Phys · endocrine
Pituitary Disease
Also known as pituitary adenoma · prolactinoma · macroprolactinoma · microprolactinoma · acromegaly · gigantism · Cushing disease · non-functioning pituitary adenoma · hypopituitarism · panhypopituitarism · pituitary apoplexy · Sheehan syndrome · craniopharyngioma · diabetes insipidus · central diabetes insipidus · growth hormone deficiency
Consultant-physician-depth guide to pituitary disease — anatomy and physiology, pituitary adenoma classification, prolactinoma, acromegaly, Cushing disease, non-functioning adenoma, hypopituitarism, pituitary apoplexy, craniopharyngioma, and diabetes insipidus — structured for FRACP DWE and DCE preparation.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Pituitary Disease
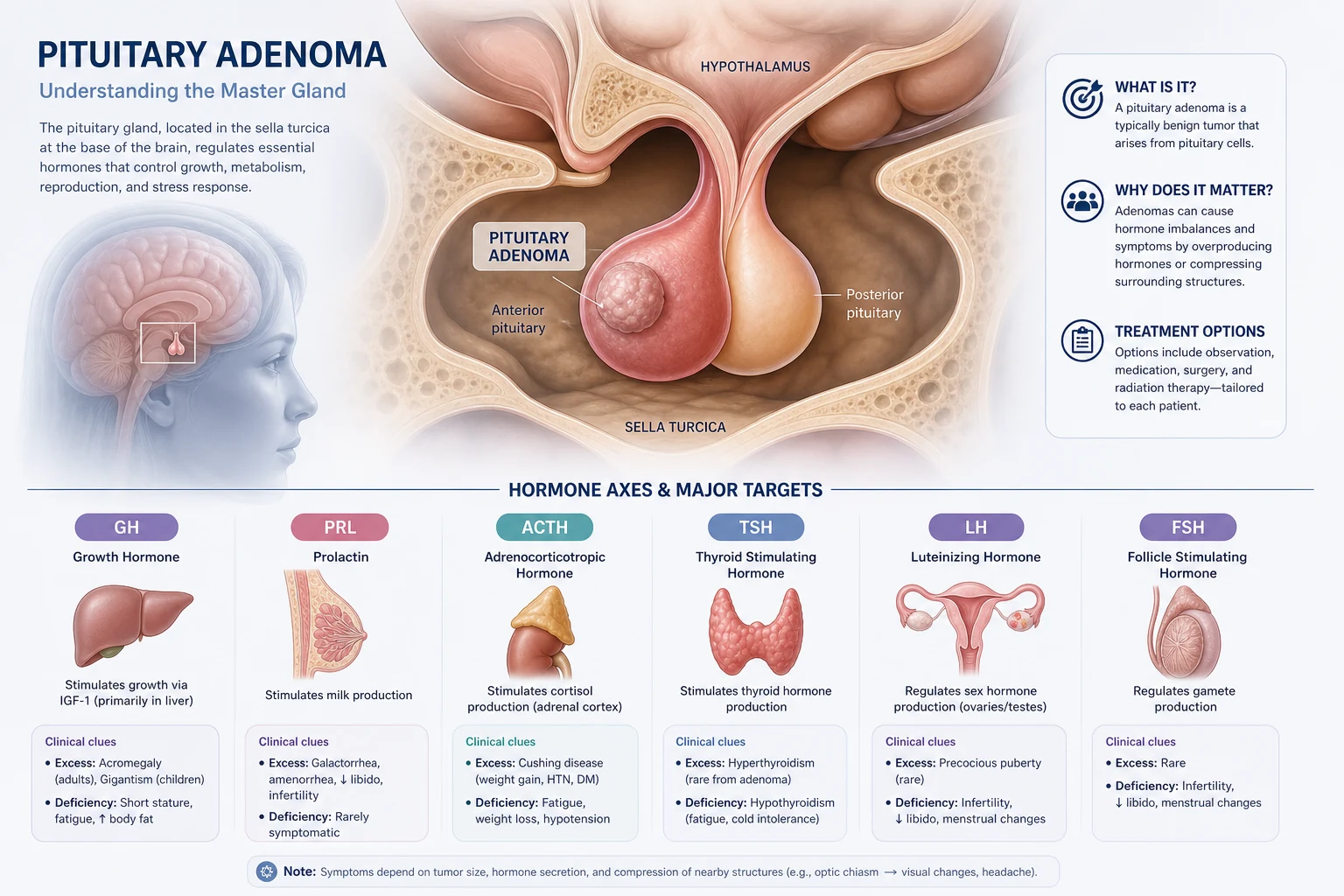
The answer first
The pituitary gland is the central regulator of the endocrine system. It sits in the sella turcica, immediately below the optic chiasm and lateral to the cavernous sinuses. This anatomy explains the three cardinal clinical syndromes of pituitary disease: hormone hypersecretion (a functioning adenoma), hormone hyposecretion (mass compression or destruction of normal gland), and mass effect (chiasm compression causing bitemporal hemianopia, or cavernous sinus invasion affecting cranial nerves III, IV, V1, V2, and VI). [1]
The single most important clinical rule: always replace cortisol before thyroid hormone. Giving levothyroxine to a patient with untreated ACTH deficiency accelerates cortisol metabolism and precipitates adrenal crisis. This is the most dangerous pituitary prescribing error and is tested in every exam format. [1]
