Phys · endocrine
Adrenal Disorders
Also known as adrenal insufficiency · Addison's disease · adrenal crisis · Cushing syndrome · Cushing disease · primary aldosteronism · Conn syndrome · phaeochromocytoma · paraganglioma · adrenal incidentaloma · hyperaldosteronism · hypocortisolism · secondary adrenal insufficiency · Waterhouse-Friderichsen syndrome
Consultant-physician-depth guide to adrenal disorders — adrenal physiology, Addison's disease (primary adrenal insufficiency) and adrenal crisis, secondary adrenal insufficiency, Cushing syndrome, primary aldosteronism (Conn syndrome), phaeochromocytoma, and the adrenal incidentaloma — structured for FRACP DWE and DCE, MRCP and ABIM preparation.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Adrenal Disorders
The answer first
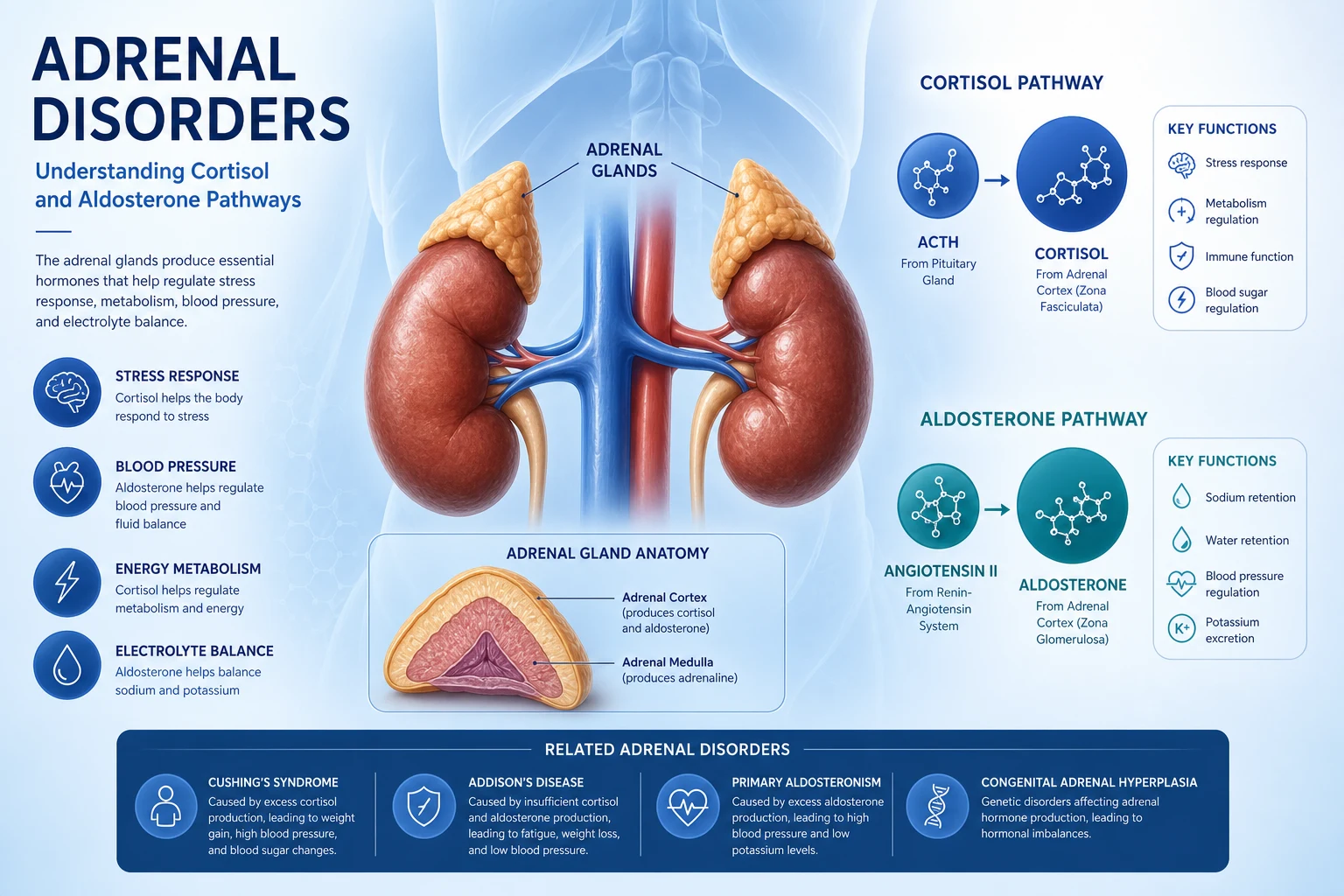
The adrenal gland produces four classes of hormone from two embryologically distinct organs fused into one: cortisol (zona fasciculata, under ACTH), aldosterone (zona glomerulosa, under angiotensin II and potassium), adrenal androgens (zona reticularis, under ACTH), and catecholamines (medulla, under sympathetic stimulation). Every adrenal disorder is a disease of one of these four hormone axes — either deficiency or excess. Master the axis, and you master the clinical reasoning. [1]
The four exam-defining syndromes, in order of exam frequency and danger: [1]
-
Primary adrenal insufficiency (Addison's disease) — deficiency of cortisol AND aldosterone from adrenal destruction. The signature is fatigue, weight loss, hyperpigmentation (from high ACTH/MSH), hyponatraemia with hyperkalaemia, and postural hypotension. Adrenal crisis is the emergency: hypotensive shock that kills within hours without parenteral hydrocortisone and fluids. [1]
-
Cushing syndrome — cortisol excess. The most discriminating signs (not just obesity) are proximal myopathy, easy bruising, purple striae wider than 1 cm, and thin skin. The diagnosis is biochemical first (confirm hypercortisolism), then localise (ACTH-dependent vs independent, then find the source). Never image first — incidental pituitary and adrenal lesions are common and will mislead you. [1]
-
Primary aldosteronism (Conn syndrome) — aldosterone excess causing hypertension, often with hypokalaemia (but frequently normokalaemic) and metabolic alkalosis. Screen with the aldosterone-to-renin ratio (ARR) in every patient with resistant hypertension. It is the commonest surgically curable cause of hypertension. [1]
-
Phaeochromocytoma — catecholamine excess. The classic triad is episodic headache, sweating, and palpitations with hypertension. Screen with plasma free metanephrines or 24h urine fractionated metanephrines. Preoperative preparation is alpha-blockade FIRST (phenoxybenzamine), then beta-blockade — never the reverse, or unopposed alpha stimulation triggers hypertensive crisis. [1]
Adrenal physiology — the axis that explains every test
The adrenal cortex is three zones — remember by GFR (glomerulosa, fasciculata, reticularis) for salt, sugar, sex (aldosterone, cortisol, androgen): [1]
- Zona glomerulosa produces aldosterone, the mineralocorticoid. It is regulated by the renin-angiotensin-aldosterone system (RAAS) and by serum potassium — NOT by ACTH. ACTH has only a permissive role. This is the single most important physiology fact for the exam: aldosterone is preserved in secondary (pituitary) adrenal insufficiency, which is why these patients do not develop hyperkalaemia.
- Zona fasciculata produces cortisol, the glucocorticoid. It is regulated by ACTH from the pituitary, which is regulated by CRH from the hypothalamus. Cortisol is under negative feedback and has a circadian rhythm — peak at 6-8 am, nadir near midnight. This rhythm is the basis for the midnight salivary cortisol test (loss of the nadir is a hallmark of Cushing).
