contractility, which lengthens diastole (improves filling) and reduces the dynamic LVOT gradient (less contractility → less SAM).
- Non-dihydropyridine calcium channel blocker (verapamil 240 to 480 mg daily, or diltiazem) is an alternative if beta-blocker is not tolerated or contraindicated. Avoid verapamil in patients with resting LVOT obstruction and severe symptoms of low output, and in those with advanced conduction disease — verapamil can cause haemodynamic collapse in severe obstruction. [1]
Step 2 — Add disopyramide for persistent obstructive symptoms despite beta-blocker or CCB. Disopyramide is a class Ia antiarrhythmic with a powerful negative inotropic effect that reduces the LVOT gradient. Typical dose 150 to 200 mg three to four times daily (or the slow-release formulation). Combine with an AV nodal blocker (beta-blocker or verapamil) because disopyramide can accelerate AV conduction if AF develops. [1]
Step 3 — Cardiac myosin inhibitor (mavacamten): for NYHA class II to III obstructive HCM despite maximally tolerated medical therapy. Mavacamten is a first-in-class allosteric inhibitor of cardiac myosin that reduces the number of myosin heads available for actin binding, decreasing contractility and the LVOT gradient. The EXPLORER-HCM trial randomised 251 patients with symptomatic obstructive HCM (LVOT gradient at least 50 mmHg, NYHA II to III) to mavacamten or placebo for 30 weeks: 37 per cent of the mavacamten group met the primary composite endpoint of improved peak VO2 and at least one NYHA class improvement, versus 17 per cent of placebo (P = 0.0005), with significant reduction in LVOT gradient and post-exercise LVOT gradient [3]. Mavacamten requires LVEF monitoring (echocardiography before initiation, at weeks 4 and 8, then every 12 weeks) because of the risk of reversible LV systolic dysfunction (LVEF below 50 per cent — interrupt therapy). It also has significant drug-drug interactions (CYP2C19 and CYP3A4) and a long half-life (6 to 9 days).
Step 4 — Septal reduction therapy for refractory obstructive symptoms (NYHA III to IV) despite maximally tolerated medical therapy: [1]
- Surgical septal myectomy (Morrow procedure) — the gold standard at experienced centres. The surgeon resects a trough of hypertrophied basal septal myocardium via an aortic approach, widening the LVOT and eliminating the SAM substrate. Performed at high-volume centres with operative mortality under 1 per cent, excellent symptom relief (over 90 per cent improve to NYHA I to II), and durable results. Preferred in younger patients, those needing concurrent cardiac surgery (e.g., mitral repair, CABG), and those with anomalous papillary muscles or mitral pathology.
- Alcohol septal ablation — a percutaneous alternative for patients at high surgical risk. Alcohol is injected into the first major septal perforator branch of the left anterior descending artery, producing a controlled myocardial infarction of the basal septum that thins over months and widens the LVOT. Gradient reduction is comparable to myectomy, but it creates a permanent scar that may carry a long-term arrhythmia risk. Requires suitable septal perforator anatomy. Preferred in older/high-risk patients who decline surgery. [1]
The choice between myectomy and ablation is a heart-team decision weighing surgical risk, anatomy, age, comorbidity, and patient preference. Neither is first-line — both are reserved for refractory obstruction after maximal medical therapy. [1]
Drugs to avoid in obstructive HCM:
- Nitrates (reduce preload → smaller LV → more obstruction)
- Dihydropyridine calcium channel blockers (amlodipine, nifedipine — vasodilation → less afterload/preload → more obstruction)
- Pure vasodilators (hydralazine, minoxidil)
- High-dose digoxin (increases contractility → more obstruction)
- Loop diuretics in obstructive HCM (cautious use only — over-diuresis reduces preload and worsens obstruction) [1]
Non-obstructive HCM
Non-obstructive HCM (no resting or provocable LVOT gradient) is managed on symptoms: beta-blocker for dyspnoea or angina from diastolic dysfunction; avoid pure vasodilators. The SCD risk stratification and ICD decisions are identical to obstructive HCM (the calculator uses the maximal gradient — zero or trivial in non-obstructive disease). End-stage burn-out (DCM-like phenotype) switches therapy to GDMT. [1]
Atrial fibrillation in HCM
AF is common (20 to 25 per cent lifetime) and poorly tolerated — the stiff ventricle depends on atrial contraction for filling, so loss of atrial kick causes acute deterioration. Management: early rhythm control (cardioversion, amiodarone, or catheter ablation), anticoagulation (CHA2DS2-VASc does not apply in HCM — all HCM patients with AF are anticoagulated regardless of score; a DOAC is acceptable), and rate control with beta-blockade. Avoid verapamil if disopyramide is being used or planned. [1]
Dilated cardiomyopathy (DCM)
Definition
DCM is left ventricular (and often right ventricular) dilatation with systolic dysfunction (LVEF below 45 per cent), not explained by abnormal loading conditions (hypertension, valvular disease) or coronary artery disease sufficient to cause the dysfunction [10][11]. The critical diagnostic step is to exclude coronary artery disease (with coronary angiography or CT coronary angiography) — ischaemic cardiomyopathy is the great mimic and is managed differently (revascularisation, secondary prevention).
Causes — the systematic hunt
The causes cluster into genetic, toxic, inflammatory/infectious, metabolic, and haemodynamic: [1]
Genetic (30 to 40 per cent of 'idiopathic' DCM): autosomal dominant in most. The genes:
- TTN (titin) — the single most common DCM gene (found in 15 to 25 per cent of familial DCM, and 10 to 15 per cent of sporadic); truncating variants.