Pituitary adenomas are classified by size — microadenoma (below 10 mm) and macroadenoma (10 mm or above) — and by function. The functional classification drives the entire investigation and management pathway. Prolactinoma is the most common functioning adenoma and is unique because medical therapy with a dopamine agonist is first-line — surgery is reserved for resistance or intolerance. All other functioning adenomas (acromegaly, Cushing disease, TSH-secreting) are primarily surgical. Non-functioning macroadenomas present with mass effect and require surgery when they compress the chiasm. [1]
Anatomy and physiology — why the mechanism drives management
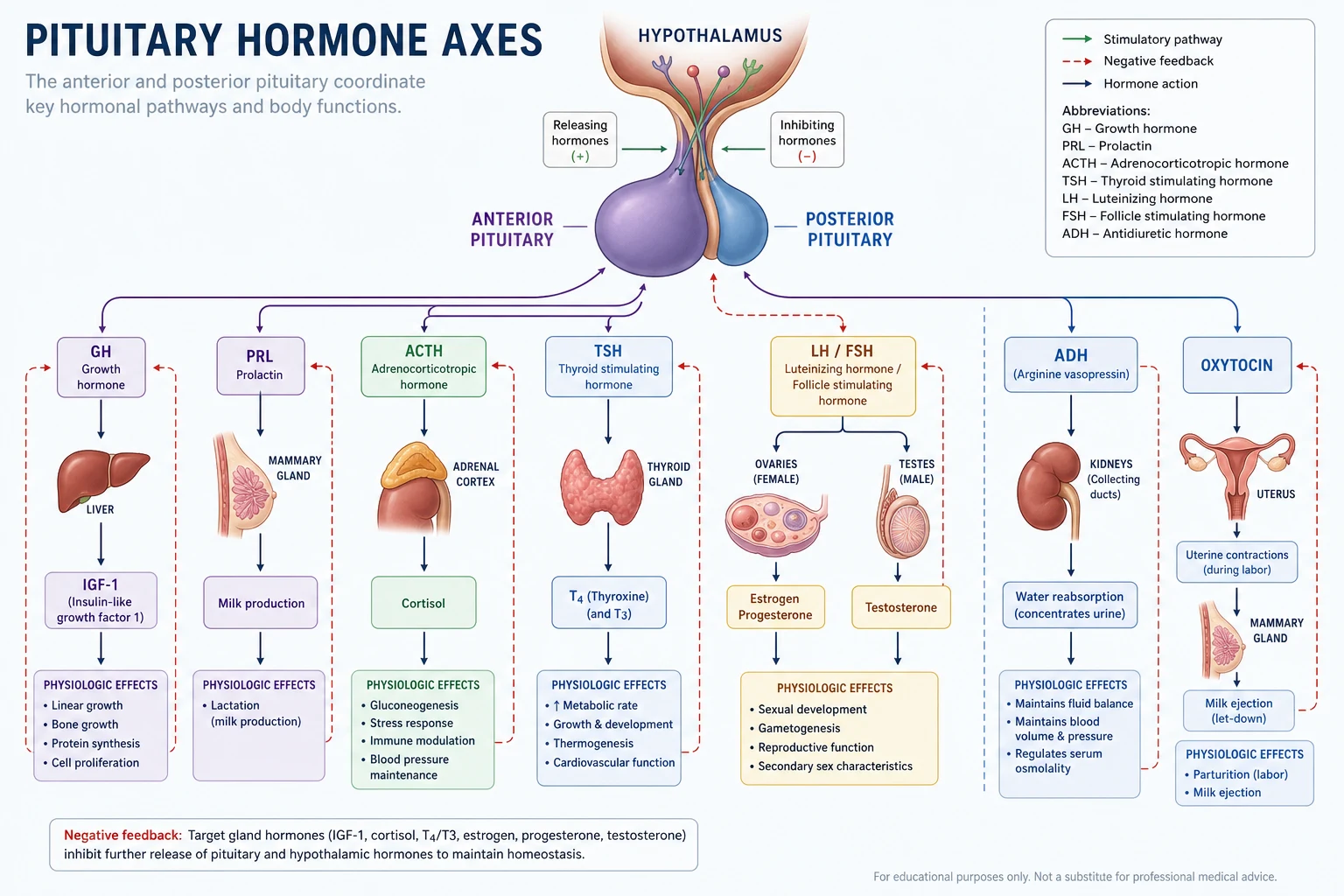
The pituitary gland has two embryologically and functionally distinct components. Understanding this distinction is the foundation for every pituitary question in the exam. [1]
The anterior pituitary (adenohypophysis) arises from Rathke's pouch (oral ectoderm). It accounts for approximately 80% of the gland. Its hormone-secreting cells are controlled by hypothalamic releasing and inhibiting factors delivered through the hypothalamic-pituitary portal venous system. The anterior pituitary secretes six hormones from five cell types: [1]
| Cell type | Hormone(s) | Hypothalamic regulator | Target |
|---|---|---|---|
| Somatotroph (50%) | Growth hormone (GH) | GHRH (stimulates), somatostatin (inhibits) | Liver (IGF-1), all tissues |
| Lactotroph (10 to 20%) | Prolactin (PRL) | Dopamine (inhibits — tonic) | Breast (lactation) |
| Corticotroph (15 to 20%) | ACTH (from POMC) | CRH and vasopressin (stimulate) | Adrenal cortex (cortisol) |
| Thyrotroph (5%) | Thyroid-stimulating hormone (TSH) | TRH (stimulates) | Thyroid (T3, T4) |
| Gonadotroph (10%) | Luteinising hormone (LH), follicle-stimulating hormone (FSH) | GnRH (stimulates, pulsatile) | Gonads (testosterone, oestradiol, gametogenesis) |