- Zona reticularis produces DHEA and androstenedione, the adrenal androgens. These are ACTH-dependent, which is why DHEA-S falls in secondary adrenal insufficiency and rises in ACTH-dependent Cushing. [1]
The adrenal medulla is a modified sympathetic ganglion producing catecholamines (80% adrenaline, 20% noradrenaline). It is not regulated by ACTH — it is driven by preganglionic sympathetic fibres. This is why catecholamine excess (phaeochromocytoma) is independent of the pituitary-adrenal axis. [1]
Cortisol metabolism and the 11-beta-HSD2 enzyme. Cortisol and aldosterone bind the mineralocorticoid receptor with equal affinity. In the kidney, 11-beta-hydroxysteroid dehydrogenase type 2 converts cortisol to inactive cortisone, protecting the mineralocorticoid receptor from cortisol. This matters clinically: in ectopic ACTH (very high cortisol), the enzyme is overwhelmed, cortisol floods the mineralocorticoid receptor, and the patient develops hypokalaemic alkalosis — an apparent mineralocorticoid excess. Licorice (glycyrrhetinic acid) inhibits this enzyme and causes a similar syndrome. [1]
DWE trap: The question "Why does the patient with ectopic ACTH have hypokalaemia but the patient with Cushing disease does not?" tests your knowledge of 11-beta-HSD2 saturation. In ectopic ACTH, cortisol levels are so high the enzyme is overwhelmed; in Cushing disease, levels are high but not extreme. [1]
Primary adrenal insufficiency (Addison's disease)
What it is
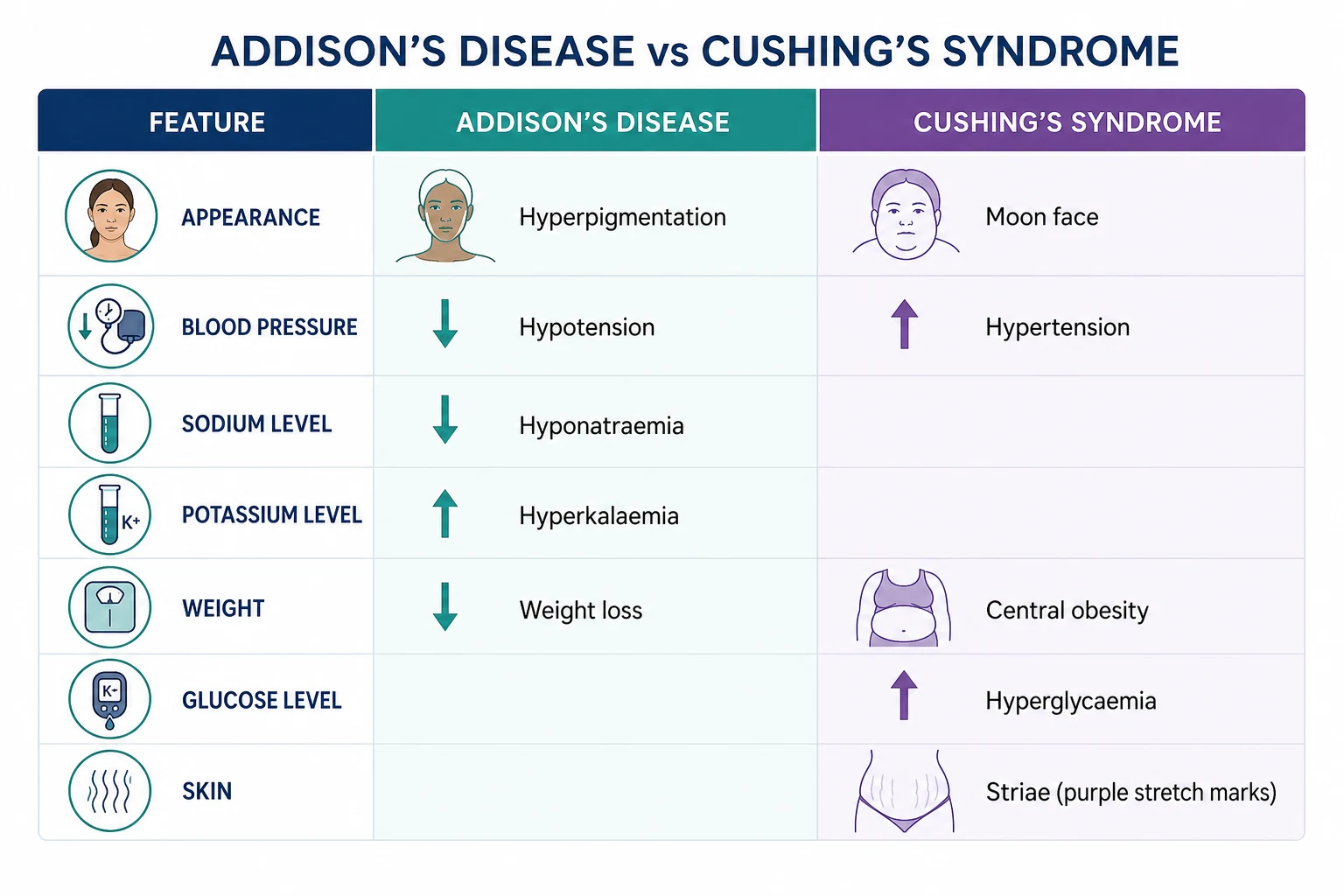
Primary adrenal insufficiency is destruction of the adrenal cortex, causing deficiency of both cortisol and aldosterone (plus adrenal androgens). The high-yield fact: because the adrenal is destroyed, ACTH from the pituitary rises unchecked (no negative feedback), and high ACTH cross-stimulates melanocyte-stimulating hormone receptors, causing hyperpigmentation — the single most discriminating sign between primary and secondary adrenal insufficiency. [1]
Causes
| Category | Specific causes | Clues |
|---|---|---|
| Autoimmune (commonest in developed world, 70-90%) | Isolated autoimmune Addison's or autoimmune polyglandular syndrome type 1 (AIRE gene, chronic mucocutaneous candidiasis, hypoparathyroidism) or type 2 (Schmidt syndrome: Addison's + autoimmune thyroid disease + type 1 diabetes) | 21-hydroxylase antibodies positive; other autoimmune conditions (vitiligo, thyroid, T1DM, pernicious anaemia, premature ovarian failure); small/atrophic adrenals on CT |
| Tuberculosis (commonest worldwide) | Disseminated TB | Adrenal enlargement or calcification; endemic area; other TB features |
| Bilateral adrenal haemorrhage | Anticoagulants (especially initiation), antiphospholipid syndrome, severe sepsis/DIC, major surgery/trauma, heparin-induced thrombocytopenia, Waterhouse-Friderichsen (meningococcaemia) | Sudden back/abdominal pain, falling haemoglobin, in an anticoagulated or septic patient; bilateral enlarged adrenals on CT |
| Infections (other than TB) | HIV/AIDS (cytomegalovirus, mycobacteria), fungal (histoplasmosis, cryptococcosis, paracoccidioidomycosis), syphilis | Immunocompromised; endemic area for fungal infection |
| Infiltrative/metastatic | Metastases (lung, breast, kidney, melanoma), lymphoma, amyloidosis, sarcoidosis, haemochromatosis | Known malignancy; bilateral adrenal masses on imaging |
| Genetic | Adrenoleukodystrophy (X-linked, very long chain fatty acids), congenital adrenal hyperplasia (21-hydroxylase deficiency), ACTH resistance | Young male with neurological features (ALD); ambiguous genitalia or salt-wasting in infancy (CAH) |
| Drugs | Adrenal enzyme inhibitors (ketoconazole, metyrapone, etomidate, mitotane), tyrosine kinase inhibitors | Drug history; especially etomidate in ICU induction |
Clinical features
The presentation is insidious in chronic disease and catastrophic in crisis. The patient has been unwell for months: [1]
- General: progressive fatigue, weakness, weight loss (often 10-15 kg), anorexia, nausea, vomiting, abdominal pain (can mimic an acute abdomen).
- Skin: hyperpigmentation — the cardinal sign. Look in sun-exposed areas, palmar creases, buccal mucosa, gums, recent scars, areolae, extensor surfaces, and skin folds. It is caused by high ACTH (and the co-secreted POMC-derived peptide beta-lipotropin, which has melanocyte-stimulating activity). Vitiligo may coexist (autoimmune).
- Cardiovascular: postural hypotension (from aldosterone deficiency causing salt wasting and volume depletion), sometimes syncope.
- Metabolic/electrolyte: hyponatraemia (from aldosterone deficiency causing renal sodium wasting, and from cortisol deficiency causing inappropriate ADH secretion), hyperkalaemia (from aldosterone deficiency — potassium is retained), metabolic acidosis, hypoglycaemia (impaired gluconeogenesis), hypercalcaemia (occasional, from increased bone resorption).
- Gastrointestinal: salt craving (a specific clue), nausea, vomiting, abdominal pain, diarrhoea.