- LMNA (lamin A/C) — the high-yield gene for the exam. LMNA DCM carries a high arrhythmia risk (conduction disease, early AF, VT, SCD) that is disproportionate to the degree of LV dysfunction — consider ICD even with moderate EF. Also causes Emery-Dreifuss muscular dystrophy and partial lipodystrophy.
- Other genes: BAG3, RBM20, FLNC, DES, SCN5A. [1]
Ischaemic — the most common cause overall; exclude with angiography/CTCA. [1]
Toxic:
- Alcohol — historically 20 to 30 per cent of 'idiopathic' DCM; reversible with abstinence (especially if caught early).
- Chemotherapy — doxorubicin (anthracycline; cumulative dose-dependent — above 400 mg/m² the risk climbs; causes permanent myocyte damage through oxidative stress and topoisomerase-II beta; may present years later); trastuzumab (HER2 antibody; often reversible on cessation; type 2 cardiotoxicity).
- Cocaine, methamphetamines — direct toxicity plus ischaemia. [1]
Inflammatory/infectious (myocarditis):
- Viral — coxsackievirus B, parvovirus B19, enterovirus, adenovirus (historically); more recently SARS-CoV-2. May cause acute myocarditis or progress to chronic DCM.
- Giant cell myocarditis — rare, aggressive, often fatal without immunosuppression and transplant evaluation. Suspect in fulminant myocarditis with ventricular arrhythmia and poor response to supportive care.
- Autoimmune — SLE, rheumatoid arthritis; eosinophilic myocarditis. [1]
Haemodynamic:
- Tachycardia-induced — AF with uncontrolled rate, permanent junctional reciprocating tachycardia, frequent PVCs (burden over 10 to 15 per cent). Reversible with rate/ectopy control (ablation for high-burden PVC). PVC-induced cardiomyopathy is an increasingly recognised and treatable cause — ablate if the burden is high and the ectopy is focal.
- Peripartum cardiomyopathy — develops in the last month of pregnancy to 5 months postpartum; bromocriptine (prolactin inhibition) may aid recovery; high recurrence risk in subsequent pregnancies.
- High-output — severe anaemia, thyrotoxicosis, large AV fistula, beriberi. [1]
Metabolic/infiltrative:
- Haemochromatosis — iron deposition in the myocardium; reversible with venesection or chelation (check iron studies, ferritin, transferrin saturation; HFE genotyping).
- Hypothyroidism and thyrotoxicosis — both can cause DCM.
- Thiamine deficiency (beriberi) — high-output HF; rare in the developed world outside alcohol use disorder, malnutrition, bariatric surgery, and prolonged diuretic use.
- Selenium deficiency, carnitine deficiency — rare. [1]
Connective tissue / neuromuscular:
- Muscular dystrophies (Duchenne, Becker — dystrophin gene), myotonic dystrophy, Friedreich ataxia (which can overlap with HCM). [1]
The Felker 2000 NEJM study of 1230 patients with initially unexplained cardiomyopathy found a probable cause in about half: lymphocytic myocarditis 9 per cent, ischaemic 8 per cent, peripartum 5 per cent, alcohol/toxic 19 per cent — underscoring that a systematic workup changes management in a large proportion [12].
Clinical presentation
DCM presents with the syndrome of heart failure with reduced ejection fraction (HFrEF): exertional dyspnoea, fatigue, orthopnoea, peripheral oedema. The examination is that of biventricular failure: displaced, diffuse apex beat; S3 gallop (a low-frequency diastolic sound from rapid early filling of the dilated, non-compliant ventricle); functional mitral and tricuspid regurgitation (from annular dilatation); bibasal crackles; raised JVP; peripheral oedema. Arrhythmia (AF, ventricular ectopy) is common and may be the presenting feature. [1]
Investigations — the DCM workup
The workup has three goals: confirm the phenotype, exclude ischaemia, and find the cause. [1]
- ECG: often non-specific — low voltage, T-wave inversion, LBBB, pathological Q waves, AF. Low voltage with a broad QRS or conduction disease raises LMNA, and low voltage with increased wall thickness on echo raises amyloidosis.
- Transthoracic echo: dilated LV (increased LV end-diastolic and end-systolic diameters), reduced EF, global or regional hypokinesis, functional MR/TR, diastolic dysfunction, possible LV thrombus (especially with severe LV dysfunction and AF — anticoagulate).
- Coronary angiography or CT coronary angiography: mandatory — to exclude ischaemic cardiomyopathy. CTCA is increasingly first-line in low-to-intermediate pretest probability.
- Cardiac MRI with late gadolinium enhancement: the single most useful test for differentiating causes. Patterns:
- Mid-wall septal LGE — suggests non-ischaemic DCM (genetic, myocarditis-recovered).
- Subendocardial or transmural LGE in a coronary distribution — suggests ischaemia (even if the patient has no chest pain history).
- Patchy, multifocal LGE — suggests myocarditis (active or healed) or sarcoidosis.
- Diffuse subendocardial or transmural LGE with difficulty nullifying the myocardium — amyloidosis.
- LGE extent also predicts prognosis (arrhythmia risk, recovery of function).
- Bloods: full blood count, U and E, LFTs, TSH, iron studies (haemochromatosis), BNP/NT-proBNP, HbA1c, lipid profile, HIV, troponin. Consider ANA, anti-Ro/La (autoimmune), viral serology (if myocarditis suspected), serum/urine electrophoresis and free light chains (amyloid AL — even in a DCM phenotype), ACE level and sarcoidosis workup.
- Holter monitoring: PVC burden (tachycardia-induced), NSVT for risk stratification.
- Cardiopulmonary exercise testing: peak VO2 for transplant evaluation and prognosis.
- Genetic testing: gene panel (especially if family history, conduction disease suggesting LMNA, or young onset).