The critical physiology to know cold: prolactin is under tonic dopamine inhibition. Any lesion or drug that disrupts the hypothalamic-pituitary stalk ("stalk effect") removes dopaminergic inhibition and causes a moderate rise in prolactin — typically up to 2000 to 3000 mU/L. This is the basis for the stalk effect and must be distinguished from true prolactinoma (typically above 3000 to 4000 mU/L and proportional to tumour size). [1]
The posterior pituitary (neurohypophysis) arises from neural ectoderm. It is not a gland but a direct neural extension of the hypothalamus. Vasopressin (ADH) and oxytocin are synthesised in the supraoptic and paraventricular nuclei of the hypothalamus, transported down axons through the pituitary stalk, and stored in nerve terminals in the posterior pituitary. This is why posterior pituitary lesions do not cause hypopituitarism of anterior hormones — they are anatomically separate systems — and why pituitary surgery rarely causes permanent diabetes insipidus (the posterior pituitary and stalk are structurally distinct; transient DI is common from stalk manipulation, permanent DI is rare and suggests high stalk or hypothalamic injury). [1]
DWE trap: A common MCQ asks why a non-functioning macroadenoma causes a moderate prolactin rise. The answer is the stalk effect — compression of the pituitary stalk removes tonic dopamine inhibition. The prolactin is typically below 3000 mU/L and is disproportionate to the tumour size. A true macroprolactinoma causes prolactin above 4000 mU/L, usually proportional to tumour size (roughly 700 mU/L per mm of maximal tumour diameter). [1]
Pituitary adenoma classification
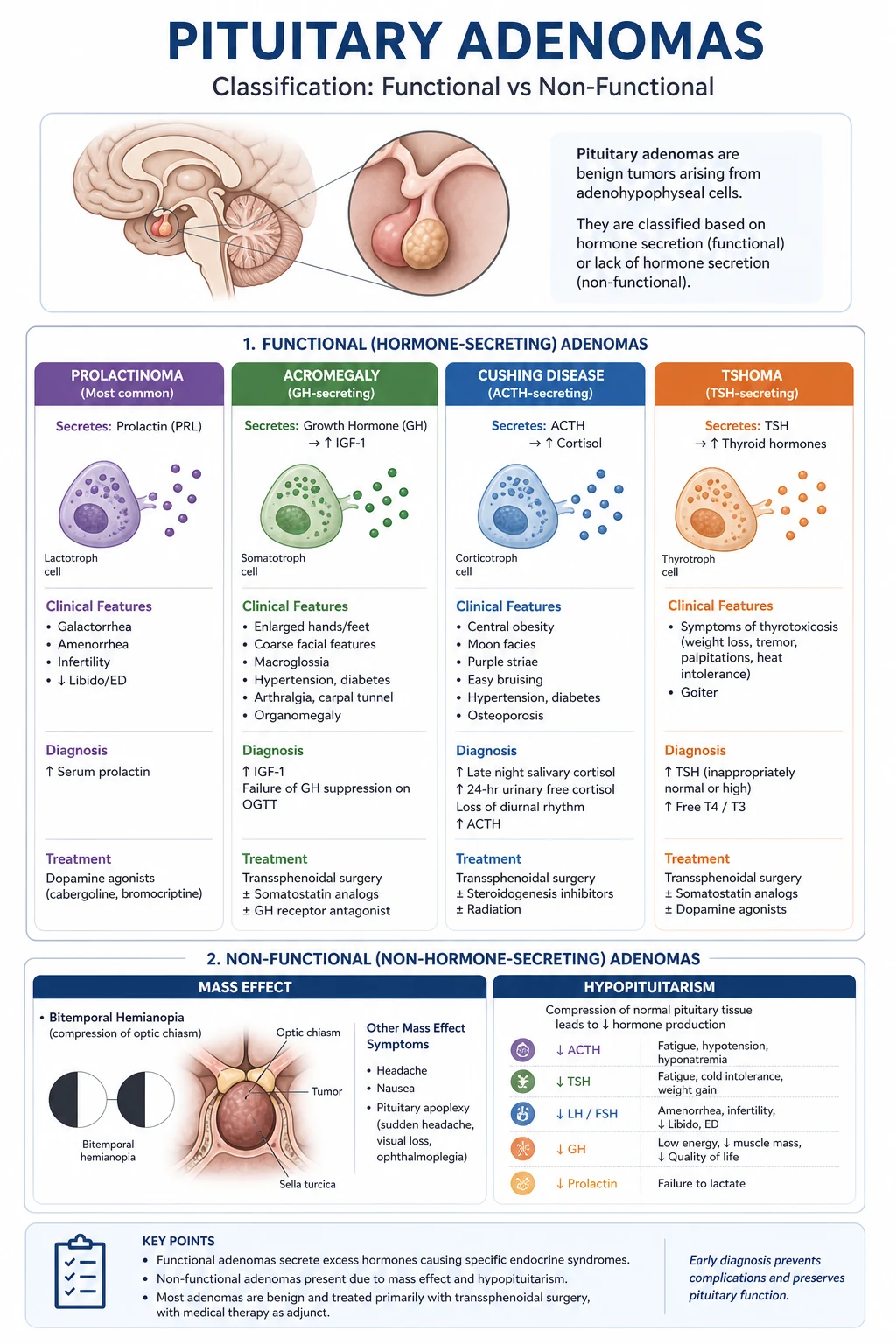
Pituitary adenomas are the most common cause of pituitary disease. They are classified along two axes — size and function — and both determine management. [1]
Size classification:
- Microadenoma: below 10 mm in greatest diameter. These are typically functioning (prolactinoma most common) and present with hormone excess, not mass effect.