- Other: loss of axillary and pubic hair in women (loss of adrenal androgens — in men, testicular androgens are preserved), amenorrhoea, reduced libido, depression. [1]
DCE insight: In the long case, if the patient is hyperpigmented with unexplained hyponatraemia, Addison's is at the top of your differential until excluded. The single test that changes management in a sick patient is a random cortisol — if it is above 500 nmol/L in a stressed patient, adrenal insufficiency is excluded and you do not need a Synacthen test to justify not giving steroids. [1]
Investigations — the diagnostic strategy
| Step | Test | What it tells you |
|---|---|---|
| 1. Screen | Morning (8-9 am) cortisol | If above 500 nmol/L, adrenal insufficiency excluded. If below 100-200 nmol/L, insufficiency likely — proceed. Between 200-500, indeterminate — do Synacthen test. |
| 2. Confirm | Short Synacthen (cosyntropin) test — 250 mcg IV/IM, cortisol at 0, 30, 60 min | Peak cortisol below 500 nmol/L confirms adrenal insufficiency. The 250 mcg dose is supraphysiological; a normal response (above 500 nmol/L) reliably excludes primary adrenal insufficiency. |
| 4. Confirm primary and assess mineralocorticoid axis | Renin (high), aldosterone (low), electrolytes (hyponatraemia, hyperkalaemia) | In primary: renin high (from volume depletion), aldosterone low (adrenal destroyed). In secondary: aldosterone normal (RAAS intact), potassium normal, renin normal. |
| 5. Find the cause | 21-hydroxylase antibodies (autoimmune), adrenal CT (atrophic in autoimmune, enlarged in TB/infiltrative, calcified in old TB) | Autoimmune if antibodies positive and adrenals small/atrophic. Refer for TB testing, HIV testing, or malignancy workup if antibody-negative with enlarged adrenals. |
The key interpretation rules:
- In an acutely unwell patient, do NOT delay treatment for tests. Take blood for cortisol, ACTH, renin, aldosterone, and electrolytes, then give hydrocortisone 100 mg IV and fluids immediately. The short Synacthen test can be done after recovery. The Bornstein 2016 Endocrine Society guideline is explicit: treat first if the patient is sick — adrenal crisis has a mortality, and a delay for testing can be fatal. [1]- Hydrocortisone cross-reacts with cortisol assays, so once you have given hydrocortisone, you cannot interpret cortisol levels until the drug is cleared (use dexamethasone if you need ongoing treatment without assay interference — but dexamethasone has no mineralocorticoid activity).
- The low-dose (1 mcg) Synacthen test is more sensitive for secondary (mild/partial) adrenal insufficiency but less validated than the standard 250 mcg test; it is mainly used in research and selected centres.
- The insulin stress (tolerance) test (ITT — insulin-induced hypoglycaemia to stress the axis) is the gold standard for diagnosing secondary adrenal insufficiency and growth hormone deficiency, but it is unpleasant and contraindicated in ischaemic heart disease, epilepsy, and pregnancy. The glucagon stimulation test is an alternative when the ITT is contraindicated. [1]
Management of chronic primary adrenal insufficiency
Glucocorticoid replacement: hydrocortisone 15-25 mg/day in 2-3 divided doses, mimicking the physiological circadian rhythm. A common regimen: 10 mg on waking, 5 mg at midday, 5 mg at 4-5 pm. Give the largest dose in the morning (the physiological peak), and the smallest dose latest (to avoid insomnia — hydrocortisone has a short half-life of about 8 hours). Alternative: prednisolone 5 mg once or twice daily (longer duration, more convenient, but harder to titrate). Avoid dexamethasone for chronic replacement (too long-acting, no mineralocorticoid effect, risk of Cushingoid features). [1]
Mineralocorticoid replacement: fludrocortisone 50-200 mcg/day (typically 100 mcg). This is ESSENTIAL in primary adrenal insufficiency — the adrenal cannot make aldosterone. Monitor with blood pressure (including postural), serum sodium, and plasma renin (aim for high-normal renin). Fludrocortisone is NOT needed in secondary adrenal insufficiency, because the zona glomerulosa and the RAAS are intact. [1]
Sick-day rules — the single most important patient education. The goal is to mimic the physiological cortisol response to stress, which in a healthy person raises cortisol output 2-10 fold: [1]
| Situation | Action |
|---|---|
| Minor illness (common cold, mild infection, afebrile) | Double the usual oral hydrocortisone for 2-3 days |
| Moderate illness (fever above 37.5, infection requiring antibiotics, vomiting/diarrhoea) | Triple the oral hydrocortisone; ensure hydration; close contact with clinician |
| Severe illness, vomiting (cannot tolerate oral), surgery, major trauma | Hydrocortisone 100 mg IM (emergency injection kit) — patients are taught to self-inject; seek urgent medical attention |
| Dental or minor procedure | Hydrocortisone 100 mg IM or IV at induction |
| Major surgery | Hydrocortisone 100 mg IM at induction, then 50 mg IM every 6 hours for 48-72 hours, tapering to maintenance |
Patient education essentials:
- Wear a MedicAlert bracelet or necklace stating "Adrenal insufficiency — requires hydrocortisone."
- Carry an emergency hydrocortisone injection kit (100 mg vial, syringe, instructions) at all times — teach the patient and family IM injection. [1]- Carry a steroid card with the diagnosis, medications, and emergency instructions.
- Never omit steroids — the patient is dependent on exogenous replacement for life.
- Educate on sick-day rules and provide written instructions.
- Consider a glucagon emergency kit if also on insulin (dual risk of hypoglycaemia). [1]
Adrenal crisis — the emergency
What it is
Acute adrenal insufficiency with shock — a medical emergency with significant mortality. It occurs in patients with known adrenal insufficiency (the commonest scenario: omission of steroids or intercurrent illness without dose escalation) or as the first presentation of adrenal insufficiency. Hahner et al. (PMID 25419882) found an incidence of 8.3 adrenal crises per 100 patient-years in educated patients with chronic adrenal insufficiency, and a mortality of approximately 6% per crisis — adrenal crisis remains a real and persistent threat despite education. [1]
Precipitants
- Infection (gastroenteritis, pneumonia, urinary tract infection) — the commonest precipitant.
- Omission or inadequate increase of steroids — the patient forgets, or does not escalate for illness.
- Surgery, trauma, major medical illness (myocardial infarction, stroke) without stress-dose cover.
- Acute adrenal destruction — bilateral adrenal haemorrhage (anticoagulation, sepsis, antiphospholipid), Waterhouse-Friderichsen (meningococcaemia).
- Drugs — rapid withdrawal of chronic exogenous steroids, enzyme inhibitors (ketoconazole, etomidate). [1]
Clinical features and the emergency response
The patient is shocked (hypotension, tachycardia, cold peripheries), often with abdominal pain (can mimic an acute abdomen and lead to unnecessary laparotomy), vomiting, confusion or reduced consciousness, and fever (from infection or the crisis itself). Biochemically: hyponatraemia, hyperkalaemia, hypoglycaemia, metabolic acidosis, elevated urea (pre-renal), and often hypercalcaemia. [1]
Emergency management — do not delay for confirmatory tests: [1]
- Take blood first (if possible, within minutes): cortisol, ACTH, renin, aldosterone, electrolytes, glucose, FBC, blood cultures. Then TREAT — do not wait for results.
- IV hydrocortisone 100 mg stat, then 100 mg every 6-8 hours (or a continuous infusion of 200 mg over 24 hours). In crisis, hydrocortisone at this dose provides sufficient mineralocorticoid activity — fludrocortisone is not needed acutely. [1]3. Aggressive IV fluid resuscitation: 1 litre of 0.9% saline stat, then 1 litre over 1 hour, then continue at 1 litre every 2-4 hours guided by clinical response, aiming to restore intravascular volume and correct hyponatraemia. Add dextrose (e.g. 5% dextrose or saline-dextrose) because hypoglycaemia is common and dangerous.
- Identify and treat the precipitant — blood and urine cultures, chest X-ray; start broad-spectrum antibiotics if infection suspected. Do not attribute fever solely to the crisis.
- Monitor: continuous ECG, blood pressure (invasive if shocked), urine output (catheterise), electrolytes and glucose every 2-4 hours, and clinical response.
- Supportive care: oxygen, treat hypoglycaemia, correct electrolyte derangements (the hyperkalaemia resolves with hydrocortisone and fluids — do not give calcium resonium or insulin-dextrose routinely unless hyperkalaemia is severe with ECG changes). [1]
After the acute phase: as the patient stabilises, taper hydrocortisone (e.g. halve the dose every 24-48 hours) to oral maintenance, introduce fludrocortisone (when hydrocortisone dose drops below approximately 50 mg/day, as higher-dose hydrocortisone provides sufficient mineralocorticoid activity), and arrange the diagnostic workup for the cause if newly diagnosed. [1]
DWE trap: "A 68-year-old woman on warfarin for atrial fibrillation presents with sudden severe back pain, hypotension, and a haemoglobin drop from 130 to 88 g/L. CT shows bilateral enlarged adrenal masses." This is bilateral adrenal haemorrhage — check cortisol and ACTH, treat with hydrocortisone and fluids, and correct the coagulopathy. Do not attribute the pain to musculoskeletal causes. [1]
Secondary adrenal insufficiency
What it is
Deficiency of ACTH from the pituitary (or CRH from the hypothalamus) leading to cortisol deficiency. The adrenal cortex itself is intact, but it is understimulated. The key differences from primary: [1]
| Feature | Primary (Addison's) | Secondary (pituitary/hypothalamic) |
|---|---|---|
| Cortisol | Low | Low |
| ACTH | High | Low or inappropriately normal |
| Aldosterone | Low (adrenal destroyed) | Normal (RAAS intact, zona glomerulosa preserved) |
| Renin | High | Normal |
| Potassium | High (from aldosterone deficiency) | Normal (aldosterone preserved) |
| Sodium | Low (salt wasting + SIADH) | Low (SIADH from cortisol deficiency alone, but less severe) |
| Hyperpigmentation | Present (high ACTH/MSH) | Absent (ACTH is low) |
| Hypoglycaemia | Common | Common (and may be the only clue) |
| Other pituitary hormones | Normal | Often deficient (TSH, LH, FSH, GH, prolactin — panhypopituitarism) |
| Response to ACTH (Synacthen test) | No response (adrenal destroyed) | Attenuated in chronic secondary (the adrenal is intact but atrophied from disuse — it may respond normally to prolonged stimulation) |
Causes
- Exogenous glucocorticoids — by far the commonest cause. Any patient on prednisolone above 5 mg daily (or equivalent) for more than 3 weeks has HPA axis suppression. The axis recovers over weeks to months after cessation; recovery can take up to a year after long-term high-dose therapy. This is the cause examiners test most.
