Phys · cardiovascular
Pulmonary Hypertension
Also known as pulmonary arterial hypertension · PAH · pulmonary vascular disease · pre-capillary pulmonary hypertension · post-capillary pulmonary hypertension · chronic thromboembolic pulmonary hypertension · CTEPH · right heart catheterisation · 6-minute walk test · right ventricular failure · idiopathic pulmonary arterial hypertension · BMPR2 mutation · sotatercept · riociguat · macitentan · selexipag · epoprostenol
Consultant-physician-depth guide to pulmonary hypertension — the updated ESC/ERS 2022 haemodynamic definition (mPAP above 20 mmHg), the WHO five-group clinical classification, Group 1 PAH workup and combination therapy, CTEPH operability and riociguat, and the three-strata risk model — structured for FRACP DWE and DCE, MRCP, and ABIM preparation.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Pulmonary Hypertension

The answer first
Pulmonary hypertension is an elevated pressure in the pulmonary circulation, defined by right heart catheterisation as a mean pulmonary artery pressure above 20 mmHg. The 2022 ESC/ERS guidelines lowered the diagnostic threshold from the previous 25 mmHg, in line with the 6th World Symposium haemodynamic definitions, and reintroduced the concept of exercise pulmonary hypertension. The diagnosis is haemodynamic, but management is determined by the clinical classification — the five WHO groups — because the groups have different causes, different treatments, and different prognoses. [1]
The four decisions examiners test again and again: [1]
- Which group is this? The single most important step is assigning the WHO group, because PAH-specific therapy (the ERA, PDE5, prostacyclin, sGC, and activin-inhibitor classes) is effective and indicated for Group 1 and Group 4 disease, and is generally harmful or unproven in Groups 2, 3, and 5.
- Confirm with right heart catheterisation. Echocardiography estimates right ventricular systolic pressure but cannot diagnose pulmonary hypertension. Right heart catheterisation is mandatory — it confirms the diagnosis, defines the haemodynamic phenotype (pre-capillary, post-capillary, or combined), and rules out left heart disease.
- Risk-stratify and treat to a low-risk target. The ESC/ERS 2022 model uses a three-strata approach (low, intermediate, high) built from functional class, 6-minute walk distance, natriuretic peptides, imaging, and haemodynamics. Treatment is escalated until the patient reaches a low-risk profile.
- Screen for the underlying cause. A cause is found in most cases — connective tissue disease (especially systemic sclerosis), congenital heart disease, portal hypertension, HIV, drugs (dasatinib, fenfluramine), and schistosomiasis — and V/Q scanning excludes chronic thromboembolic disease, which is potentially curable. [1]
DWE high-yield: When a stem describes a young woman with exertional dyspnoea, a loud P2, and a raised estimated RVSP on echo, the next investigation is a full PAH workup culminating in right heart catheterisation. If the question asks for the best test to exclude chronic thromboembolic pulmonary hypertension, the answer is a V/Q scan, not a CTPA. [1]