- Macroadenoma: 10 mm or above. These may function or be non-functioning. Macroadenomas above 20 mm, or those with suprasellar extension, risk optic chiasm compression. Giant adenomas (above 40 mm) are aggressive and carry a poor prognosis. [1]
Functional classification — frequency and presentation: [1]
| Type | Frequency | Presenting syndrome | First-line treatment |
|---|---|---|---|
| Prolactinoma | ~40% (most common) | Galactorrhoea, amenorrhoea, infertility, low libido, erectile dysfunction | Medical — dopamine agonist |
| Non-functioning | ~30% | Mass effect (visual field defect, hypopituitarism, headache) | Surgery (transsphenoidal) |
| Acromegaly (GH) | ~10% | Acral enlargement, prognathism, visceromegaly, cardiomyopathy | Surgery first; somatostatin analogue, pegvisomant |
| Cushing disease (ACTH) | ~10% | Cushingoid features, central obesity, hypertension, diabetes | Surgery first (see adrenal topic for workup) |
| TSH-secreting | ~1% | Hyperthyroidism with inappropriately normal or high TSH | Surgery first; somatostatin analogue |
The practical rule: a prolactinoma is the only adenoma treated primarily with medication. All others are surgical first-line, with medical therapy as adjuvant or when surgery is not curative. The exception is the patient with a macroprolactinoma and visual field compromise — even here, cabergoline is first-line because it shrinks the tumour rapidly. Surgery is reserved for dopamine agonist resistance or intolerance, or for the rare patient with rapidly progressive visual loss despite medical therapy. [1]
Prolactinoma — the most common functioning adenoma
Prolactinoma is the most common functioning pituitary adenoma and the most common cause of pathological hyperprolactinaemia. It presents differently in men and women because of a detection bias: women present earlier with menstrual disturbance and galactorrhoea, while men present later with macroadenomas and mass effect (visual loss, headache, hypogonadism). [1]
Clinical features of hyperprolactinaemia:
- Women: galactorrhoea, oligomenorrhoea or amenorrhoea, infertility, low libido, vaginal dryness, osteopenia from chronic hypogonadism
- Men: low libido, erectile dysfunction, infertility (reduced spermatogenesis), gynaecomastia (uncommon — prolactin does not directly cause gynaecomastia but coexists with hypogonadism), galactorrhoea (rare)
- Macroadenoma: headache, visual field defect (bitemporal hemianopia), cranial nerve palsies (cavernous sinus invasion), hypopituitarism from stalk compression [1]
The prolactin level matters. A single serum prolactin is diagnostic — dynamic testing is not needed. Interpret by the level:
- Mild elevation (below 2000 mU/L): often the stalk effect from a non-prolactinoma, pregnancy, lactation, stress, or a drug. Check the medication list first.
- Moderate elevation (2000 to 4000 mU/L): may be a microprolactinoma or stalk effect. Image with MRI.
- Marked elevation (above 4000 mU/L): almost certainly a prolactinoma. The level is proportional to tumour size — roughly 700 mU/L per mm of maximal diameter. A macroadenoma with a prolactin of 200 mU/L is NOT a prolactinoma — it is a non-functioning adenoma with stalk effect, and surgery is the treatment, not a dopamine agonist. [1]
The differential of hyperprolactinaemia — always exclude the reversible causes first: [1]
| Cause | Mechanism | Typical PRL |
|---|---|---|
| Pregnancy and lactation | Physiological | Variable, may be very high |
| Drugs (antipsychotics, metoclopramide, domperidone, methyldopa, opiates, verapamil) | Dopamine D2 receptor blockade or depletion | Up to 3000 mU/L (risperidone can exceed) |
| Stress, exercise, sleep | Transient | Mild, below 1500 mU/L |
| Stalk effect (non-functioning adenoma, craniopharyngioma) | Loss of tonic dopamine inhibition | 1000 to 3000 mU/L |
| Hypothyroidism (primary) | TRH stimulates prolactin | Mild, below 2000 mU/L |
| Renal failure, hepatic failure | Reduced clearance | Mild to moderate |
| Macroprolactin (biologically inactive complex) | Assay interference | Any level — always check if asymptomatic |
| Prolactinoma | Autonomous secretion | Above 4000 mU/L, proportional to size |
Macroprolactin is a biologically inactive complex of prolactin with IgG that is detected by immunoassays but does not bind to prolactin receptors. It causes an apparent hyperprolactinaemia in an asymptomatic patient with normal menstrual function. The diagnosis is made by polyethylene glycol (PEG) precipitation — the laboratory reports the monomeric (active) prolactin after removing the macroprolactin complex. Always request PEG precipitation when the prolactin is elevated but the patient has no symptoms of hyperprolactinaemia and normal gonadal function. The key trap: treating macroprolactin with a dopamine agonist is wrong and exposes the patient to unnecessary medication. [1]
DWE high-yield: "A 45-year-old woman on risperidone for bipolar disorder has a prolactin of 2800 mU/L with galactorrhoea." The medication is the cause. Switch to aripiprazole (a partial dopamine agonist with low hyperprolactinaemia risk) in consultation with psychiatry, and re-check. Do not rush to MRI or cabergoline unless the level persists or is disproportionate to the drug. [1]
Management of prolactinoma: [1]
Dopamine agonists are first-line for all prolactinomas, including macroadenomas with visual compromise — unlike any other pituitary adenoma. The Endocrine Society hyperprolactinaemia guideline (Melmed 2011, PMID 21296991) recommends cabergoline over bromocriptine based on superior efficacy, tolerability, and tumour shrinkage. [1]
Cabergoline — a selective D2 receptor agonist — is first-line. Start at 0.5 mg once or twice weekly and titrate to achieve normoprolactinaemia (typical maintenance 0.5 to 2.0 mg weekly, occasionally higher for macroadenomas). It normalises prolactin in 80 to 90% of microadenomas and shrinks macroadenomas in 70 to 80% within weeks to months. Visual field defects improve in most patients with macroprolactinoma within days to weeks of starting cabergoline. [1]
Bromocriptine is an alternative (2.5 to 15 mg daily, divided). It is less well tolerated (nausea, orthostatic hypotension, nasal congestion) and less effective than cabergoline, but has the largest pregnancy safety database. It is preferred by some centres when pregnancy is planned, though cabergoline is increasingly used pre-conception and stopped at confirmation of pregnancy. [1]
Cardiac valve surveillance: Cabergoline at the high doses used for Parkinson disease (above 3 mg daily) is associated with cardiac valve regurgitation. The doses used for prolactinoma (below 2 mg weekly) are far lower and the risk is very low. The Endocrine Society recommends echocardiographic monitoring only for patients on high cumulative doses or those on doses above 2 mg weekly for prolonged periods. [1]
When is surgery indicated?