- Pituitary disease — pituitary adenoma, pituitary surgery, pituitary apoplexy, Sheehan syndrome (postpartum pituitary infarction), infiltrative (hemochromatosis, sarcoidosis, lymphocytic hypophysitis), traumatic brain injury, cranial irradiation.
- Hypothalamic disease — tumours, infiltration, radiation. [1]
Why this matters
The exogenous-steroid patient is the highest-yield exam scenario. Any patient on chronic steroids who presents with intercurrent illness, surgery, or sepsis is at risk of adrenal crisis from HPA suppression — they need stress-dose hydrocortisone. The patient who abruptly stops long-term steroids (often because of side-effects or "I felt better") is at risk of crisis. Conversely, the patient with secondary adrenal insufficiency does NOT need fludrocortisone (the RAAS is intact). [1]
The Bornstein 2016 guideline (PMID 26760044) recommends that patients on long-term glucocorticoids (above 5 mg prednisolone equivalent for more than 3 weeks) be considered to have adrenal suppression and managed with stress-dose steroids during illness and surgery. [1]
Cushing syndrome
What it is
Cushing syndrome is the clinical state of cortisol excess from any cause. Cushing disease is a specific subtype — cortisol excess from an ACTH-secreting pituitary corticotroph adenoma (typically a microadenoma). The distinction is critical because the treatment differs: Cushing disease is treated by transsphenoidal pituitary surgery, ectopic ACTH by finding and resecting the tumour, and adrenal sources by adrenalectomy. [1]
Causes
| Category | Cause | Proportion of endogenous cases |
|---|---|---|
| Exogenous (iatrogenic) | Glucocorticoid therapy (oral, inhaled, topical, injected), immunosuppression post-transplant | By far the commonest overall cause — always take a drug history |
| ACTH-dependent | Cushing disease (pituitary corticotroph adenoma) | ~70% of endogenous |
| Ectopic ACTH (small cell lung cancer, bronchial/pancreatic/gut carcinoid, medullary thyroid cancer, phaeochromocytoma, ovarian tumours) | ~15% of endogenous | |
| ACTH-independent | Adrenal adenoma | ~10% of endogenous |
| Adrenal carcinoma | ~5% of endogenous | |
| Bilateral macronodular adrenal hyperplasia, primary pigmented nodular adrenocortical disease (PPNAD, part of Carney complex), McCune-Albright syndrome | Rare |
Clinical features — and what really discriminates
The patient with Cushing looks Cushingoid — but the key is to distinguish Cushing from simple obesity, which is far commoner. The features that best discriminate Cushing from simple obesity and the metabolic syndrome are: [1]
- Proximal myopathy — the single best discriminator. Ask the patient to rise from a squat without using their hands, or to hold their arms outstretched; they cannot. This reflects the catabolic effect of cortisol on skeletal (type II) muscle.
- Easy bruising and thin, fragile skin (cortisol thins the dermis and capillaries).
- Purple striae wider than 1 cm (cortisol-induced skin thinning and rapid weight gain). Distinguish from the white/silver striae of simple obesity or pregnancy.
- Osteoporosis with fragility fractures (cortisol suppresses osteoblasts).
- Hypertension and glucose intolerance/diabetes (cortisol and mineralocorticoid effects). [1]
Other features: central (truncal) obesity with sparing of the limbs, moon face (fat redistribution), buffalo hump (cervical fat pad), plethora (thin skin over cheeks), hirsutism and acne (adrenal androgen excess in ACTH-dependent disease), amenorrhoea/impotence (cortisol suppresses GnRH), psychiatric disturbance (depression, anxiety, emotional lability, psychosis), hypokalaemic alkalosis (especially ectopic ACTH — see 11-beta-HSD2 above), hyperpigmentation (in ectopic ACTH and Nelson syndrome — the ACTH is very high), nephrolithiasis, and increased infection risk. [1]
DCE short-case trap: The examiner asks you to examine a patient with a "round face." The discriminator between Cushing and simple obesity is proximal myopathy. Examine it explicitly: "I would now like to assess for proximal muscle weakness by asking the patient to rise from a squatting position and to hold the arms outstretched." Examiners are testing whether you know this is the key sign. [1]
Investigation strategy — biochemical first, then localise
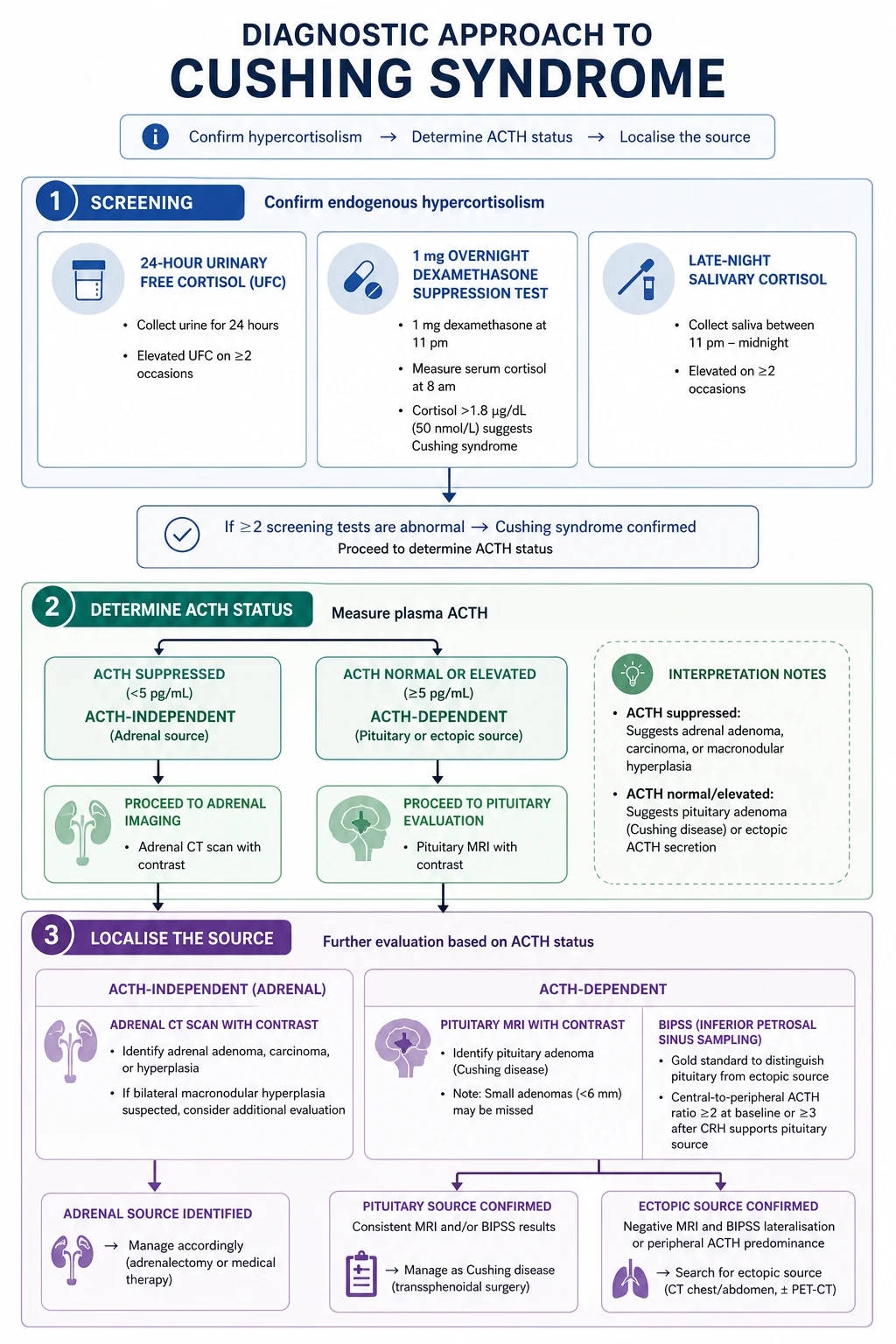
The Nieman 2008 Endocrine Society guideline (PMID 18334580) mandates a three-step strategy. The cardinal rule: never image first. Incidental pituitary lesions are found in 10% of normal people, and incidental adrenal adenomas in 5-7%. Imaging before confirming hypercortisolism biochemically will mislead you. [1]
Step 1 — Confirm hypercortisolism. Use at least two of the three first-line tests: [1]
| Test | How | Positive result | Notes |
|---|---|---|---|
| 1 mg overnight dexamethasone suppression test (ONDST) | Dexamethasone 1 mg orally at 11 pm; measure cortisol at 8-9 am | Cortisol above 50 nmol/L (1.8 mcg/dL) fails to suppress — suggests Cushing | The most widely used screen. Oestrogen (oral contraceptive pill/HRT) raises cortisol-binding globulin and causes false positives — stop OCP 6 weeks before or use a non-oestrogen method. Drugs that accelerate dexamethasone metabolism (phenytoin, carbamazepine, phenobarbital, rifampicin) cause false positives. |
| 24-hour urine free cortisol | Collect all urine for 24 hours; measure urinary free cortisol | Above the upper limit of normal (assay-dependent, typically above 100-150 mcg/24h or above 3x ULN for clear positivity) | A measure of integrated cortisol output over 24h. Requires a complete collection. Less sensitive for mild Cushing. Repeat 2-3 times. |
| Late-night (midnight) salivary cortisol | Patient collects saliva at home at 11 pm-midnight on 2 separate nights | Above the assay-specific upper limit | Loss of the midnight cortisol nadir is an early and sensitive hallmark of Cushing. Convenient, non-invasive, reliable. The preferred screen in many centres. |
Step 2 — Determine ACTH-dependent vs ACTH-independent. Measure plasma ACTH at 8-9 am (on at least 2 occasions, handled correctly — on ice, processed immediately): [1]
- ACTH low or undetectable = ACTH-independent (adrenal source: adenoma, carcinoma, hyperplasia). Proceed to adrenal CT/MRI.