- Endomyocardial biopsy: reserved for suspected myocarditis (especially giant cell), sarcoidosis, or amyloidosis when non-invasive testing is equivocal. [1]
Management — GDMT four pillars + cause treatment
The management of DCM with HFrEF is guideline-directed medical therapy (GDMT), identical to the HFrEF of any cause. The four pillars: [1]
- ARNI (angiotensin receptor-neprilysin inhibitor) — sacubitril/valsartan 97/103 mg twice daily (target). The PARADIGM-HF trial established ARNI superiority over enalapril: the primary composite of cardiovascular death or HF hospitalisation was reduced by 20 per cent (HR 0.80, 95 per cent CI 0.73 to 0.87, P less than 0.001), and all-cause mortality was also reduced (HR 0.84) [8]. If ARNI is unavailable or not tolerated, use an ACE inhibitor (ramipril, enalapril, lisinopril) or ARB (candesartan, valsartan).
- Beta-blocker — one of the four mortality-reducing agents: bisoprolol, carvedilol, metoprolol succinate, or nebivolol. Start low, titrate to target.
- MRA (mineralocorticoid receptor antagonist) — spironolactone 12.5 to 50 mg daily (or eplerenone if gynaecomastia). Reduces mortality and hospitalisation.
- SGLT2 inhibitor — dapagliflozin 10 mg daily or empagliflozin 10 mg daily, regardless of diabetes status. The DAPA-HF trial showed dapagliflozin reduced the primary composite of worsening HF or cardiovascular death by 26 per cent (HR 0.74, 95 per cent CI 0.65 to 0.85, P less than 0.001) [9].
These four should be initiated promptly (the modern approach starts all four early, often in sequence over weeks, rather than waiting for up-titration of each). Add a loop diuretic for symptom control (furosemide, torsemide) — diuretics do not reduce mortality but control congestion. [1]
Device therapy:
- Cardiac resynchronisation therapy (CRT) — for LBBB with QRS at least 150 ms and LVEF at most 35 per cent despite at least 3 months of optimal GDMT. CRT improves symptoms and survival.
- ICD for primary prevention — the nuanced area. For ischaemic cardiomyopathy: ICD if LVEF at most 35 per cent at least 40 days post-MI and at least 90 days post-revascularisation, NYHA II to III, life expectancy over 1 year with good functional status. For non-ischaemic DCM: the DANISH trial (Kober 2016 NEJM) showed that routine prophylactic ICD implantation did NOT reduce all-cause mortality in non-ischaemic systolic HF, though it halved the risk of SCD [7]. Guidelines now recommend ICD only if LVEF at most 35 per cent persists despite at least 3 months of optimal GDMT, with careful consideration of life expectancy and comorbidity. This is a high-yield exam contrast — DANISH challenged the reflexive ICD for all non-ischaemic DCM.
Cause-specific treatment:
- Alcohol: abstinence — often improves EF.
- Tachycardia-induced: rate control (AV nodal ablation if refractory AF), PVC ablation if burden high.
- Iron overload (haemochromatosis): venesection or chelation.
- Thyroid dysfunction: correct.
- Anthracycline: stop or switch cardioprotective strategies (dexrazoxane); may be partially reversible.
- Peripartum: bromocriptine, standard GDMT (avoid ACEi/ARB/MRA if breastfeeding or pregnant — switch to nitrates/hydralazine and beta-blocker in pregnancy).
- Autoimmune/sarcoid: corticosteroids or immunosuppression.
- Revascularisation for ischaemia if viable myocardium present. [1]
Advanced HF: refer for transplant evaluation or LVAD if refractory despite optimal GDMT and device therapy. [1]
Arrhythmogenic right ventricular cardiomyopathy (ARVC)
Definition and genetics
ARVC (increasingly called arrhythmogenic cardiomyopathy because LV involvement is common) is an inherited heart muscle disease characterised by progressive fibrofatty replacement of the myocardium, predominantly affecting the right ventricle, predisposing to ventricular arrhythmia and sudden cardiac death [5].
The inheritance is autosomal dominant with variable penetrance. The mutations are in genes encoding desmosomal proteins — the cell-cell adhesion complexes that bind cardiac myocytes:
- PKP2 (plakophilin-2) — the most common (25 to 40 per cent of genotype-positive cases).
- DSG2 (desmoglein-2), DSC2 (desmocollin-2), DSP (desmoplakin), JUP (junction plakoglobin).
- Rarer: TMEM43, PLN (phospholamban — high arrhythmia risk in the Netherlands founder mutation). [1]
Recessive forms cause the syndromic cardiocutaneous diseases Naxos disease (PKP2/strakon — woolly hair, palmoplantar keratoderma, ARVC) and Carvajal syndrome (DSP — similar triad with LV-dominant disease). [1]
Pathophysiology — the exercise hypothesis
The desmosomal defect weakens myocyte-cell adhesion. Mechanical stress (especially intense exercise) causes myocyte detachment, necrosis, and apoptosis; the healing response replaces dead myocardium with fibrous and fatty tissue. This produces the characteristic histology and the triangle of dysplasia (the inflow tract, outflow tract, and apex of the RV are most affected). The fibrotic substrate supports re-entrant ventricular arrhythmias. The 'exercise hypothesis' explains why ARVC presents in athletes, why it is more severe and earlier in competitive athletes, and why sport restriction is a core management decision — continued intense exercise accelerates disease progression and arrhythmia risk. [1]
Clinical presentation
ARVC classically presents in a young adult (often male, ages 20 to 40) with:
- Palpitations, syncope, or aborted SCD from ventricular ectopy or sustained VT.
- The arrhythmia is classically of LBBB morphology (RV origin) — left bundle branch block pattern, often with a superior axis (RV outflow tract origin) or inferior axis depending on the site.