- Dopamine agonist resistance (failure to normalise prolactin or shrink tumour despite escalating doses)
- Dopamine agonist intolerance (severe nausea, hypotension, psychiatric effects, impulse control disorders)
- Tumour growth despite medical therapy with visual compromise
- Cerebrospinal fluid rhinorrhoea (tumour shrinkage unmasked a skull base defect)
- Patient preference or non-adherence to long-term medical therapy [1]
Pregnancy and prolactinoma: Stop cabergoline at confirmation of pregnancy. Oestrogen-driven pituitary hyperplasia may enlarge the tumour. For microadenoma, the risk of symptomatic enlargement is below 2%. For macroadenoma, the risk is 15 to 35% (higher if the tumour was previously invasive or abutted the chiasm). For macroprolactinoma, continue bromocriptine or cabergoline through pregnancy if the tumour was previously invasive or close to the chiasm, and monitor with visual fields each trimester. [1]
Acromegaly — growth hormone excess
Acromegaly is caused by chronic GH hypersecretion, almost always from a somatotroph adenoma of the anterior pituitary. The clinical features are insidious and the diagnosis is delayed by an average of 7 to 10 years, by which time the patient has sustained irreversible cardiovascular, respiratory, and metabolic damage. Excess mortality in acromegaly (standardised mortality ratio approximately 1.7) is driven by cardiovascular disease, respiratory failure, and cancer — and is reduced to population norms with biochemical control. [1]
Clinical features — the acromegalic phenotype: [1]
The features are due to GH and IGF-1 excess acting on somatic tissues. Recognise them on inspection — the DCE short case is built around these signs. [1]
| System | Features |
|---|---|
| Acral (extremities) | Enlarged hands and feet (patient notices increasing shoe, glove, ring size), frontal bossing, macrognathia with dental malocclusion, interdental spaces, prognathism, large nose, oily skin, skin tags |
| Cardiovascular | Cardiomyopathy (concentric left ventricular hypertrophy, diastolic then systolic dysfunction), hypertension, arrhythmia, valvular regurgitation, premature coronary disease |
| Respiratory | Obstructive sleep apnoea (from macroglossia and pharyngeal soft tissue thickening) — screen all patients, mandatory pre-operatively |
| Metabolic | Impaired glucose tolerance or type 2 diabetes (GH is anti-insulin), hypertriglyceridaemia, hypercalciuria |
| Musculoskeletal | Carpal tunnel syndrome (bilateral, from soft tissue oedema), arthropathy (degenerative, large and small joints), proximal myopathy |
| Endocrine | Headache, hyperhidrosis, hypogonadism (co-secretion or stalk compression), goitre, colonic polyps and increased colon cancer risk |
| Local (tumour) | Visual field defect, cranial nerve palsies, hypopituitarism |
Screening and diagnosis: [1]
The Endocrine Society acromegaly guideline (Katznelson 2014, PMID 25356808) recommends a structured biochemical approach. Do not rely on random GH — it is pulsatile and varies minute to minute. [1]
Step 1 — Serum IGF-1 (screening). IGF-1 is the integrative marker of GH action — it is stable, reflects the average GH exposure over days to weeks, and is age- and sex-matched. A normal age- and sex-matched IGF-1 excludes acromegaly. An elevated or equivocal IGF-1 requires confirmation. [1]
Step 2 — Oral glucose tolerance test (OGTT) with GH nadir (confirmation). Give 75 g oral glucose and measure GH at 0, 30, 60, 90, and 120 minutes. In health, glucose suppresses GH to a nadir below 1.0 microgram per litre (below 0.4 with modern sensitive assays). Failure of GH to suppress below 1.0 microgram per litre after glucose confirms acromegaly. The tumour escapes glucose suppression because somatotroph adenomas are dysregulated. [1]
Step 3 — MRI pituitary. Once biochemically confirmed, image the pituitary to localise the adenoma. Almost all acromegaly is pituitary. Rare ectopic causes (GHRH-secreting pancreatic or bronchial neuroendocrine tumour) are considered when the pituitary MRI is normal and IGF-1 and GH are very high. [1]
DWE trap: "A 48-year-old man with acral enlargement has IGF-1 twice the upper limit of normal and a GH nadir of 0.6 microgram per litre on OGTT." If the assay is the modern sensitive type (threshold 0.4), this is acromegaly. If it is the older standard assay (threshold 1.0), this is not. The assay matters — examiners test whether you know the assay-dependent threshold. [1]
Management of acromegaly — surgery first, medical therapy adjuvant: [1]
Transsphenoidal surgery is first-line for all patients with acromegaly. An experienced neurosurgeon achieves biochemical remission in 75 to 95% of microadenomas and 40 to 70% of macroadenomas. Cure is defined as a random GH below 1.0 microgram per litre, an OGTT GH nadir below 0.4 (modern assay), and a normal age-matched IGF-1. Failure to achieve cure, or recurrence, triggers medical therapy. [1]
Somatostatin analogues (SSA) are first-line medical therapy. They bind somatostatin receptor subtype 2 (sst2) on somatotroph adenomas and suppress GH secretion and tumour growth. First-generation agents are octreotide LAR (20 to 30 mg intramuscularly every 4 weeks) and lanreotide autogel (90 to 120 mg deep subcutaneously every 4 weeks). They normalise IGF-1 in approximately 50 to 60% of patients and shrink tumour in 40 to 60%. [1]