- ACTH high-normal or elevated = ACTH-dependent (pituitary Cushing disease or ectopic ACTH). Proceed to Step 3 to localise. [1]
Step 3 — Localise the source in ACTH-dependent disease. [1]
- Pituitary MRI (dynamic contrast-enhanced) — identifies a pituitary adenoma in about 60-70% of Cushing disease. But 30-40% of corticotroph adenomas are microadenomas not seen on MRI, and 10% of normal people have incidental pituitary lesions. MRI alone is not enough.
- Bilateral inferior petrosal sinus sampling (BIPSS) — the gold standard for confirming Cushing disease versus ectopic ACTH. Catheterise both inferior petrosal sinuses and a peripheral vein; measure ACTH at baseline and after CRH stimulation. A central-to-peripheral ACTH ratio above 2 at baseline, or above 3 after CRH stimulation, confirms pituitary (Cushing disease). A ratio below 2 at both times suggests ectopic ACTH. Oldfield et al. (PMID 1652686) established the technique and thresholds: a basal IPS:P ratio at or above 2 had 95% sensitivity and 100% specificity, and post-CRH ratio at or above 3 achieved 100% sensitivity and specificity. BIPSS is indicated when ACTH-dependent Cushing is confirmed but pituitary MRI is normal, equivocal, or shows a lesion that could be incidental.
- High-dose dexamethasone suppression test (HDDST) — dexamethasone 8 mg overnight or 2 mg every 6 hours for 48 hours. Cushing disease suppresses (cortisol falls by at least 50% — the adenoma retains some feedback); ectopic ACTH and adrenal sources do not. This test is less reliable than BIPSS and is now second-line, but it is still tested in exams.
- For ectopic ACTH localisation: CT chest/abdomen/pelvis (small cell lung cancer is the commonest source), octreotide scintigraphy or Ga-68 DOTATATE PET (for neuroendocrine tumours), and a careful search for carcinoid tumours. [1]
Step 3 for ACTH-independent disease (low ACTH):
- Adrenal CT (or MRI) — identifies the adrenal lesion. Use Hounsfield units to characterise: a benign adenoma is at or below 10 HU on non-contrast CT (lipid-rich). See the incidentaloma section below for the full imaging algorithm. [1]
Management
Exogenous (iatrogenic) Cushing — the commonest form. Reduce and withdraw the glucocorticoid slowly (over weeks to months) to allow HPA axis recovery. Treat the underlying disease with steroid-sparing agents where possible. NEVER stop suddenly — adrenal crisis from HPA suppression can be fatal. [1]
Cushing disease (pituitary):
- First-line: transsphenoidal resection of the pituitary adenoma. Selective adenomectomy by an experienced surgeon achieves remission in 70-90% of microadenomas and 50-65% of macroadenomas. Postoperatively, the patient develops transient secondary adrenal insufficiency (the suppressed normal corticotrophs and the contralateral adrenal need months to recover) and requires glucocorticoid replacement until the HPA axis recovers.
- Second-line: stereotactic radiotherapy or radiosurgery (gamma knife) for residual or recurrent disease, or bilateral adrenalectomy for refractory disease.
- Medical therapy while awaiting definitive treatment or for refractory disease: steroidogenesis inhibitors (metyrapone, ketoconazole, osilodrostat), pasireotide (somatostatin analogue targeting somatostatin receptor subtype 5 on corticotroph adenomas — Colao et al. PMID 22397653 showed 15-26% normalisation of urinary free cortisol, with hyperglycaemia in 73% as the key adverse effect), mifepristone (glucocorticoid receptor antagonist, for hyperglycaemia and hypertension, but does not lower cortisol — monitor other markers).
- Bilateral adrenalectomy — curative but commits the patient to lifelong glucocorticoid and mineralocorticoid replacement and carries a 10-30% risk of Nelson syndrome (growth of the pituitary corticotroph adenoma after loss of cortisol feedback, causing high ACTH, hyperpigmentation, and potential mass effect). Monitor with pituitary MRI and ACTH. [1]
Ectopic ACTH — find and resect the tumour (e.g. bronchial carcinoid). If the tumour cannot be found or resected, control cortisol excess medically (metyrapone, ketoconazole, osilodrostat) or with bilateral adrenalectomy. [1]
Adrenal adenoma — unilateral laparoscopic adrenalectomy is curative. Postoperatively, the contralateral adrenal is suppressed and needs months to recover — give glucocorticoid replacement and taper. [1]
Adrenal carcinoma — surgical resection (open approach for large tumours) plus adjuvant mitotane (adrenolytic) for residual or metastatic disease. Prognosis is poor (5-year survival 20-40%). [1]
Manage the comorbidities throughout: treat hypertension, hyperglycaemia, osteoporosis (bisphosphonate or denosumab — DEXA scan), provide VTE prophylaxis (Cushing is a prothrombotic state — consider prophylactic anticoagulation in severe cases), and screen for and treat infection. Untreated Cushing carries a 50% 5-year mortality from cardiovascular disease, infection, and thromboembolism. [1]
Primary aldosteronism (Conn syndrome)
What it is
Autonomous aldosterone secretion from the adrenal zona glomerulosa, independent of the RAAS, causing hypertension (and classically hypokalaemia and metabolic alkalosis). It accounts for 5-10% of all hypertension — far higher than the 1% historically quoted — and is the commonest surgically curable cause of hypertension. The message for exams: screen every patient with resistant hypertension. [1]
Why it matters
Primary aldosteronism is not just another cause of high blood pressure. Patients with primary aldosteronism have higher cardiovascular morbidity and mortality than age- and blood-pressure-matched patients with essential hypertension — aldosterone has direct fibrotic and inflammatory effects on the heart (left ventricular hypertrophy, heart failure, atrial fibrillation) and kidneys (albuminuria, CKD). Curing or treating the aldosterone excess reduces this excess risk beyond blood pressure lowering alone. [1]
Who to screen
The Funder 2016 Endocrine Society guideline (PMID 26934393) recommends screening with the ARR in: [1]
- Resistant hypertension (above 140/90 on 3 antihypertensives including a diuretic, or controlled on 4 or more).
- Hypertension with spontaneous or diuretic-induced hypokalaemia.
- Hypertension with an adrenal incidentaloma.
- Hypertension with a family history of early-onset stroke (below age 40).