- Some are detected through family screening of a proband. [1]
Diagnosis — the 2010 Revised Task Force Criteria (Marcus)
The diagnosis integrates structural, histological, ECG, arrhythmic, and familial findings, each categorised as major or minor [5]. Definite ARVC requires two major, or one major and two minor, or four minor criteria from different categories:
Structural (imaging — echo, cardiac MRI, or angiography):
- Major: regional RV akinesia, dyskinesia, or aneurysm plus RV dilatation or RV dysfunction (RVOT diameter or RV area above thresholds).
- Minor: regional RV akinesia/dyskinesia/aneurysm with mild dilatation or dysfunction. [1]
Histological:
- Major: residual myocytes less than 60 per cent by morphometric analysis, with fibrous replacement of the RV free wall, with or without fatty replacement.
- Minor: residual myocytes 60 to 75 per cent. [1]
ECG repolarisation:
- Major: T-wave inversion in V1 to V3 (or beyond) in the absence of complete RBBB, in patients older than 14.
- Minor: T-wave inversion in V1 to V2, or in V4 to V6, or in V1 to V4 with complete RBBB. [1]
ECG depolarisation / conduction:
- Major: epsilon waves — distinct low-amplitude deflections at the end of the QRS complex in V1 to V3 (pathognomonic but insensitive — present in about 30 per cent).
- Minor: late potentials on signal-averaged ECG; or terminal activation duration of QRS (from nadir of S wave to end of QRS, including R prime or epsilon wave) of at least 55 ms in V1 to V3 (in the absence of complete RBBB). [1]
Arrhythmia:
- Major: non-sustained or sustained ventricular tachycardia of LBBB morphology with superior axis (highly specific for RV dysplasia).
- Minor: non-sustained or sustained VT of LBBB morphology with inferior or unknown axis, or more than 500 PVCs per 24 hours. [1]
Family history:
- Major: ARVC confirmed in a first-degree relative (by Task Force criteria or pathogenic desmosomal gene mutation).
- Minor: history of ARVC in a first-degree relative when that relative cannot be formally evaluated; SCD under 35 in a first-degree relative; ARVC confirmed pathologically at surgery or autopsy in a second-degree relative. [1]
DWE trap — epsilon waves. Epsilon waves are the most specific ECG finding of ARVC but are insensitive. Their absence does not exclude ARVC. The T-wave inversion in V1 to V3 is more common and the first thing to look for. T-wave inversion in V1 to V3 in a young person without RBBB is ARVC until proven otherwise — get a cardiac MRI. [1]
Investigations
- ECG: T-wave inversion V1 to V3 (without RBBB); epsilon waves; terminal activation delay; PVCs with LBBB morphology.
- Signal-averaged ECG: late potentials (a research/adjunctive tool).
- Echo: regional RV wall motion abnormalities (akinesia, dyskinesia, aneurysm), RV dilatation. May be normal early.
- Cardiac MRI: the most sensitive test — RV dilatation, regional RV akinesia/dyskinesia, intramyocardial fat (on T1-weighted imaging with fat suppression), late gadolinium enhancement of the RV free wall and (in advanced disease) the LV. Also detects LV involvement (now part of the broader 'arrhythmogenic cardiomyopathy' spectrum).
- Holter: quantify PVC burden, detect NSVT/sustained VT, characterise morphology (LBBB with superior axis is major).
- Exercise testing: may provoke arrhythmia.
- Genetic testing: desmosomal gene panel — a pathogenic variant is a major family-history criterion and enables cascade screening.
- Endomyocardial biopsy: rarely needed (risk of RV free wall perforation); histology shows fibrofatty replacement. [1]
Management
- Beta-blocker first-line for symptom and arrhythmia control.
- Sport restriction — competitive and intensive endurance sport is excluded; this is a core, non-pharmacological management decision. Leisure moderate exercise is permitted.
- ICD for sustained VT or high-risk features (extensive RV or LV involvement, syncope, family history of SCD, inducible VT at EP study). The decision balances the high arrhythmia risk of ARVC against the lifetime risk of ICD complications (especially in young, active patients — lead fractures, inappropriate shocks).
- Catheter ablation for recurrent VT (often palliative, not curative — ARVC is progressive with multiple circuits over time).
- Heart failure therapy in advanced disease with biventricular failure.
- Family screening — first-degree relatives need clinical evaluation (ECG, echo, Holter, cardiac MRI) and, if a pathogenic variant is known, targeted genetic testing. Genotype-positive/phenotype-negative relatives need lifelong surveillance. [1]
Restrictive cardiomyopathy
Definition
Restrictive cardiomyopathy is the rarest subtype: non-dilated, non-hypertrophied (or only mildly thickened) ventricles with restrictive (stiff) diastolic filling, bi-atrial enlargement, and preserved or near-preserved systolic function. The haemodynamic problem is high filling pressures — the ventricles cannot relax and fill, so atrial pressures rise and the atria dilate. [1]
Causes
The causes are categorised as infiltrative, storage diseases, endomyocardial, and idiopathic: [1]
Infiltrative:
- Cardiac amyloidosis — the dominant cause in the developed world.
- AL amyloidosis (primary, from a plasma cell dyscrasia) — light-chain deposition; multi-system (nephrotic syndrome, neuropathy, macroglossia, periorbital purpura, easy bruising). Cardiac involvement drives the prognosis. Diagnosis: serum free light chains, serum and urine immunofixation, bone marrow biopsy; cardiac biopsy confirms Congo red-positive amyloid. Treatment is haematological (bortezomib-based chemotherapy, autologous stem cell transplant in selected patients) — the heart often improves with a good haematological response.
- ATTR amyloidosis (transthyretin) — deposition of misfolded transthyretin.