Definition and haemodynamics — the 2022 ESC/ERS update
The haemodynamic definition of pulmonary hypertension was updated by the 2022 ESC/ERS guidelines (PMID 36017548), building on the 6th World Symposium definitions (PMID 30545968). The threshold for mean pulmonary artery pressure (mPAP) was lowered from 25 to above 20 mmHg, because the upper limit of normal mPAP is approximately 19 to 20 mmHg and values previously labelled borderline carry excess risk. [1]
The full haemodynamic classification, all measured at right heart catheterisation: [1]
| Haemodynamic category | mPAP | PAWP | PVR | Clinical group |
|---|---|---|---|---|
| Pre-capillary PH | above 20 | 15 or below | above 2 WU | Groups 1, 3, 4, 5 |
| Isolated post-capillary PH | above 20 | above 15 | 2 or below | Group 2 |
| Combined post- and pre-capillary PH | above 20 | above 15 | above 2 WU | Group 2 (with mixed picture) |
PAWP is the pulmonary artery wedge pressure (also called pulmonary capillary wedge pressure, PCWP), the catheter estimate of left atrial pressure. PVR is pulmonary vascular resistance, expressed in Wood units (1 WU equals 80 dyn per sec per cm to the fifth power). The 2022 update lowered the PVR threshold from 3 to above 2 WU, recognising that a normal PVR is under 2. [1]
Examiner insight: The shift from 25 to 20 mmHg is the single most tested fact about the modern definition. Be ready to state the new thresholds for mPAP (above 20), PAWP (15 as the pre-capillary cut-point), and PVR (above 2 WU). The exercise-PH concept (mPAP divided by cardiac output slope above 3 mmHg per litre per minute between rest and exercise) was reintroduced for symptomatic patients with normal resting pressures. [1]
The WHO five-group clinical classification
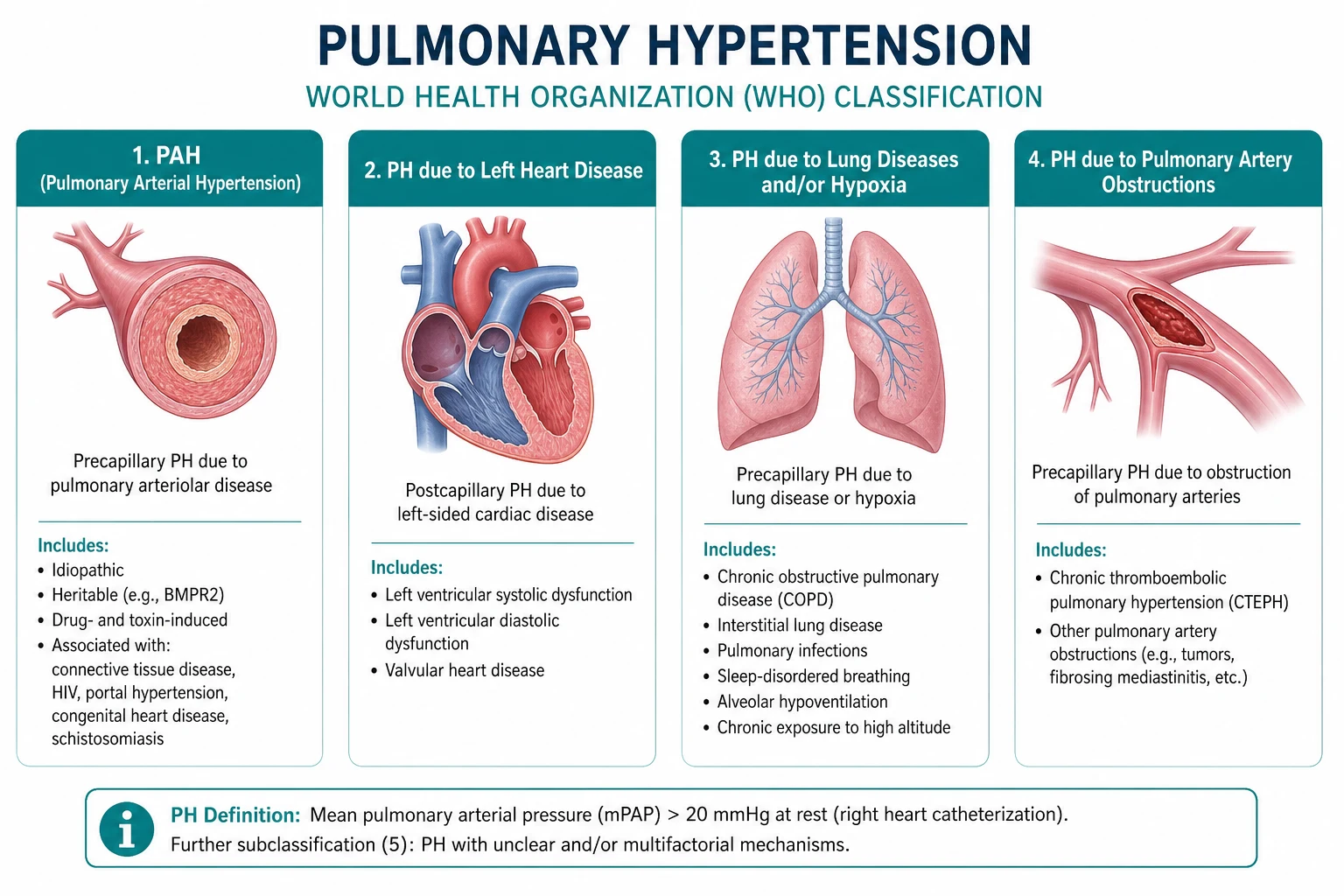
The clinical classification, first established at the 2003 Venice symposium and refined through the 6th World Symposium (PMID 30545968), categorises pulmonary hypertension into five groups based on shared pathophysiology and response to treatment. Assigning the group is the pivotal diagnostic step. [1]

Group 1 — pulmonary arterial hypertension (PAH)
PAH is a disease of the small pulmonary arterioles characterised by vasoconstriction, vascular remodelling, and in-situ thrombosis, producing a pre-capillary haemodynamic profile (mPAP above 20, PAWP 15 or below, PVR above 2). The histological hallmark is the plexiform lesion — a disorganised proliferation of endothelial cells. [1]
Subtypes: [1]
| Subtype | Key points |
|---|---|
| Idiopathic PAH | Diagnosis of exclusion; mean age around 50; female predominance 2 to 1; no family history or identified trigger |
| Heritable PAH | Autosomal dominant; BMPR2 mutations in approximately 70 percent of familial and 20 percent of idiopathic cases; also ALK1 (ACVRL1), ENG, SMAD9, KCNK3; incomplete penetrance |
| Drug- and toxin-induced PAH | Anorexigens (fenfluramine, dexfenfluramine); dasatinib (the tyrosine kinase inhibitor — increasingly recognised, often reversible on cessation); toxic rapeseed oil; amphetamines |
| PAH associated with CTD | Systemic sclerosis is the leading cause — screen annually; also SLE, mixed connective tissue disease, rheumatoid arthritis |