- Hypertension at a young age (below 40). [1]
The 2025 Endocrine Society update goes further, recommending screening of all patients with hypertension — a shift toward universal screening that reflects the high prevalence and the availability of an effective test. [1]
Clinical features
The patient is hypertensive, often resistant to multiple agents. Hypokalaemia (below 3.5 mmol/L) with metabolic alkalosis is the classic biochemical picture — but most patients are normokalaemic at presentation, and hypokalaemia is a late finding. Other features: nocturia and polyuria (hypokalaemic nephrogenic diabetes insipidus), muscle weakness and cramps (hypokalaemia), and headache. [1]
Investigation — screen, confirm, subtype
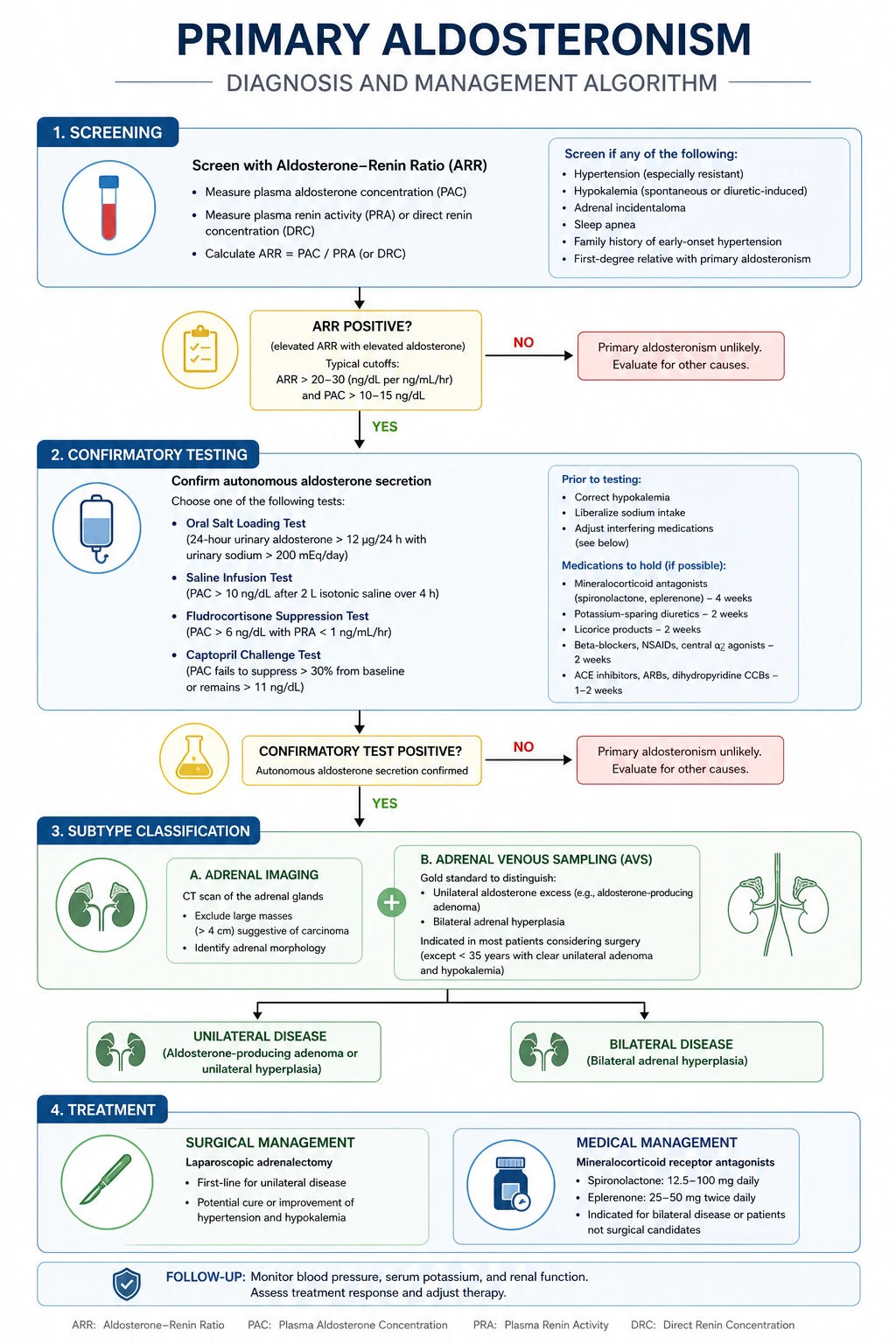
Step 1 — Screen with the aldosterone-to-renin ratio (ARR). [1]
- Measure plasma aldosterone and plasma renin activity (or direct renin concentration) in the morning, with the patient seated for 5-15 minutes, normokalaemic, and off interfering drugs.
- A positive screen: ARR above 30 (with aldosterone above 15 ng/dL / 416 pmol/L and suppressed renin). The ARR is sensitive but not specific — it is a screen, not a diagnostic test.
- Drug withdrawal before testing: stop spironolactone and eplerenone for 6 weeks (they markedly raise renin and aldosterone and interfere with the result); stop ACE inhibitors, ARBs, and diuretics for at least 2 weeks (they raise renin); stop beta-blockers (they lower renin and can cause false positives). Replace antihypertensives with non-interfering agents (verapamil slow-release plus hydralazine, or alpha-blockers like doxazosin). Correct hypokalaemia (it suppresses aldosterone secretion and causes false negatives). [1]
Step 2 — Confirm autonomous aldosterone secretion. [1]
A positive ARR is not diagnostic — it must be confirmed by demonstrating that aldosterone is not suppressible by salt loading. Four accepted confirmatory tests: [1]
| Test | How | Confirms primary aldosteronism if |
|---|---|---|
| Oral salt loading test | High-sodium diet (over 200 mmol/day) plus sodium chloride tablets for 3 days, then 24h urine for aldosterone, sodium and potassium | Urinary aldosterone above 12 mcg/24h (with adequate sodium loading, urine sodium above 200 mmol/24h) |
| Fludrocortisone suppression test | Fludrocortisone 0.1 mg every 6 hours for 4 days plus salt loading; measure upright plasma aldosterone | Aldosterone not suppressed (above 6 ng/dL) |
| Captopril suppression test | Captopril 25 mg orally; measure aldosterone at 2 hours | Aldosterone not suppressed (above criteria vary) |
Step 3 — Subtype: unilateral (surgically curable) vs bilateral (medical). [1]
- Adrenal CT — identifies a unilateral adenoma versus bilateral hyperplasia, and excludes carcinoma. But CT is unreliable for subtype determination: it misses small adenomas, misidentifies microadenomas, and cannot distinguish an aldosteronoma from a non-functioning incidentaloma. CT alone is not enough.
- Adrenal venous sampling (AVS) — the gold standard for subtype determination. Catheterise both adrenal veins (the right is technically difficult) and compare aldosterone:cortisol ratios from each side versus the periphery. Unilateral lateralisation (ratio above 2-4 on one side, suppressed on the other) indicates a surgically curable unilateral adenoma. AVS is indicated in all patients considering surgery, especially those above 40 (higher risk of incidental non-functioning adenoma) and when CT is normal, equivocal, or bilateral. [1]
The subtype decision:
- Unilateral aldosterone-producing adenoma (aldosteronoma, ~30% of cases) — laparoscopic adrenalectomy cures or improves hypertension in most patients (biochemical cure in 95-100%, hypertension cure in 30-60%, improvement in most). Cure is more likely in younger patients, shorter duration of hypertension, fewer antihypertensives, no family history of hypertension, and a single adenoma on AVS.
- Bilateral idiopathic hyperplasia (bilateral adrenal hyperplasia, ~60% of cases) — medical therapy with a mineralocorticoid receptor antagonist. Surgery does NOT cure bilateral disease. Spironolactone (start 12.5-25 mg daily, titrate to 100-400 mg) is the most effective agent but causes gynaecomastia, impotence, and menstrual irregularity (it also blocks androgen receptors). Eplerenone (50-100 mg daily) is more selective (fewer sex-steroid side effects) but less potent and more expensive. Add amiloride for resistant hypokalaemia.