Pasireotide is a second-generation SSA with broader receptor affinity (sst1, 2, 3, and 5). The PAOLA trial (Colao 2014, PMID 25260838) demonstrated that pasireotide LAR achieves superior biochemical control compared to octreotide in patients inadequately controlled on first-generation SSA. The trade-off is a higher rate of hyperglycaemia — pasireotide inhibits insulin and incretin secretion, causing new or worsened diabetes in 50 to 60%. [1]
Pegvisomant is a genetically engineered GH receptor antagonist — it blocks GH action at the receptor rather than reducing GH secretion. The Trainer trial (PMID 10770982) showed pegvisomant normalises IGF-1 in 89% of patients at the 20 mg daily dose. Because it blocks the receptor, serum GH rises — this is expected and is not tumour growth. Pegvisomant is used as monotherapy or in combination with an SSA, particularly for patients resistant to SSA. Monitor liver function (transaminitis) — the main adverse effect. [1]
Dopamine agonists (cabergoline) have modest efficacy in acromegaly, normalising IGF-1 in approximately 10 to 30%. They are useful as combination therapy with an SSA, especially when co-prolactin is elevated, or for mild residual disease. They are not first-line monotherapy. [1]
Radiotherapy (stereotactic radiosurgery or conventional fractionated) is reserved for patients not controlled by surgery and medical therapy, or who cannot tolerate medical therapy. It is slow-acting (GH and IGF-1 fall over years), carries a risk of hypopituitarism (50 to 60% at 5 to 10 years), and may affect cognitive function. It is third-line. [1]
Surveillance after treatment:
- Colonoscopy at diagnosis — acromegaly increases colonic polyp and cancer risk. Repeat at 3 to 5 years.
- Echocardiogram — screen for cardiomyopathy and valvular disease at baseline and periodically.
- Sleep study — screen for obstructive sleep apnoea; it affects up to 70% of patients and is an independent cause of mortality.
- Visual fields — if macroadenoma with suprasellar extension.
- Pituitary function — assess anterior pituitary axes at baseline and after treatment (surgery, radiotherapy, SSA may cause hypopituitarism).
- Biochemical control — monitor IGF-1 every 3 to 6 months. Aim for normal IGF-1 and GH below 1.0 microgram per litre. Biochemical control restores mortality to population norms. [1]
Cushing disease — pituitary ACTH excess
Cushing disease is ACTH-dependent Cushing syndrome caused by a corticotroph adenoma of the pituitary. It accounts for approximately 70% of endogenous Cushing syndrome in adults (the remainder is adrenal adenoma/carcinoma and ectopic ACTH). The full diagnostic workup for Cushing syndrome is covered in the adrenal topic; here we focus on the pituitary-specific aspects. [1]
The diagnostic pathway for Cushing disease:
- Confirm endogenous hypercortisolism — at least two of: elevated 24-hour urine free cortisol, non-suppression on 1 mg overnight dexamethasone suppression test (cortisol above 50 nmol/L at 8 AM), elevated late-night salivary cortisol. This is per the Endocrine Society Cushing guideline (Nieman 2008, PMID 18334580). [1]2. Determine ACTH dependence — measure plasma ACTH. If elevated or inappropriately normal, the hypercortisolism is ACTH-dependent. If suppressed, it is adrenal.
- Distinguish pituitary from ectopic ACTH — high-dose dexamethasone suppression test (8 mg overnight or 2 mg every 6 hours for 48 hours): cortisol suppression by more than 50% suggests pituitary Cushing disease (corticotroph adenomas retain partial glucocorticoid feedback). Failure to suppress suggests ectopic ACTH. This test has imperfect sensitivity and specificity and is now supplemented by inferior petrosal sinus sampling (IPSS) — the gold standard for confirming pituitary ACTH source. IPSS with CRH stimulation measures central-to-peripheral ACTH gradient; a ratio above 2 at baseline or above 3 after CRH confirms pituitary Cushing disease.
- MRI pituitary — identify the corticotroph adenoma. Up to 40% are not visible on MRI (microadenomas below 2 to 3 mm). [1]
Management of Cushing disease:
- Transsphenoidal surgery is first-line. An experienced surgeon achieves remission in 70 to 90% of microadenomas. Immediate post-operative cortisol should be low (the suppressed normal corticotrophs and adrenal axis take months to recover) — persistently high cortisol indicates persistent disease.
- Second-line for surgical failure or recurrence: repeat surgery, radiotherapy, or bilateral adrenalectomy (for refractory disease — carries a risk of Nelson syndrome, pituitary corticotroph tumour growth from loss of cortisol feedback).
- Medical therapy to control cortisol while awaiting definitive treatment: steroidogenesis inhibitors (metyrapone, ketoconazole, mitotane, osilodrostat), mifepristone (glucocorticoid receptor antagonist for hyperglycaemia). [1]
Non-functioning pituitary adenoma — mass effect
Non-functioning adenomas do not secrete clinically significant hormone excess. They are the second most common pituitary adenoma after prolactinoma and typically present as macroadenomas with mass effect. [1]
Clinical presentation:
- Visual field defect — the classic feature. Suprasellar extension compresses the central decussating fibres of the optic chiasm, causing a bitemporal hemianopia (often superior first, as the inferior chiasmal fibres are more posterior). This is a short-case examination staple.
- Headache — from stretching of the diaphragma sellae or cavernous sinus invasion.
- Hypopituitarism — progressive loss of anterior pituitary axes from compression. The order of loss is characteristic: GH first, then LH/FSH, then TSH, then ACTH (prolactin may rise from stalk effect). This order reflects the spatial arrangement of cell types and their sensitivity to compression.