| PAH associated with congenital heart disease | Eisenmenger syndrome; systemic-to-pulmonary shunts; corrected defects with late PH |
| PAH associated with portal hypertension | Portopulmonary hypertension; distinct from hepatopulmonary syndrome |
| PAH associated with HIV | Screen all HIV patients with unexplained dyspnoea |
| PAH associated with schistosomiasis | The most common cause of PAH worldwide in endemic regions |
DWE high-yield discriminator: Systemic sclerosis is the connective tissue disease most strongly associated with PAH, and annual screening with echocardiography and DLCO is recommended for all systemic sclerosis patients. The combination of a falling DLCO (under 60 percent predicted) with relatively preserved lung volumes is a red flag for pulmonary vascular disease in scleroderma. Dasatinib is an increasingly tested drug cause — ask about tyrosine kinase inhibitor exposure in any new PAH diagnosis. [1]
Group 2 — PH due to left heart disease
The most common form of PH worldwide. Left-sided systolic or diastolic dysfunction, and valvular disease (especially mitral), raise left atrial pressure, which is transmitted backward to produce a post-capillary profile. Treatment is of the underlying heart disease — PAH-specific therapy is not indicated and trials such as riociguat in left-heart PH were stopped for harm or absence of benefit. [1]
Group 3 — PH due to lung disease and/or hypoxia
Chronic obstructive pulmonary disease, interstitial lung disease, sleep-disordered breathing, and chronic exposure to high altitude produce PH through hypoxic vasoconstriction and loss of the vascular bed. Treatment is of the underlying lung disease plus long-term oxygen therapy for hypoxaemic patients. PAH-specific therapy has limited evidence and is reserved for selected patients with disproportionate PH referred to a specialist centre. [1]
Group 4 — PH due to pulmonary artery obstructions (CTEPH)
Chronic thromboembolic pulmonary hypertension is the only potentially curable form of PH. Organised thromboembolic material obstructs the proximal pulmonary arteries, producing a pre-capillary profile. Pulmonary endarterectomy (PEA) is the definitive treatment for operable proximal disease; riociguat and balloon pulmonary angioplasty (BPA) are options for inoperable or distal disease. [1]
Group 5 — PH with unclear or multifactorial mechanisms
A heterogeneous group including haematological disorders (myeloproliferative neoplasms, chronic haemolytic anaemia, splenectomy), systemic disorders (sarcoidosis, vasculitis), metabolic disorders (glycogen storage disease, thyroid disease), and chronic renal failure with haemodialysis. [1]
Pathophysiology
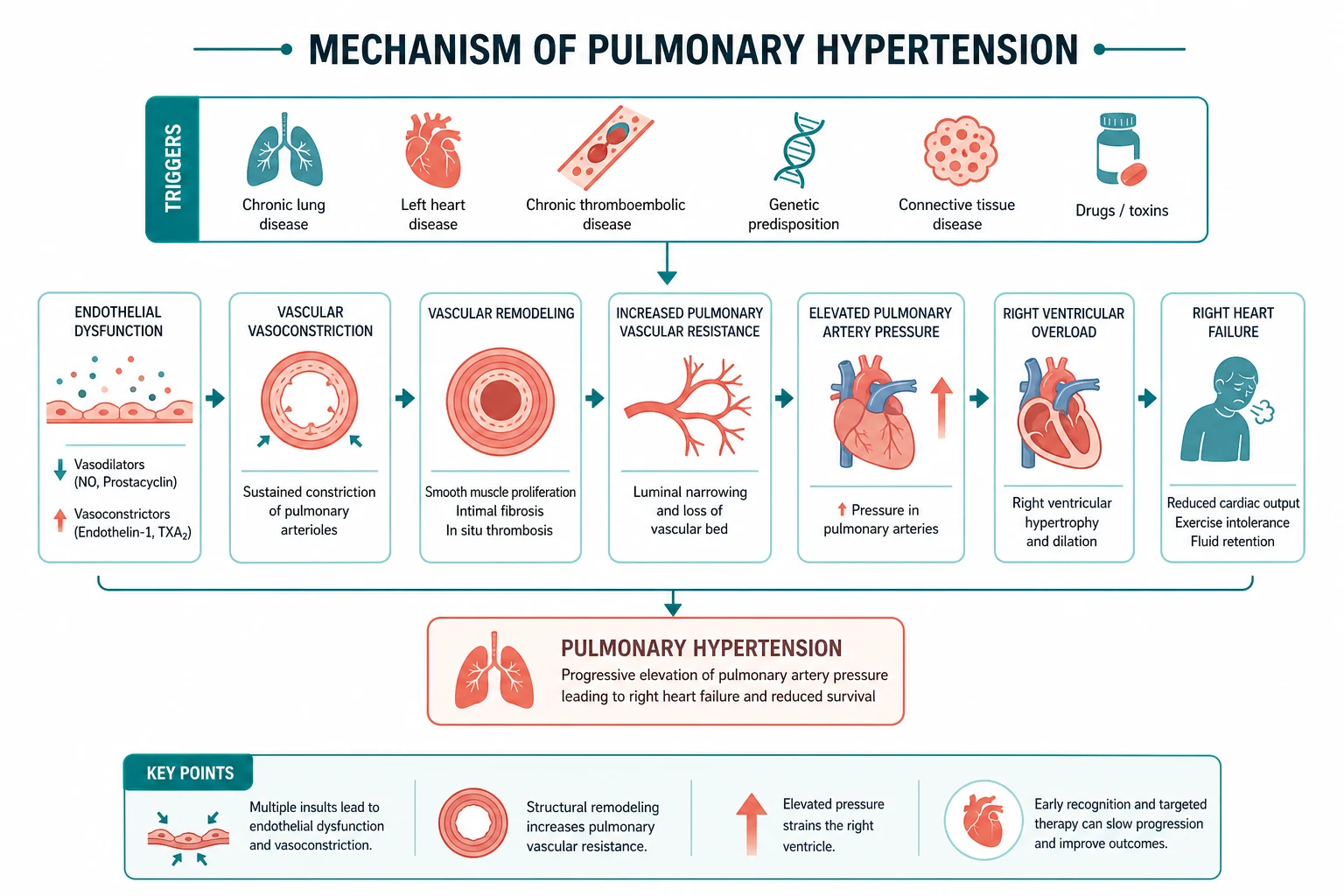
PAH is fundamentally a disease of pulmonary vascular remodelling. Three interrelated processes drive the rise in pulmonary vascular resistance: [1]
- Vasoconstriction — sustained pulmonary arterial vasoconstriction from an imbalance of vasoactive mediators. Endothelial dysfunction reduces the vasodilators prostacyclin and nitric oxide while increasing the potent vasoconstrictor endothelin-1.