- Wild-type ATTR (senile systemic) — older men (typically over 65); strong associations with carpal tunnel syndrome (often bilateral, years before the cardiac presentation) and aortic stenosis. The heart is the dominant organ; neuropathy is mild or absent.
- Hereditary ATTR — autosomal dominant TTR gene mutation (Val122Ile is common in people of West African descent; Thr60Ala in Irish/UK populations; others). Neuropathy and cardiomyopathy coexist in many variants.
- Diagnosis: 99mTc-PYP (pyrophosphate), DPD, or HMDP scintigraphy — diffuse myocardial uptake grades 2 to 3 is diagnostic of ATTR if light chains are negative (no need for biopsy). Cardiac MRI shows diffuse subendocardial or transmural LGE with difficulty nullifying the myocardium and an elevated extracellular volume fraction. Echocardiogram shows increased wall thickness with a 'sparkling' granular appearance and a low voltage ECG (the discordance pattern — thick walls, low voltage).
- Treatment: tafamidis (transthyretin stabiliser) 80 mg (or 61 mg Vyndaqel) daily — the ATTR-ACT trial (Maurer 2018 NEJM) showed tafamidis reduced the hierarchical composite of all-cause mortality and cardiovascular hospitalisations, reduced all-cause mortality (29.5 per cent vs 42.9 per cent; HR 0.70), and slowed the decline in functional capacity and quality of life over 30 months in 441 patients with ATTR-CM [6]. Note: tafamidis is for ATTR, not AL. Other emerging therapies: patisiran (gene silencing by lipid nanoparticle siRNA) and inotersen (antisense oligonucleotide) for hereditary ATTR with polyneuropathy; acoramidis (new stabiliser).
- Sarcoidosis — granulomatous infiltration; can mimic any cardiomyopathy phenotype (conduction disease, VT, DCM-like, restrictive). Cardiac sarcoid is an exam favourite — look for hilar lymphadenopathy, hypercalcaemia, elevated ACE, and uptake on FDG-PET or late gadolinium enhancement on MRI. Treat with corticosteroids.
Storage diseases:
- Haemochromatosis (iron — more commonly a DCM phenotype but can be restrictive).
- Glycogen storage diseases (Danon disease — LAMP2, X-linked, with intellectual disability and skeletal myopathy; Pompe disease — acid alpha-glucosidase deficiency).
- Fabry disease (alpha-galactosidase A deficiency, X-linked) — more commonly an HCM phenotype but can be restrictive; enzyme replacement therapy is available — a treatable phenocopy of HCM. [1]
Endomyocardial:
- Endomyocardial fibrosis — tropical, endemic in equatorial Africa, India, South America; eosinophil-mediated fibrosis of the RV and LV apices and inflow tracts; presents with restrictive physiology, AV valve regurgitation, and thrombus.
- Loeffler endocarditis (hypereosinophilic syndrome) — eosinophilic infiltration and degranulation damaging the endocardium, with mural thrombus formation and restrictive filling. Associated with the FIP1L1-PDGFRA fusion gene (responds to imatinib) and other hypereosinophilic causes. Treat the eosinophilia (corticosteroids, imatinib if FIP1L1-PDGFRA positive) and anticoagulate.
- Radiation-induced — years after chest radiotherapy; fibrosis of the myocardium, pericardium, and valves; restrictive physiology with concurrent valvular and pericardial disease. [1]
Idiopathic: rare; a diagnosis of exclusion after the above are excluded. [1]
Differentiation from constrictive pericarditis — the exam favourite
Restrictive cardiomyopathy and constrictive pericarditis produce identical clinical syndromes (right heart failure, raised JVP with prominent x and y descents, Kussmaul sign, hepatomegaly, ascites, peripheral oedema) but opposite management — constriction is treated with pericardiectomy, restrictive cardiomyopathy with cause-specific therapy. The differentiation is an exam staple: [1]
| Feature | Restrictive cardiomyopathy | Constrictive pericarditis |
|---|---|---|
| Pericardium (CT/MRI) | Normal thickness | Thickened (often at least 3 to 4 mm; may be normal in some) |
| BNP | Markedly elevated | Normal or mildly elevated |
| Cardiac MRI tissue characterisation | Amyloid (diffuse LGE, difficulty nullifying); infiltrative patterns | Often normal myocardium; pericardial LGE |
| Echo — tissue Doppler (e prime) | Reduced (myocardial disease) | Preserved or exaggerated (the myocardium is normal; restriction is extrinsic) — the annulus paradoxus |
| Cardiac catheterisation — diastolic pressures | LV and RV diastolic pressures elevated and equalised, but with a difference of at least 5 mmHg that persists | LV and RV diastolic pressures equalised (within 5 mmHg) and concordant |
| Respiratory variation (cath) | Concordant (both ventricular pressures rise and fall together with respiration) | Discordant (inspiration increases RV pressure but decreases LV pressure, and vice versa) — ventricular interdependence |
| Square root sign (dip-and-plateau) | Present in both | Present in both |
| Septal motion | Usually normal | Septal bounce — abrupt septal shift toward the LV with inspiration |
| Apex | May be occluded (Loeffler, endomyocardial fibrosis) | Preserved |
DCE long-case trap: The single most reliable non-invasive discriminator is the tissue Doppler annular e prime velocity on echo — preserved in constriction (the myocardium is normal), reduced in restrictive cardiomyopathy (the myocardium is diseased). Combined with pericardial thickness on CT/MRI and BNP, this usually settles the diagnosis. If still uncertain, cardiac MRI and cardiac catheterisation with respiratory variation are definitive. [1]
Approach to the newly diagnosed cardiomyopathy
Every newly diagnosed cardiomyopathy deserves a structured workup. The sequence: [1]
- Confirm the phenotype. Echocardiography defines morphology, EF, diastolic function, valve function, and (in HCM) the LVOT gradient at rest and with provocation.