- Familial hyperaldosteronism — rare (type 1, glucocorticoid-remediable aldosteronism, is treated with dexamethasone; type 2 and 3 have other genetics). Screen young patients and those with a family history. [1]
Phaeochromocytoma
What it is
A catecholamine-secreting tumour from the adrenal medulla (phaeochromocytoma) or extra-adrenal paraganglia (paraganglioma). The clinical danger is from episodic or sustained catecholamine surges causing hypertensive crisis, arrhythmia, myocardial infarction, stroke, and multi-organ failure — all preventable with correct biochemical diagnosis and meticulous preoperative alpha-blockade. [1]
Clinical features — the triad that demands a test
The classic presentation is the triad of episodic headache, sweating, and palpitations with hypertension. The episodes last minutes to an hour and may be triggered by physical exertion, posture change, abdominal palpation, micturition (bladder paraganglioma), anaesthesia, certain foods/cheese (tyramine), or drugs (MAOIs, tricyclics, metoclopramide, contrast media). Other features: pallor (not flushing — pallor is from alpha-mediated vasoconstriction; flushing suggests carcinoid), anxiety and panic, tremor, nausea and vomiting, weight loss (hypermetabolic state), hyperglycaemia (catecholamine-induced insulin suppression), orthostatic hypotension (volume depletion), dilated cardiomyopathy (catecholamine-induced), and rarely hypertensive crisis with encephalopathy, MI, or stroke. [1]
The key teaching point: the triad of headache, sweating, and palpitations with hypertension has a very high positive predictive value for phaeochromocytoma. The triad is present in only a tiny fraction of hypertensive patients without phaeo, but in most with phaeo. Any patient with paroxysmal symptoms and hypertension must have metanephrines measured — do not attribute the symptoms to anxiety or panic until phaeo is excluded. [1]
Hypertension is paroxysmal in about 50% and sustained in 50%. Some patients are normotensive between episodes. Sustained hypertension that is resistant to multiple agents is another clue. [1]
Investigation — biochemistry first
Screening tests (choose based on sensitivity and pre-test probability): [1]
| Test | Sensitivity | Specificity | Notes |
|---|---|---|---|
| Plasma free metanephrines (metanephrine and normetanephrine) | Highest (96-100%) | Moderate (85-89%) | The most sensitive single test. Preferred in high-risk patients (known hereditary syndrome, adrenal incidentaloma with features, family history). Affected by: caffeine, smoking, severe illness, physical stress, tricyclic antidepressants, phenoxybenzamine, acetaminophen (interferes with the assay). Draw supine, resting, after an overnight fast. |
| 24-hour urine fractionated metanephrines (metanephrine and normetanephrine, plus catecholamines) | High (86-97%) | Highest (69-95%) | More specific than plasma; preferred in low-to-moderate-risk patients to reduce false positives. Requires a complete 24h collection, acidified. |
The 2014 Endocrine Society guideline (Lenders et al. PMID 24893135) recommends plasma free metanephrines OR 24h urine fractionated metanephrines as first-line, chosen by pre-test probability. Levels above 3-4 times the upper limit of normal are highly suggestive of phaeochromocytoma; mildly elevated levels require repeat testing and consideration of a clonidine suppression test (clonidine normally suppresses catecholamine release from normal adrenal medulla but not from a tumour). [1]
Localisation — only after biochemical confirmation:
- CT abdomen/pelvis (contrast-enhanced) — the first-line imaging. Identifies the adrenal mass (90% are adrenal). Non-contrast density helps characterise, but phaeochromocytomas are variable.
- MRI — preferred in pregnancy and in patients with contrast allergy; T2-hyperintense ("light bulb") appearance is suggestive.
- Functional imaging — MIBG (meta-iodobenzylguanidine) scintigraphy (I-123) for noradrenaline-producing tumours and to detect metastatic or multifocal disease; FDG-PET or Ga-68 DOTATATE PET for metastatic/SDH-related disease and when MIBG is negative.
- Never biopsy an adrenal mass before excluding phaeochromocytoma — biopsy can precipitate a fatal hypertensive crisis. [1]
Preoperative preparation — alpha first, always
The principle: alpha-blockade first, then beta-blockade, then volume expansion. This order is non-negotiable. The Lenders 2014 guideline is explicit. [1]
- Alpha-blockade first — phenoxybenzamine (irreversible, non-selective alpha-blocker) is the preferred agent. Start 10-14 days before surgery: 10 mg twice daily, titrate every 2-3 days to control blood pressure and heart rate, aiming for a supine blood pressure below 130/80 (seated below 90 systolic — but individualise), and evidence of alpha-blockade (nasal congestion, orthostatic hypotension, ejaculation dysfunction). Typical dose 40-80 mg daily, up to 1 mg/kg/day. Alternatively, doxazosin or prazosin (selective alpha-1 antagonists) — competitive and shorter-acting; easier to titrate but less complete blockade. Phenoxybenzamine is preferred because its irreversible blockade prevents intraoperative catecholamine surges more reliably.
- Beta-blockade — ONLY AFTER full alpha-blockade is established (typically 3-4 days after starting phenoxybenzamine), add a beta-blocker (propranolol, metoprolol, atenolol) to control the reflex tachycardia from alpha-blockade and any catecholamine-driven arrhythmia. NEVER give a beta-blocker before alpha-blockade — unopposed alpha stimulation causes severe vasoconstriction and hypertensive crisis. This is the most dangerous error in phaeo management.
- Volume expansion — high-salt diet and liberal oral fluids, or IV saline in the days before surgery, to correct the chronic volume depletion from catecholamine-induced vasoconstriction (which has been "unmasked" by alpha-blockade). This prevents profound postoperative hypotension (blood pressure drops when the catecholamine source is removed).
- Blood transfusion is NOT needed — but have cross-matched blood available for adrenalectomy. [1]
Target the preoperative state: blood pressure well controlled, no significant orthostatic hypotension (or mild), nasal congestion present (sign of adequate alpha-blockade), heart rate below 90, and no more than 1-2 premature ventricular contractions per minute. Surgery is performed by an experienced anaesthetic and surgical team in a high-volume centre. Laparoscopic adrenalectomy is standard for most adrenal phaeochromocytomas. [1]
The Rule of 10s — and its modern update
The traditional rule: 10% bilateral, 10% malignant, 10% extra-adrenal (paraganglioma), 10% familial. This is a useful mnemonic but underestimates the hereditary fraction. [1]
The modern update, driven by genetic testing: at least 30-40% of all phaeochromocytomas and paragangliomas carry a germline mutation, making this the most heritable of all tumour types. The hereditary syndromes: [1]
| Syndrome | Gene | Features |
|---|---|---|
| MEN2A and MEN2B | RET | Medullary thyroid cancer, hyperparathyroidism (2A), mucosal neuromas and marfanoid habitus (2B); phaeo is bilateral in 50% |
| Von Hippel-Lindau (VHL) | VHL | Retinal and CNS haemangioblastomas, renal cell carcinoma, pancreatic neuroendocrine tumours, epididymal cysts; noradrenaline-producing phaeo |
| Neurofibromatosis type 1 (NF1) | NF1 | Cafe-au-lait spots, neurofibromas, Lisch nodules; phaeo in 1-5% |
| Familial paraganglioma syndromes | SDHB, SDHC, SDHD (succinate dehydrogenase complex) | Extra-adrenal paragangliomas; SDHB carries a high risk of malignancy and metastasis |
All patients with phaeochromocytoma or paraganglioma should be offered genetic testing (the guideline recommendation), and undergo lifelong surveillance for recurrence (annual or biennial metanephrines) because of the risk of malignancy (especially SDHB) and metachronous tumours. [1]
The adrenal incidentaloma
What it is
An adrenal mass discovered incidentally on imaging performed for another reason (the definition excludes patients being staged for cancer, who are a separate group). Found in 4-7% of all abdominal CT scans, and the prevalence rises with age (above 7% in those over 70). The two questions the physician must answer for every incidentaloma: (1) is it functioning (secreting a hormone)? and (2) is it malignant? The answer to both determines management. [1]
The workup — function and malignancy, in parallel
1. Functional assessment — test every incidentaloma for three hormones: [1]
| Hormone | Test | Positive result |
|---|---|---|
| Cortisol (autonomous secretion) | 1 mg overnight dexamethasone suppression test | Cortisol above 50 nmol/L (1.8 mcg/dL) at 8-9 am post-dexamethasone. The 2023 ESE guideline (Fassnacht, PMID 37318239) uses the term mild autonomous cortisol secretion (MACS) for this finding (replacing "subclinical Cushing"), reflecting that these patients have morbidity (hypertension, diabetes, osteoporosis) even without overt Cushing signs. |
| Aldosterone (in hypertensive or hypokalaemic patients) | ARR with suppressed renin | ARR above 30 with suppressed renin (as for Conn syndrome) |
| Catecholamines (every patient, as phaeo can be asymptomatic or atypical) | Plasma free metanephrines or 24h urine fractionated metanephrines | Above the reference range (above 3-4x ULN is diagnostic of phaeo) |
2. Malignancy risk — imaging characteristics and size: [1]
| Feature | Benign (adenoma) | Suspicious (needs further workup or surgery) |
|---|---|---|
| Non-contrast CT Hounsfield units (HU) | At or below 10 HU (lipid-rich adenoma) | Above 20 HU (indeterminate 10-20) |
| Contrast washout (on contrast-enhanced CT) | Absolute washout above 60% at 15 minutes (lipid-poor adenoma) | Below 60% |
| Size | Below 4 cm (most adenomas are small) | Above 4 cm — higher risk of adrenocortical carcinoma (above 6 cm, risk approaches 25%) |
| Morphology | Smooth, homogeneous, round/oval | Irregular, heterogeneous, necrotic, calcified, invading adjacent structures |
| Growth | Stable on follow-up | Growth above 5 mm in 6 months |
The 2023 ESE guideline update: a homogeneous mass at or below 10 HU on non-contrast CT is considered benign regardless of size — the previous size criterion of below 4 cm for benignity has been removed, as the lipid content (HU) is more reliable than size. [1]
When to operate
- Functional tumour: overt Cushing syndrome (adrenalectomy), MACS with significant comorbidity (consider surgery), aldosteronoma (adrenalectomy after AVS confirms unilateral disease), phaeochromocytoma (adrenalectomy after alpha-blockade).