- Cranial nerve palsies — cavernous sinus invasion affects cranial nerves III, IV, V1, V2, and VI. CN III palsy (ptosis, diplopia, impaired eye movement) is most common.
- Apoplexy — sudden haemorrhage into the adenoma (see below). [1]
Investigation of a pituitary incidentaloma or macroadenoma:
- Full anterior pituitary panel — 9 AM cortisol, ACTH, TSH, free T4, LH, FSH, oestradiol or testosterone, prolactin, IGF-1. The prolactin must always be checked before surgery — a macroprolactinoma is treated medically, not surgically.
- Visual fields — formal perimetry (Humphrey) for any lesion abutting the chiasm.
- MRI pituitary with contrast — define size, invasion (cavernous sinus, sphenoid sinus), and relationship to the chiasm.
- If Cushing suspected — 24-hour urine free cortisol, 1 mg overnight dexamethasone suppression, late-night salivary cortisol.
- If acromegaly suspected — IGF-1 (and OGTT GH if elevated). [1]
Management:
- Surgery (transsphenoidal) for macroadenomas with visual compromise, growth on surveillance, significant hypopituitarism, or apoplexy.
- Conservative surveillance for microincidentalomas without mass effect or hormone excess — repeat MRI at 1 year, then every 2 years if stable. Visual fields annually if near the chiasm.
- There is no effective medical therapy for non-functioning adenoma — dopamine agonists and somatostatin analogues have minimal shrinkage effect. [1]
Hypopituitarism — hormone replacement and the cortisol-first rule
Hypopituitarism is deficiency of one or more anterior pituitary hormones. It may be caused by a pituitary lesion, pituitary surgery or radiotherapy, infarction (apoplexy, Sheehan), or infiltrative disease. Panhypopituitarism refers to deficiency of all anterior pituitary hormones. [1]
Causes of hypopituitarism: [1]
| Mechanism | Examples |
|---|---|
| Mass lesion | Pituitary adenoma, craniopharyngioma, Rathke cleft cyst, metastasis (breast, lung), meningioma |
| Vascular | Pituitary apoplexy, Sheehan syndrome (postpartum pituitary infarction from hypovolaemic shock — the pituitary is enlarged and vulnerable in pregnancy), sickle cell |
| Iatrogenic | Pituitary surgery, cranial radiotherapy (delayed, years later) |
| Infiltrative / inflammatory | Lymphocytic hypophysitis (autoimmune, often in pregnancy or post-partum, or with ipilimumab and other checkpoint inhibitors), sarcoidosis, haemochromatosis, Langerhans cell histiocytosis, IgG4 disease |
| Infectious | Tuberculosis, syphilis, fungal |
| Genetic | PROP1, POU1F1 mutations (combined pituitary hormone deficiency) |
| Developmental | Septo-optic dysplasia, congenital hypopituitarism |
| Functional | Severe chronic illness, severe weight loss (but always exclude structural disease) |
The cortisol-first rule — never break it: [1]
When replacing pituitary hormones, always replace cortisol first, before thyroid hormone. Giving levothyroxine to a patient with untreated ACTH deficiency accelerates cortisol clearance by increasing hepatic 11-beta-hydroxysteroid dehydrogenase type 2 activity, precipitating acute adrenal crisis. This is the single most dangerous pituitary prescribing error. [1]
Hormone replacement regimen for hypopituitarism: [1]
| Axis | Replacement | Notes |
|---|---|---|
| ACTH (cortisol) | Hydrocortisone 15 to 25 mg daily in divided doses (e.g., 10 mg AM, 5 mg at noon, 5 mg at 4 PM) | Replace first. Patient education for sick day rules (double dose for illness, emergency IM hydrocortisone injection, MedicAlert). Avoid over-replacement — it contributes to the excess mortality and osteoporosis. Prednisone 3 to 5 mg daily is an alternative with a longer duration of action. |