- Vascular remodelling — proliferation of all three vascular cell layers (intima, media, adventitia). In heritable PAH, loss of BMPR2 signalling derepresses proliferative pathways and impairs apoptosis, allowing smooth-muscle and endothelial proliferation that narrows the arteriolar lumen. The plexiform lesion is the advanced hallmark.
- In-situ thrombosis — the remodelled, dysfunctional endothelium is prothrombotic, contributing to further vascular obstruction. [1]
These three pathways map directly onto the three classical drug targets: the prostacyclin, nitric oxide, and endothelin pathways. The newer activin-signalling inhibitor sotatercept adds a fourth — an anti-remodelling mechanism that restores the balance between proliferative (activin) and anti-proliferative (BMPR2) signalling. [1]
The clinical consequence is rising pulmonary vascular resistance, which imposes a pressure load on the right ventricle. The right ventricle initially hypertrophies to compensate, but it is structurally adapted for the low-pressure pulmonary circulation and tolerates acute and chronic pressure overload poorly. Progressive right ventricular dilation, tricuspid regurgitation, falling cardiac output, and systemic congestion follow. Right ventricular failure is the mode of death in PAH, and the health of the right ventricle — not the pulmonary pressure — is the strongest determinant of survival. [1]
Clinical features
The presentation is insidious. The dominant symptom is progressive exertional dyspnoea, present in virtually all patients, often present for months or years before diagnosis. The nonspecific nature of breathlessness and the subtlety of early signs mean the median time from symptom onset to diagnosis is still unacceptably long. [1]
Symptoms:
- Exertional dyspnoea — the universal symptom
- Fatigue and reduced exercise capacity
- Exertional chest pain — from right ventricular ischaemia
- Presyncope and syncope on exertion — an ominous sign of advanced disease and low cardiac output
- Palpitations, cough, haemoptysis
- Hoarseness — Ortner syndrome, from compression of the left recurrent laryngeal nerve by a dilated pulmonary artery [1]
Signs — the physical examination: [1]
| Sign | Mechanism |
|---|---|
| Loud pulmonary component of S2 (loud P2) | Forceful closure of the pulmonary valve over a high-pressure pulmonary artery — the cardinal auscultatory finding |
| Right ventricular heave (left parasternal lift) | Hypertrophied, pressure-loaded right ventricle |
| Systolic murmur at lower left sternal edge | Tricuspid regurgitation from annular dilation of the failing right ventricle |
| Early diastolic murmur (Graham Steell) | Pulmonary regurgitation from dilation of the pulmonary valve annulus — rare |
| Raised JVP with prominent a wave | Reduced right ventricular compliance; a prominent v wave appears with tricuspid regurgitation |
| Fourth heart sound (right-sided S4) | Atrial contraction into a stiff, hypertrophied right ventricle |
| Peripheral oedema, ascites, hepatomegaly | Systemic venous congestion from right ventricular failure |
| Cool peripheries, low pulse volume | Low cardiac output in advanced disease |
DCE short-case strategy: Present the findings in a logical sequence. "On examination the patient is comfortable at rest at 45 degrees, with no cyanosis but evidence of right heart failure. The pulse is regular, the JVP is elevated three centimetres with a prominent v wave. There is a right ventricular heave at the lower left sternal edge. On auscultation the second heart sound has a loud pulmonary component, there is a pansystolic murmur at the lower left sternal edge consistent with tricuspid regurgitation, and there is a right ventricular fourth sound. There is peripheral oedema to the mid-shin." Then conclude: "These findings are consistent with pulmonary hypertension with right ventricular failure. I would confirm with echocardiography and right heart catheterisation." [1]
Investigations — the diagnostic algorithm
The investigation strategy has two phases: non-invasive screening to build suspicion and identify the cause, and right heart catheterisation to confirm the diagnosis and define the haemodynamic phenotype. [1]

Non-invasive investigations
| Investigation | What it shows |
|---|---|
| ECG | Right ventricular hypertrophy, right axis deviation, p-pulmonale (tall peaked P waves in II, III, aVF), right bundle branch block |
| Transthoracic echocardiography | Estimated RVSP from the tricuspid regurgitation jet velocity; right ventricular size, thickness, and function; pericardial effusion (a prognostic marker); left heart structure and function to exclude Group 2 |
| BNP or NT-proBNP | Marker of right ventricular wall stress; used for diagnosis, prognosis, and risk stratification |
| Pulmonary function tests | Mildly reduced lung volumes and a disproportionately reduced DLCO — a DLCO under 60 percent predicted with preserved volumes suggests pulmonary vascular disease |