- Size above 4-6 cm or indeterminate imaging with suspicious features — adrenalectomy (the cancer risk justifies surgery).
- Growth above 5 mm in 6 months on follow-up imaging — adrenalectomy. [1]
When to follow up
For a non-operative incidentaloma: repeat imaging at 6-12 months (non-contrast CT or MRI), and annual biochemical screening (1 mg DST, ARR, metanephrines) for up to 4 years (Fassnacht 2023). After 4 years with no change, the risk of developing function or malignancy is very low. [1]
DWE trap: "A 55-year-old man has a 3 cm right adrenal mass on CT performed for renal colic. Non-contrast density is 8 HU. What is the next step?" The mass is benign by imaging (HU at or below 10). But you MUST still assess function — do a 1 mg DST, an ARR (if hypertensive), and plasma metanephrines. Do not dismiss an incidentaloma as "just an adenoma" without the biochemical workup. [1]
High-yield exam discriminators and common traps
| Scenario | The trap | The correct answer |
|---|---|---|
| Fatigue, hyperpigmentation, hyponatraemia, hyperkalaemia | Diagnosing SIADH or "dehydration" and missing Addison's | Check cortisol and ACTH; a morning cortisol below 200 nmol/L with high ACTH and a failed Synacthen test is primary adrenal insufficiency |
| Hypertensive patient on 4 drugs | Treating as "resistant essential hypertension" and never screening for Conn | Screen with the ARR; 5-10% of hypertension is primary aldosteronism, and much is curable |
| Episodic headache, sweating, palpitations with hypertension | Diagnosing anxiety or panic disorder | Measure plasma free metanephrines or 24h urine fractionated metanephrines; the triad demands a biochemical test |
| Patient with phaeo given beta-blocker for tachycardia | Giving beta-blockade first — unopposed alpha stimulation causes hypertensive crisis | Alpha-blockade (phenoxybenzamine) first, then beta-blockade, then volume expansion |
| Suspected Cushing, first test is a pituitary MRI | Imaging before confirming hypercortisolism biochemically | Confirm hypercortisolism (ONDST, 24h UFC, midnight salivary cortisol), then ACTH level, then localise |
| Patient on long-term prednisolone presents with sepsis | Not giving stress-dose steroids — HPA suppression means they cannot mount a cortisol response | Give hydrocortisone 100 mg IV (stress dose) along with sepsis management |
| Adrenal incidentaloma, 6 cm, 35 HU, heterogeneous | Following it up with "repeat CT in 6 months" | The features (large, high HU, heterogeneous) suggest adrenocortical carcinoma — refer for surgical resection |
| Patient with Addison's on hydrocortisone 20 mg and fludrocortisone 100 mcg | Not educating on sick-day rules and not providing an emergency injection kit | Adrenal crisis still occurs in educated patients (Hahner 2015) — education, MedicAlert, and emergency IM hydrocortisone are essential |
How this is tested
DWE MCQ: Expect vignettes on (1) the diagnostic strategy for Cushing (biochemical first, then ACTH level, then localise), (2) the Synacthen test interpretation, (3) the preoperative preparation of phaeochromocytoma (alpha first), (4) the sick-day rules and adrenal crisis management, (5) the ARR in resistant hypertension, (6) distinguishing primary from secondary adrenal insufficiency (hyperpigmentation, potassium, aldosterone), and (7) the incidentaloma workup. The answer is almost always the guideline-concordant step, not a rare zebronkey. [1]
DCE long case: The classic long case is the Cushing patient (integrated diagnostic and management plan, including the three-step investigation strategy and the transsphenoidal surgery decision) or the Addison patient presenting in crisis (emergency management, then chronic replacement and patient education). The examiner probes your understanding of the HPA axis, the steroid replacement regimens, and the comorbidity management. [1]
DCE short case: Face and hands. The Cushingoid patient (moon face, plethora, striae, buffalo hump — examine the proximal myopathy). The Addisonian patient (hyperpigmentation in palmar creases, buccal mucosa, scars; vitiligo; postural blood pressure). The key is to state the syndrome confidently from the signs, demonstrate the discriminating sign (proximal myopathy for Cushing, hyperpigmentation and postural drop for Addison), and offer the next investigation. [1]
Guidelines and regional anchoring
- Endocrine Society (US, primary): Bornstein 2016 (PMID 26760044) for primary adrenal insufficiency; Nieman 2008 (PMID 18334580) for Cushing diagnosis; Funder 2016 (PMID 26934393) for primary aldosteronism; Lenders 2014 (PMID 24893135) for phaeochromocytoma. The 2025 Endocrine Society primary aldosteronism update extends screening to all hypertensive patients.
- European Society of Endocrinology (ESE): Fassnacht 2023 (PMID 37318239) for adrenal incidentaloma, with the MACS definition and the HU criterion for benignity.
- ANZ primary: Endocrine Society of Australia guidance; the Australian Therapeutic Guidelines and local endocrine society protocols align with the Endocrine Society and ESE guidelines. Hydrocortisone and fludrocortisone are the standard ANZ replacement agents. Phenoxybenzamine is available and preferred for phaeo preparation.
- UK secondary: NICE guidance on hypertension (screening for secondary causes including primary aldosteronism), adrenal incidentaloma (NICE NG on adrenal tumours in development), and Addison's disease (NICE CKS). The Society for Endocrinology endorses the Endocrine Society guidelines.
- ANZ/UK/US deltas: The drug doses (hydrocortisone 15-25 mg, fludrocortisone 50-200 mcg, phenoxybenzamine 20-80 mg) are consistent across regions. The 1 mg DST cortisol threshold for MACS (50 nmol/L / 1.8 mcg/dL) and the HU threshold for benignity (10 HU) are internationally consistent per the ESE 2023 guideline. The ARR threshold varies slightly by assay (above 20-30 depending on units and method). The extension of ARR screening to all hypertensive patients is the 2025 Endocrine Society update and may not yet be reflected in all regional guidelines. [1]
References
- [1]Bornstein SR, Allolio B, Arlt W, et al. Diagnosis and Treatment of Primary Adrenal Insufficiency: An Endocrine Society Clinical Practice Guideline J Clin Endocrinol Metab, 2016.PMID 26760044
- [2]Nieman LK, Biller BMK, Findling JW, et al. The diagnosis of Cushing's syndrome: an Endocrine Society Clinical Practice Guideline J Clin Endocrinol Metab, 2008.PMID 18334580
- [3]Funder JW, Carey RM, Mantero F, et al. The Management of Primary Aldosteronism: Case Detection, Diagnosis, and Treatment: An Endocrine Society Clinical Practice Guideline J Clin Endocrinol Metab, 2016.PMID 26934393
- [4]Lenders JWM, Duh QY, Eisenhofer G, et al. Pheochromocytoma and paraganglioma: an endocrine society clinical practice guideline J Clin Endocrinol Metab, 2014.PMID 24893135
- [5]Fassnacht M, Tsagarakis S, Terzolo M, et al. European Society of Endocrinology clinical practice guidelines on the management of adrenal incidentalomas, in collaboration with the European Network for the Study of Adrenal Tumors Eur J Endocrinol, 2023.PMID 37318239
- [6]Hahner S, Spindler M, Fassnacht M, et al. High incidence of adrenal crisis in educated patients with chronic adrenal insufficiency: a prospective study J Clin Endocrinol Metab, 2015.PMID 25419882
- [7]Oldfield EH, Doppman JL, Nieman LK, et al. Petrosal sinus sampling with and without corticotropin-releasing hormone for the differential diagnosis of Cushing's syndrome N Engl J Med, 1991.PMID 1652686
- [8]Colao A, Petersenn S, Newell-Price J, et al. A 12-month phase 3 study of pasireotide in Cushing's disease N Engl J Med, 2012.PMID 22397653