- Cardiac MRI with late gadolinium enhancement. The single most useful test for phenotype, cause, and prognosis. The LGE pattern often points to the diagnosis (see the cardiac MRI patterns figure).
- Exclude ischaemia. Coronary angiography or CT coronary angiography in any DCM or mixed phenotype. Do not assume ischaemia has been excluded by a negative troponin or the absence of chest pain.
- Find the cause. Targeted bloods — full blood count, U and E, LFTs, TSH, iron studies, BNP, HbA1c, lipid profile, HIV, and (if amyloid suspected) serum/urine electrophoresis and free light chains. Consider viral serology, autoimmune screen, ACE level, and a sarcoidosis workup in selected cases.
- Take a three-generation family history. Sudden cardiac death (especially under 50), heart failure, cardiomyopathy, pacemaker or ICD, unexplained death, skeletal myopathy (LMNA, dystrophinopathies), and conduction disease. A positive family history is itself a diagnostic and prognostic signal.
- Genetic testing and family screening. Refer to an inherited cardiac conditions service. Test the proband with a gene panel; if a pathogenic variant is identified, cascade genetic testing of first-degree relatives enables targeted surveillance of genotype-positive relatives and discharge of genotype-negative relatives (who need no further follow-up for that condition).
- Risk stratify and treat. SCD risk stratification (HCM Risk-SCD or major risk factors; DANISH-informed ICD decision for DCM), GDMT initiation, device evaluation, and the cause-specific therapy.
- Counsel. Sport restriction (HCM, ARVC), pregnancy planning (GDMT drugs are teratogenic — plan pre-conception), driving restrictions, advance care planning in advanced disease, and family communication. [1]
Cardiac MRI late gadolinium enhancement patterns

| Pattern | Distribution | Diagnosis |
|---|---|---|
| Ischaemic | Subendocardial or transmural, in a coronary territory (LAD, RCA, LCx distribution) | Prior infarction; ischaemic cardiomyopathy |
| HCM | Patchy, multifocal, at the RV insertion points (the septum–RV free wall junction) and within hypertrophied segments | HCM; extent predicts SCD |
| DCM (non-ischaemic) | Mid-wall septal LGE (non-coronary distribution) | Idiopathic or genetic DCM |
| Myocarditis | Patchy, multifocal, often subepicardial or mid-wall, lateral or inferolateral; may have pericardial and pleural effusion | Acute or healed myocarditis (Lake Louise criteria) |
| ARVC | RV free wall LGE with fatty replacement; LV subepicardial in advanced disease | ARVC / arrhythmogenic cardiomyopathy |
| Cardiac amyloidosis | Diffuse subendocardial or transmural LGE with difficulty nullifying the myocardium (the blood pool stays bright); elevated extracellular volume fraction; often bi-atrial dilatation and pleural effusions | AL or ATTR amyloidosis |
| Cardiac sarcoidosis | Patchy, multifocal, often basal and septal or lateral; may have lymphadenopathy on whole-body imaging | Cardiac sarcoidosis |
| Chagas disease | Typically apical aneurysm with thinning and LGE; basal inferolateral LGE | Chronic Chagas cardiomyopathy |
DWE trap — the 'subendocardial' rule. Subendocardial LGE is almost always ischaemic (the subendocardium is the most vulnerable layer to ischaemia, being the furthest from the epicardial coronary arteries). Mid-wall, subepicardial, and patchy multifocal patterns are non-ischaemic. The pattern alone often points to the diagnosis. [1]
DCE long-case approach
Opening statement (SASPOP)
"Mr K is a 34-year-old electrician presenting after a witnessed syncope during a game of basketball, on a background of exertional breathlessness for 6 months and a paternal uncle who died suddenly at 38. His echocardiogram shows asymmetrical septal hypertrophy with a maximal wall thickness of 24 mm, an LVOT gradient of 70 mmHg on Valsalva, systolic anterior motion of the mitral valve, and preserved EF. His Holter captured a run of non-sustained VT. The HCM Risk-SCD calculator estimates his 5-year risk of sudden cardiac death at 7.2 per cent. [1]
His problems are:
- Symptomatic obstructive hypertrophic cardiomyopathy — the primary problem
- High risk of sudden cardiac death (HCM Risk-SCD 7.2 per cent; family history of SCD; NSVT; abnormal BP response on exercise) — ICD indicated for primary prevention
- Possible autosomal dominant inheritance — genetic testing and family screening required
- Competitive sport participation — needs restriction
- Psychological and vocational impact of a chronic cardiac diagnosis in a young working man"* [1]
Integrated management plan
- Confirm phenotype and risk: cardiac MRI (for wall thickness, apical assessment, LGE extent for prognostication); exercise test (blood pressure response); consider genetic testing (sarcomeric panel).
- Symptom control: beta-blocker (bisoprolol 5 mg daily, uptitrate) for the obstruction and symptoms; add disopyramide if symptoms persist; consider mavacamten if refractory to beta-blocker plus disopyramide; septal reduction therapy (myectomy preferred in a young patient at an experienced centre) if refractory despite maximal medical therapy. [1]3. SCD prevention: ICD for primary prevention (HCM Risk-SCD at least 6 per cent; family history of SCD; NSVT) — shared decision-making discussion of the benefits and lifetime risks (inappropriate shocks, lead complications, infection, psychological impact).
- Family screening: refer to an inherited cardiac conditions service; genetic testing of the proband; cascade genetic testing and clinical screening of first-degree relatives (parents, siblings, children).
- Lifestyle: competitive sport exclusion; leisure moderate exercise permitted; driving restrictions (especially after ICD); pregnancy counselling for female relatives (and the patient's partner if relevant).