| Arterial blood gas | Hypoxaemia, often with a respiratory alkalosis; widened A-a gradient |
| V/Q scan | The investigation of choice to exclude chronic thromboembolic pulmonary hypertension — at least one segmental perfusion defect warrants CTEPH assessment |
| CTPA | Visualises proximal thrombus and assesses lung parenchyma; may miss distal CTEPH — do not rely on it to exclude CTEPH |
| Autoimmune screen | ANA, anti-centromere and anti-Scl70 (systemic sclerosis), anti-U1 RNP (mixed connective tissue disease), anti-dsDNA (SLE) |
| HIV serology | HIV-associated PAH |
| Liver function and hepatitis serology | Portal hypertension and portopulmonary PH |
| Schistosomiasis serology | In endemic regions or travellers |
| Six-minute walk test | Objective functional capacity and prognostic marker; desaturation during the test suggests right-to-left shunt or severe disease |
| Cardiopulmonary exercise testing | Reduced peak oxygen consumption with low anaerobic threshold |
| High-resolution CT and pulmonary angiography | Lung parenchyma (interstitial lung disease, emphysema) and pulmonary artery anatomy (dilated main PA, mosaic perfusion in CTEPH) |
DWE high-yield discriminator: The V/Q scan — not CTPA — is the recommended test to exclude chronic thromboembolic pulmonary hypertension. V/Q is more sensitive for the perfusion defects of CTEPH because it is a pure perfusion-versus-ventilation comparison, whereas CTPA can miss the webs, bands, and distal obstructions of chronic disease. A normal V/Q scan effectively excludes CTEPH. This is one of the most frequently tested facts in this topic. [1]
Right heart catheterisation — the definitive test
Right heart catheterisation is mandatory in every patient with suspected PAH. It confirms the diagnosis, defines the haemodynamic phenotype, excludes significant post-capillary left heart disease, and assesses severity and prognosis. [1]
Key measurements:
- mPAP — the diagnostic pressure
- PAWP (PCWP) — distinguishes pre-capillary (15 or below) from post-capillary (above 15)
- Cardiac output (by thermodilution or Fick) and cardiac index
- Pulmonary vascular resistance — calculated, above 2 WU defines pre-capillary
- Right atrial pressure — a key prognostic marker; elevated RAP signals right ventricular failure
- Mixed venous oxygen saturation (SvO2) — low values reflect impaired cardiac output and tissue extraction [1]
Vasoreactivity testing is performed at catheterisation in selected patients (idiopathic, heritable, or drug-associated PAH) using inhaled nitric oxide, intravenous epoprostenol, or intravenous adenosine. A positive response is a fall in mPAP of at least 10 mmHg to a value of 40 mmHg or below, with normal or increased cardiac output. Only vasoreactive patients — about 6 to 15 percent of idiopathic PAH — respond to high-dose calcium channel blockers, and they have a markedly better prognosis. Vasoreactivity testing is not useful in associated PAH (CTD, congenital heart disease) or Groups 2 to 5. [1]
Examiner insight: Echo estimates right ventricular systolic pressure, it does NOT diagnose pulmonary hypertension. Right heart catheterisation is the only test that confirms the diagnosis. A candidate who recommends starting PAH therapy on the basis of echo alone will fail. State this explicitly in a long-case defence: "The echo raises the suspicion; the right heart catheter confirms the diagnosis and defines the phenotype before I commit to therapy." [1]
Risk stratification — the ESC/ERS 2022 three-strata model
The 2022 ESC/ERS guidelines (PMID 36017548) use a simplified three-strata risk model — low, intermediate, and high estimated one-year mortality — built from a multi-parameter assessment. This replaced the earlier four-strata model and is applied at baseline and at every follow-up visit to guide treatment intensity. The therapeutic goal is to achieve and maintain a low-risk profile. [1]
| Parameter | Low risk | Intermediate | High risk |
|---|---|---|---|
| WHO functional class | I or II | III | IV |
| 6-minute walk distance | over 440 m | 165 to 440 m | under 165 m |
| NT-proBNP | under 300 | 300 to 1100 | over 1100 |
| Right atrial pressure | under 8 | 8 to 14 | over 14 |
| Cardiac index | 2.5 or above | 2.0 to 2.4 | under 2.0 |
| Mixed venous SvO2 | 65 or above | 60 to 64 | under 60 |
| Imaging | RV normal, no effusion | RV mild dysfunction | RV severe dysfunction or pericardial effusion |
Clinical principle: Treat to target. Every patient is reassessed at three to six monthly intervals, and therapy is escalated — adding a second or third agent, or switching to parenteral prostacyclin — until the patient reaches a low-risk profile. A patient who remains intermediate or high risk despite dual oral therapy needs escalation to triple therapy or consideration of transplant. This goal-directed strategy has transformed survival: modern registry data show three-year survival above 80 percent with combination therapy, compared with a median survival of 2.8 years in the pre-treatment era. [1]
Treatment of Group 1 PAH
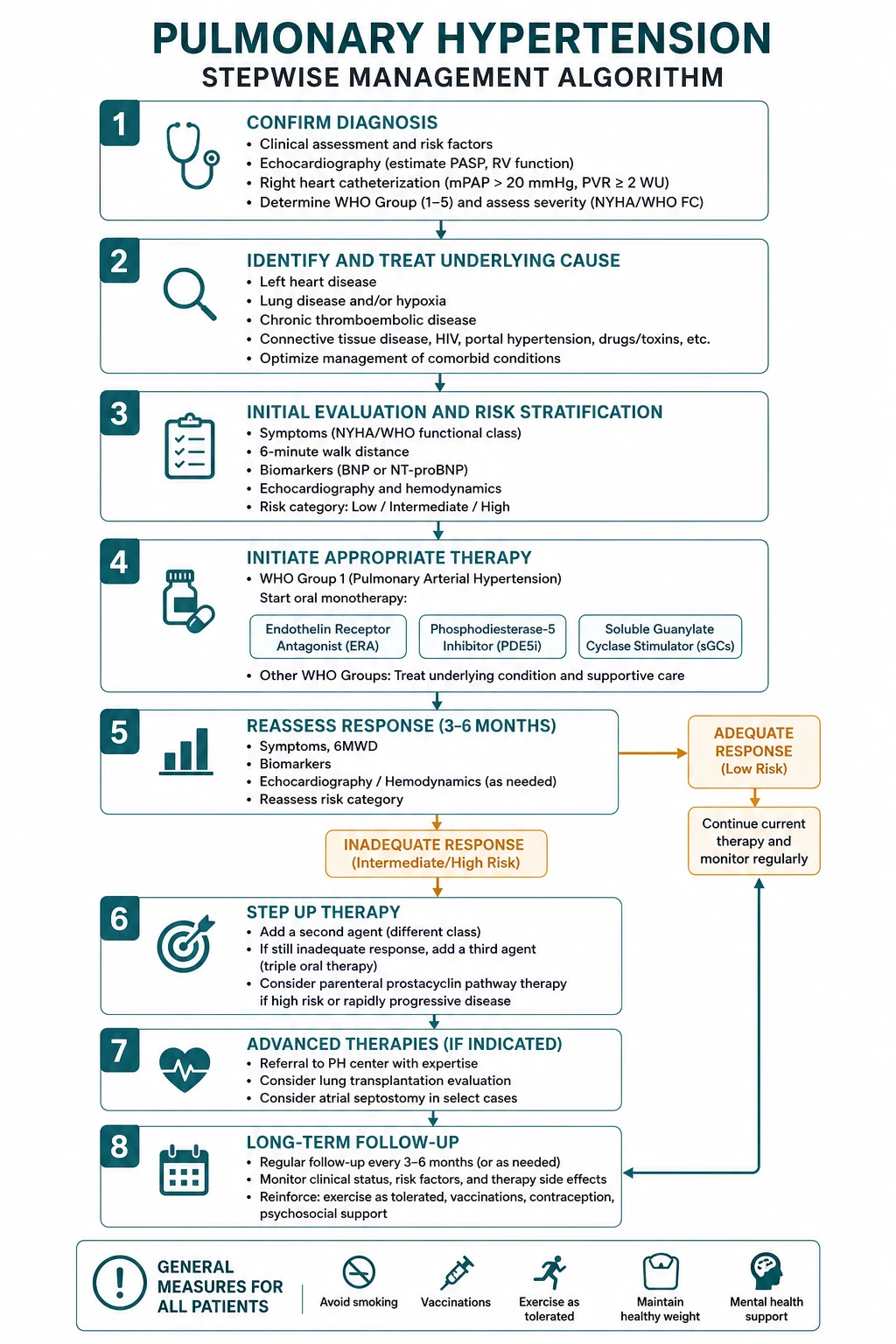
PAH therapy targets the three classical vasoactive pathways — prostacyclin, nitric oxide, and endothelin — plus the newer activin-signalling pathway. The modern standard, established by the AMBITION trial, is initial oral combination therapy for most patients at low or intermediate risk, with escalation guided by serial risk assessment. [1]

Endothelin receptor antagonists (ERA)
Endothelin-1 is a potent vasoconstrictor and mitogen over-expressed in PAH. The ERAs block its action: [1]
| Drug | Receptor selectivity | Key trial | Monitoring |
|---|---|---|---|
| Bosentan | Dual ETA and ETB | BREATHE | Monthly liver function tests — hepatotoxicity; teratogenic |
| Ambrisentan | Selective ETA | ARIES | Less hepatotoxic; can cause peripheral oedema; teratogenic |
| Macitentan | Dual ETA and ETB | SERAPHIN (PMID 23984728) | Less hepatotoxic; teratogenic; anaemia |
The SERAPHIN trial, Pulido and colleagues in the New England Journal of Medicine 2013, was the first morbidity-mortality endpoint trial in PAH. In 742 patients, macitentan 10 mg reduced the composite of death or PAH-related morbidity by 45 percent versus placebo. All ERAs are teratogenic and require robust contraception. [1]
Phosphodiesterase-5 inhibitors (PDE5)
These inhibit PDE5, the enzyme that degrades cyclic GMP, thereby potentiating the nitric-oxide pathway and producing pulmonary vasodilation:
- Sildenafil — 20 mg three times daily
- Tadalafil — 40 mg once daily [1]
Soluble guanylate cyclase stimulator — riociguat
Riociguat directly stimulates soluble guanylate cyclase, increasing cyclic GMP through a mechanism independent of, and synergistic with, endogenous nitric oxide. The PATENT-1 trial (PMID 23883378), Ghofrani and colleagues in NEJM 2013, showed a 36-metre improvement in 6-minute walk distance versus placebo. Riociguat is also approved for inoperable or persistent CTEPH (Group 4). [1]
Critical safety point: Riociguat must NEVER be combined with a PDE5 inhibitor. Both act on the nitric-oxide to cyclic-GMP pathway, and combined use risks profound and potentially fatal hypotension. This is a high-yield exam question and an absolute contraindication. [1]
Prostacyclin pathway
Prostacyclin is the most potent endogenous pulmonary vasodilator and also has anti-platelet and anti-proliferative effects. The prostacyclin analogues are the most potent PAH therapies and are first-line for severe disease (WHO functional class IV): [1]
| Drug | Route | Notes |
|---|---|---|
| Epoprostenol | Continuous intravenous | First-line for WHO FC IV; requires a permanent central line and infusion pump; improves survival in idiopathic PAH |
| Treprostinil | Subcutaneous, intravenous, inhaled, oral | Long half-life; the most flexible route options |
| Selexipag | Oral selective IP receptor agonist | GRIPHON (PMID 26699168) — oral prostacyclin with 40 percent reduction in composite morbidity-mortality |
The GRIPHON trial, Sitbon and colleagues in NEJM 2015, randomised 1156 patients to the oral selective IP-receptor agonist selexipag or placebo. Selexipag reduced the composite primary endpoint of death or PAH complication by 40 percent (hazard ratio 0.60), making it a well-tolerated oral option for add-on therapy. [1]
Activin signalling inhibitor — sotatercept
Sotatercept (brand name Winrevair) is a first-in-class activin-signalling inhibitor — a fusion protein that acts as a ligand trap for activin and related growth factors, restoring the balance between the pro-proliferative activin pathway and the anti-proliferative BMPR2 pathway. Unlike the vasoactive drugs, sotatercept is an anti-remodelling agent. [1]
The STELLAR trial (PMID 36877098), Hoeper and colleagues in NEJM 2023, randomised 323 patients with PAH on stable background therapy to sotatercept or placebo. Sotatercept improved the 6-minute walk distance by 40.8 metres versus placebo at week 24 and improved 8 of 9 secondary endpoints, including pulmonary vascular resistance, NT-proBNP, WHO functional class, and time to death or clinical worsening. It was approved by the FDA in 2024 as add-on therapy for Group 1 PAH. [1]
Initial combination therapy — the AMBITION paradigm
The AMBITION trial (PMID 26308684), Galie and colleagues in NEJM 2015, was the pivotal study establishing upfront dual oral combination therapy. In 500 treatment-naive patients, initial ambrisentan plus tadalafil reduced the risk of clinical failure by 50 percent versus pooled monotherapy (hazard ratio 0.50), driven mainly by fewer PAH hospitalisations. The 2022 ESC/ERS guidelines now recommend initial oral combination with an ERA plus a PDE5 inhibitor as the standard for most patients with low or intermediate risk PAH, with escalation to triple therapy (adding a prostacyclin) for those who do not achieve a low-risk profile. [1]