- Communication: explain the diagnosis and its inheritance; address the psychological impact; discuss the ICD decision; clarify goals and follow-up. [1]
DCE examiner probing questions you must anticipate:
- "Why an ICD in this man?" → HCM Risk-SCD at least 6 per cent (high risk); plus the major risk factors of family history of SCD, NSVT, and abnormal exercise BP response. The Maron registry established ICD efficacy in high-risk HCM.
- "Myectomy or alcohol ablation?" → Myectomy at a high-volume centre — he is young, low surgical risk, and myectomy has lower long-term arrhythmia risk than the scar of alcohol ablation. The heart team decides.
- "What do you tell him about exercise?" → Competitive sport is excluded; leisure moderate aerobic exercise is permitted; avoid heavy resistance training. The exercise restriction in ARVC is even more stringent.
- "What about his brothers and his children?" → All first-degree relatives need clinical screening (ECG, echo, Holter, cardiac MRI) and, once the proband's pathogenic variant is known, targeted genetic testing. Genotype-positive/phenotype-negative relatives need lifelong surveillance; genotype-negative relatives can be discharged. [1]
DCE short-case approach: cardiovascular examination (HCM)
Instruction: "Examine this patient's cardiovascular system." [1]
Systematic routine — the murmurs station with dynamic manoeuvres
- End of bed: comfortable, no cachexia; look for stigmata of syndromic or phenocopy conditions (Fabry — angiokeratomas; Noonan — short stature, webbed neck; amyloid — periorbital purpura, macroglossia).
- Hands and pulse: rate and rhythm (AF in advanced HCM); character — the jerky, bisferiens carotid (rapid upstroke then transient decline from dynamic obstruction then a second wave) distinguishes HCM from the slow-rising pulse of AS. Assess both carotids.
- Blood pressure: may be normal; a narrow pulse pressure suggests severe obstruction.
- JVP: usually normal; raised JVP suggests advanced disease or a phenocopy (amyloid).
- Apex beat: the bifid apex — a palpable forceful atrial contraction followed by the ventricular contraction, in the 4th or 5th intercostal space, non-displaced (pressure-overloaded, not dilated). This is highly specific for HCM.
- Praecordium: a parasternal thrill may be palpable at the lower left sternal edge.
- Auscultation: the heart of the exam.
- First heart sound: normal.
- Ejection systolic murmur at the lower left sternal edge and apex, radiating poorly (no carotid radiation — subvalvular obstruction).
- Possible mitral regurgitant murmur at the apex from SAM.
- No diastolic murmur (distinguishes from AS, which has no diastolic component either — but AR would).
- Dynamic manoeuvres — the discriminating step:
- Valsalva (strain phase) or squat-to-stand: murmur gets louder.
- Squatting or handgrip: murmur gets softer.
- Passive leg raise: murmur gets softer.
- These responses distinguish HCM from AS (which gets softer with Valsalva) and from MR (which gets louder with handgrip, opposite to HCM).
- Back and abdomen: basal crackles (decompensation), peripheral oedema. [1]
Presentation template (hypertrophic cardiomyopathy)
"I examined Mr K's cardiovascular system. He is comfortable at rest at 45 degrees. [1]
The radial pulse is regular at 68 beats per minute, of normal volume. The carotid pulse is jerky in character, with a rapid upstroke and a transient decline. The blood pressure is 125 over 75, with a normal pulse pressure. [1]
The apex beat is in the 4th intercost space at the mid-clavicular line and is bifid in character, with a palpable atrial impulse followed by the ventricular impulse. There is a parasternal thrill at the lower left sternal edge. [1]
On auscultation, the first heart sound is normal and the second heart sound is preserved. There is an ejection systolic murmur at the lower left sternal edge and apex, which does not radiate to the carotids. With the Valsalva manoeuvre, the murmur becomes louder; with squatting and with handgrip, the murmur becomes softer. There is no diastolic murmur. [1]
These findings are consistent with hypertrophic cardiomyopathy with left ventricular outflow tract obstruction. I would confirm the diagnosis with echocardiography, perform cardiac MRI with late gadolinium enhancement, and stratify the sudden cardiac death risk with the HCM Risk-SCD calculator and Holter monitoring." [1]
Key DWE MCQ patterns
- Ejection systolic murmur louder on Valsalva/standing, softer on squatting → HCM (not AS — AS gets softer on Valsalva).
- Young athlete with syncope and a family history of SCD → HCM; calculate HCM Risk-SCD; ICD if at least 6 per cent.
- Symptomatic obstructive HCM refractory to beta-blocker → add disopyramide; if still refractory, septal reduction therapy (myectomy preferred in younger/low-risk).
- Non-ischaemic DCM with LVEF 30 per cent despite optimal GDMT → consider ICD (DANISH says mortality benefit is not proven, but SCD is reduced; decision individualised with life expectancy over 1 year).
- LMNA mutation DCM with conduction disease → ICD risk is disproportionate to EF; consider early device therapy.
- PVC burden over 10 to 15 per cent with DCM → tachycardia-induced cardiomyopathy; ablate.
- T-wave inversion V1 to V3 in a young athlete → suspect ARVC; restrict sport; cardiac MRI.
- Elderly man with carpal tunnel syndrome, heart failure, low-voltage ECG, and thick walls on echo → ATTR amyloidosis; 99mTc-PYP scan; tafamidis.
- Restrictive filling with bi-atrial enlargement and nephrotic syndrome → AL amyloidosis; serum free light chains and bone marrow biopsy; chemotherapy (not tafamidis — that is for ATTR).
- Low-voltage ECG with increased wall thickness on echo → cardiac amyloidosis, not HCM (the discordance is the clue).