General measures and supportive therapy
- Oxygen therapy to maintain saturation above 90 percent or more at all times
- Diuretics for right heart failure and volume overload
- Anticoagulation — recommended in idiopathic PAH, heritable PAH, and CTEPH; not routinely in associated PAH
- Vaccination — influenza and pneumococcal
- Pregnancy avoidance — PAH carries a 30 to 56 percent maternal mortality; effective contraception is mandatory
- Supervised exercise rehabilitation improves functional capacity and quality of life
- Psychosocial support for a chronic, life-limiting disease [1]
Treatment of the other groups
Group 2 — left heart disease
Treat the underlying cardiac condition. Optimise guideline-directed medical therapy for heart failure, address valvular disease, manage atrial fibrillation. Diurese for volume overload. PAH-specific therapy is not indicated and may be harmful — it can worsen outcomes by increasing cardiac output into a failing left heart. [1]
Group 3 — lung disease and hypoxia
Treat the underlying lung disease and correct hypoxia. Long-term oxygen therapy improves survival in hypoxaemic COPD. PAH-specific therapy has limited evidence and is reserved for selected patients with disproportionately severe PH compared with their lung disease, managed at a specialist centre. [1]
Group 4 — CTEPH
Chronic thromboembolic pulmonary hypertension is the only potentially curable form of PH and every patient must be assessed by a multidisciplinary CTEPH team: [1]
- Pulmonary endarterectomy (PEA) — the definitive treatment for operable proximal disease. Performed under deep hypothermic circulatory arrest; removes organised thromboembolic material. Operability depends on thrombus accessibility and the expertise of the surgical centre. PEA can be curative.
- Riociguat — the only approved drug for inoperable CTEPH or persistent/recurrent PH after PEA. The CHEST-1 trial (PMID 23883377), Ghofrani and colleagues in NEJM 2013, showed a 46-metre improvement in 6-minute walk distance.
- Balloon pulmonary angioplasty (BPA) — staged percutaneous dilation of segmental and subsegmental pulmonary arteries for inoperable distal disease; improves haemodynamics and symptoms.
- Lifelong anticoagulation for all CTEPH patients; investigate for thrombophilia. [1]
Group 5 — multifactorial
Manage the underlying systemic or haematological condition. Refer to a specialist centre for consideration of PAH-specific therapy on a case-by-case basis. [1]
High-risk referral and transplantation
All patients with confirmed PAH or CTEPH should be managed at, or in shared care with, a specialist pulmonary hypertension centre. The indications for aggressive escalation or transplant referral: [1]
- WHO functional class IV at presentation — intravenous epoprostenol and urgent transplant assessment
- Persistent high-risk profile despite maximal combination therapy
- Clinical deterioration — falling 6-minute walk distance, rising NT-proBNP, signs of right heart failure
- Recurrent syncope or refractory right heart failure
- Atrial septostomy or Potts shunt (creating a right-to-left shunt to decompress the right heart) as palliative bridge-to-transplant in refractory cases [1]
Lung or heart-lung transplantation is the final therapeutic option. Bilateral lung transplant is preferred for PAH; heart-lung transplant is reserved for complex congenital heart disease with Eisenmenger physiology. [1]
Pulmonary hypertension in systemic sclerosis — the high-yield long case
Systemic sclerosis is the connective tissue disease most strongly associated with PAH, with a prevalence of approximately 8 to 12 percent. PAH is the leading cause of death in scleroderma, and annual screening is recommended because early disease is asymptomatic and treatment improves outcomes. [1]
Screening strategy for systemic sclerosis:
- Annual transthoracic echocardiogram to estimate RVSP and assess RV function
- Annual pulmonary function tests — a falling DLCO (under 60 percent predicted) with preserved lung volumes is the earliest sign of pulmonary vascular involvement
- Serum NT-proBNP as a screening biomarker
- Right heart catheterisation to confirm any case with a TR jet velocity above 2.8 m/s or unexplained dyspnoea [1]
In a long case, the candidate must distinguish scleroderma-associated PAH (Group 1) from the other pulmonary complications of systemic sclerosis: interstitial lung disease with secondary PH (Group 3), and left heart disease from myocardial fibrosis (Group 2). The right heart catheterisation is essential to make this distinction — a PAWP above 15 indicates left heart disease, not PAH. [1]