- Restrictive cardiomyopathy vs constrictive pericarditis → preserved annular e prime velocity on echo and pericardial thickening on CT/MRI favour constriction (pericardiectomy); reduced e prime and normal pericardium favour restrictive cardiomyopathy. [1]
Comorbidity and special situations
- Pregnancy: GDMT drugs (ACEi, ARB, ARNI, MRA) are teratogenic — discontinue before conception and switch to labetalol, nitrates/hydralazine, and a beta-blocker in pregnancy. SGLT2 safety in pregnancy is not established. HCM in pregnancy is generally well tolerated if obstruction is controlled; peripartum cardiomyopathy requires bromocriptine and standard postpartum GDMT.
- Elderly and frailty: the SGLT2 inhibitors and ARNI are generally well tolerated; avoid excessive diuretic use (hypotension, renal dysfunction); DAPA-HF and EMPEROR-Reduced included older patients.
- CKD: adjust ARNI/ACEi/ARB/MRA dose; SGLT2 inhibitor efficacy holds even with moderate CKD; monitor potassium.
- Athletes: distinguishing HCM from athlete's heart can be difficult — the athlete's heart has concentric mild hypertrophy (usually under 13 to 14 mm), normal diastolic function, no LGE on MRI, and deconditions with detraining (3 months off training reduces wall thickness). HCM has asymmetric hypertrophy, abnormal diastolic function, LGE, and a family history.
- End of life: device deactivation discussions (especially ICD — inappropriate shocks at end of life are distressing); LVAD and transplant candidacy; advance care planning. [1]
References
[1] Ommen et al. (Circulation 2020) — 2020 AHA/ACC HCM Guideline. [2] O'Mahony et al. (Eur Heart J 2014) — HCM Risk-SCD risk prediction model. [3] Olivotto et al. (Lancet 2020) — EXPLORER-HCM mavacamten for obstructive HCM. [4] Maron et al. (N Engl J Med 2000) — ICD efficacy for SCD prevention in HCM. [5] Marcus et al. (Circulation 2010) — Revised Task Force Criteria for ARVC. [6] Maurer et al. (N Engl J Med 2018) — ATTR-ACT tafamidis for ATTR amyloid cardiomyopathy. [7] Kober et al. (N Engl J Med 2016) — DANISH trial of ICD in non-ischaemic systolic HF. [8] McMurray et al. (N Engl J Med 2014) — PARADIGM-HF sacubitril/valsartan in HFrEF. [9] McMurray et al. (N Engl J Med 2019) — DAPA-HF dapagliflozin in HFrEF. [10] Elliott et al. (Eur Heart J 2008) — ESC classification of the cardiomyopathies. [11] Weintraub, Semsarian, Macdonald (Lancet 2017) — Dilated cardiomyopathy review. [12] Felker et al. (N Engl J Med 2000) — Causes and survival in unexplained cardiomyopathy.
2020 AHA/ACC HCM Guideline (Ommen, Circulation 2020); ESC classification of the cardiomyopathies (Elliott, Eur Heart J 2008); NHFA/CSANZ heart failure and inherited cardiac conditions guidance; ATTR-ACT (Maurer, NEJM 2018); DANISH (Kober, NEJM 2016). [1]
References
- [1]Ommen SR, Mital S, Burke MA, et al. 2020 AHA/ACC Guideline for the Diagnosis and Treatment of Patients With Hypertrophic Cardiomyopathy: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines Circulation, 2020.PMID 33215931
- [2]O'Mahony C, Jichi F, Pavlou M, et al. A novel clinical risk prediction model for sudden cardiac death in hypertrophic cardiomyopathy (HCM risk-SCD) Eur Heart J, 2014.PMID 24126876
- [3]Olivotto I, Oreziak A, Barriales-Villa R, et al. Mavacamten for treatment of symptomatic obstructive hypertrophic cardiomyopathy (EXPLORER-HCM): a randomised, double-blind, placebo-controlled, phase 3 trial Lancet, 2020.PMID 32871100
- [4]Maron BJ, Shen WK, Link MS, et al. Efficacy of implantable cardioverter-defibrillators for the prevention of sudden death in patients with hypertrophic cardiomyopathy N Engl J Med, 2000.PMID 10666426
- [5]Marcus FI, McKenna WJ, Sherrill D, et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: proposed modification of the Task Force Criteria Eur Heart J, 2010.PMID 20172912
- [6]Maurer MS, Schwartz JH, Gundapaneni B, et al. Tafamidis Treatment for Patients with Transthyretin Amyloid Cardiomyopathy N Engl J Med, 2018.PMID 30145929
- [7]Kober L, Thune JJ, Nielsen JC, et al. Defibrillator Implantation in Patients with Nonischemic Systolic Heart Failure N Engl J Med, 2016.PMID 27571011
- [8]McMurray JJV, Packer M, Desai AS, et al. Angiotensin-neprilysin inhibition versus enalapril in heart failure N Engl J Med, 2014.PMID 25176015
- [9]McMurray JJV, Solomon SD, Inzucchi SE, et al. Dapagliflozin in Patients with Heart Failure and Reduced Ejection Fraction N Engl J Med, 2019.PMID 31535829
- [10]Elliott P, Andersson B, Arbustini E, et al. Classification of the cardiomyopathies: a position statement from the European Society Of Cardiology Working Group on Myocardial and Pericardial Diseases Eur Heart J, 2008.PMID 17916581
- [11]Weintraub RG, Semsarian C, Macdonald P Dilated cardiomyopathy Lancet, 2017.PMID 28190577
- [12]Felker GM, Thompson RE, Hare JM, et al. Underlying causes and long-term survival in patients with initially unexplained cardiomyopathy N Engl J Med, 2000.PMID 10760308