DCE long-case integration: "This 48-year-old woman with limited cutaneous systemic sclerosis (CREST syndrome) presents with a six-month history of exertional dyspnoea. Her DLCO has fallen to 52 percent with preserved lung volumes, her echo estimates an RVSP of 55 mmHg with RV dilation, and her NT-proBNP is elevated. My provisional diagnosis is scleroderma-associated PAH. My next step is right heart catheterisation to confirm the diagnosis and exclude post-capillary involvement from myocardial fibrosis. If confirmed as pre-capillary PAH, I will initiate initial oral combination therapy with an ERA and a PDE5 inhibitor and treat to a low-risk target, with early transplant referral if she does not respond." [1]
Exam synthesis — high-yield discriminators

| Question | Discriminator |
|---|---|
| What is the diagnostic threshold for PH? | mPAP above 20 mmHg by right heart catheterisation (2022 ESC/ERS update from 25) |
| What confirms the diagnosis? | Right heart catheterisation — echo only estimates RVSP |
| What distinguishes pre- from post-capillary PH? | PAWP: 15 or below is pre-capillary, above 15 is post-capillary; PVR above 2 WU defines elevated resistance |
| Best test to exclude CTEPH? | V/Q scan, not CTPA |
| Which CTD causes PAH? | Systemic sclerosis — screen annually |
| Which drug class needs monthly LFT monitoring? | Bosentan (and all ERAs — teratogenic, check LFTs) |
| Which combination is absolutely contraindicated? | Riociguat plus a PDE5 inhibitor — hypotension risk |
| What is the new anti-remodelling agent? | Sotatercept — activin-signalling inhibitor (STELLAR) |
| Which PAH therapy is first-line for WHO FC IV? | Intravenous epoprostenol |
| What is the curative treatment for operable CTEPH? | Pulmonary endarterectomy |
| What is the standard initial PAH therapy? | Oral combination ERA plus PDE5 (AMBITION) |
Regional guideline anchoring
- 2022 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension (Humbert et al, PMID 36017548) — the primary guideline. Lowered the mPAP threshold to above 20 mmHg, introduced the simplified three-strata risk model, and codified initial oral combination therapy as standard. Updated in real time by the European Pulmonary Hypertension registry.
- 6th World Symposium on Pulmonary Hypertension (Simonneau et al, PMID 30545968) — the haemodynamic definitions and updated clinical classification that underpin the 2022 guideline.
- Australian and New Zealand practice follows the ESC/ERS framework, with PH managed through designated specialist centres and subsidised access to ERA, PDE5, prostacyclin, and sGC therapies via specialist-initiated prescribing. [1]
Summary of verified references
- Humbert M, et al. 2022 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Heart J 2022;43(38):3618-3731. PMID 36017548.
- Simonneau G, et al. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur Respir J 2019;53(1):1801913. PMID 30545968.
- Hoeper MM, et al. Phase 3 Trial of Sotatercept for Treatment of Pulmonary Arterial Hypertension (STELLAR). N Engl J Med 2023;388(16):1478-1490. PMID 36877098.
- Galie N, et al. Initial Use of Ambrisentan plus Tadalafil in Pulmonary Arterial Hypertension (AMBITION). N Engl J Med 2015;373(9):834-844. PMID 26308684.
- Sitbon O, et al. Selexipag for the Treatment of Pulmonary Arterial Hypertension (GRIPHON). N Engl J Med 2015;373(25):2438-2450. PMID 26699168.
- Pulido T, et al. Macitentan and Morbidity and Mortality in Pulmonary Arterial Hypertension (SERAPHIN). N Engl J Med 2013;369(10):909-818. PMID 23984728.
- Ghofrani HA, et al. Riociguat for the Treatment of Pulmonary Arterial Hypertension (PATENT-1). N Engl J Med 2013;369(4):330-340. PMID 23883378.
- Ghofrani HA, et al. Riociguat for the Treatment of Chronic Thromboembolic Pulmonary Hypertension (CHEST-1). N Engl J Med 2013;369(4):319-329. PMID 23883377. [1]
All PMIDs were verified live via web search before entry. Primary guideline sources: ESC/ERS 2022; 6th World Symposium definitions 2019; STELLAR, AMBITION, GRIPHON, SERAPHIN, PATENT-1, and CHEST-1 trials. [1]
References
- [1]Humbert M, Kovacs G, Hoeper MM, et al. 2022 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension Eur Heart J, 2022.PMID 36017548
- [2]Simonneau G, Montani D, Celermajer DS, et al. Haemodynamic definitions and updated clinical classification of pulmonary hypertension Eur Respir J, 2019.PMID 30545968
- [3]Hoeper MM, Badesch DB, Ghofrani HA, et al. Phase 3 Trial of Sotatercept for Treatment of Pulmonary Arterial Hypertension N Engl J Med, 2023.PMID 36877098
- [4]Galie N, Barbera JA, Frost AE, et al. Initial Use of Ambrisentan plus Tadalafil in Pulmonary Arterial Hypertension N Engl J Med, 2015.PMID 26308684
- [5]Sitbon O, Channick R, Chin KM, et al. Selexipag for the Treatment of Pulmonary Arterial Hypertension N Engl J Med, 2015.PMID 26699168
- [6]Pulido T, Adzerikho I, Channick RN, et al. Macitentan and morbidity and mortality in pulmonary arterial hypertension N Engl J Med, 2013.PMID 23984728
- [7]Ghofrani HA, Galiè N, Grimminger F, et al. Riociguat for the treatment of pulmonary arterial hypertension N Engl J Med, 2013.PMID 23883378
- [8]Ghofrani HA, D'Armini AM, Grimminger F, et al. Riociguat for the treatment of chronic thromboembolic pulmonary hypertension N Engl J Med, 2013.PMID 23883377