| LH/FSH (gonadal) | Men: testosterone (gel daily or depot injection every 10 to 14 weeks). Women: oestradiol/progestogen replacement (HRT or oral contraceptive until average menopause age ~50) | Replace after cortisol and thyroid. Monitor testosterone or oestradiol. Fertility requires gonadotropin therapy (hCG, FSH) — refer to reproductive endocrinology. |
| GH | Somatropin (recombinant human GH) 0.2 to 0.5 mg subcutaneously daily, titrate to IGF-1 | Reserved for confirmed GH deficiency with impaired quality of life, after other axes replaced. Improves body composition, lipids, bone density, and quality of life. Contraindicated in active malignancy. |
| ADH (posterior) | Desmopressin 100 to 200 microgram orally two to three times daily, or 10 to 40 microgram intranasally daily | Treat diabetes insipidus (see below). |
Mortality in hypopituitarism: The West Midlands Prospective Hypopituitarism Study (Tomlinson 2001, PMID 11273062) demonstrated a standardised mortality ratio of 1.87 in patients with hypopituitarism, driven by cardiovascular, respiratory, and cerebrovascular causes. The meta-analysis by Pappachan (PMID 25658016) confirmed an overall weighted SMR of 1.99. Key modifiable contributors include over-replacement of glucocorticoids, untreated GH deficiency, and undertreated gonadal failure. A key insight from Murad (PMID 27817141): higher hydrocortisone replacement doses are associated with increased mortality, supporting a lower-dose hydrocortisone strategy (below 25 mg daily). [1]
Pituitary apoplexy — the endocrine emergency
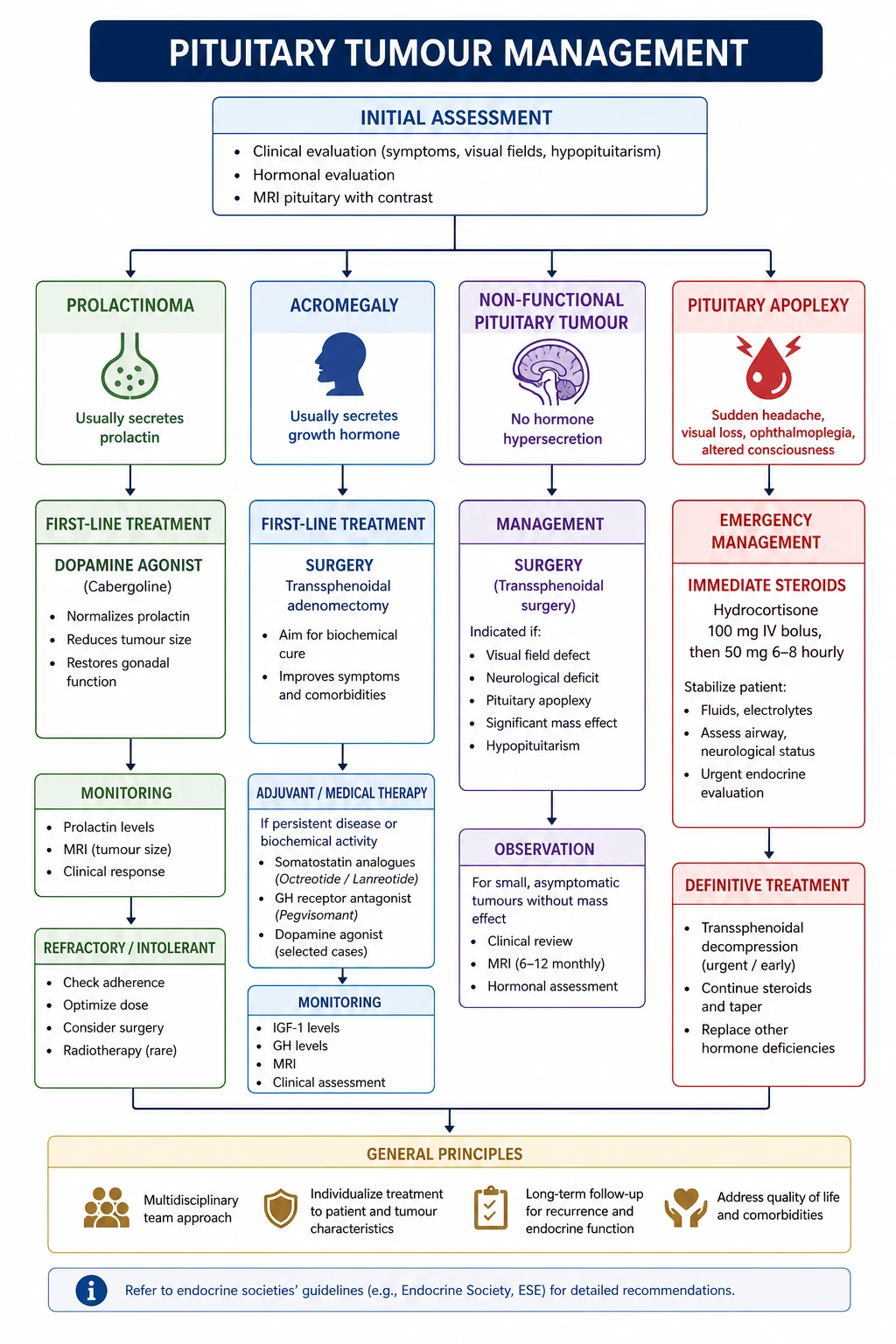
Pituitary apoplexy is acute haemorrhage or infarction of a pituitary adenoma. It is a medical and potentially surgical emergency that every physician must recognise. The UK guidelines for pituitary apoplexy (Rajasekaran 2011, PMID 21044119) are the standard reference. [1]
Clinical features — the classic presentation:
- Sudden severe headache (often thunderclap, may mimic subarachnoid haemorrhage)
- Nausea and vomiting
- Visual loss — sudden reduction in visual acuity or new visual field defect from chiasm or optic nerve compression
- Ophthalmoplegia — cranial nerve III, IV, or VI palsy from cavernous sinus compression or involvement
- Altered consciousness — from acute adrenal crisis (ACTH deficiency) or hypothalamic compression
- Meningism — blood or necrotic tissue in the CSF may cause neck stiffness [1]
Precipitants: Apoplexy may be spontaneous but is associated with anticoagulation, pregnancy (Sheehan overlap), bromocriptine initiation, head trauma, general anaesthesia, hypertension, and pituitary irradiation. [1]
Immediate management — the cortisol-first principle saves lives: [1]
- Assess and resuscitate — airway, breathing, circulation. The patient may be hypotensive from acute ACTH deficiency.
- Empirical high-dose hydrocortisone immediately — hydrocortisone 100 to 200 mg intravenously, followed by 50 to 100 mg every 6 hours intramuscularly or 2 to 4 mg per hour intravenous infusion. This treats presumptive adrenal insufficiency and reduces vasogenic oedema. Do NOT wait for a cortisol result. [1]3. Blood tests — cortisol, ACTH, full pituitary panel, electrolytes (hyponatraemia from cortisol and/or ADH deficiency), glucose.
- MRI pituitary (urgent) — the diagnostic study. CT may show a sellar mass but MRI is more sensitive for haemorrhage and infarction.
- Visual fields and acuity — formal ophthalmology assessment within 24 hours. This is the key determinant of surgical decision-making.
- Fluid and electrolyte management — the patient may be hyponatraemic from cortisol deficiency, or hypernatraemic from developing diabetes insipidus.
- Neurosurgical and endocrine referral — to a specialist pituitary centre. [1]
Surgery versus conservative management: [1]
The UK guidelines recommend surgical decompression (transsphenoidal) within 1 to 2 weeks for patients with:
- Severely reduced visual acuity
- Severe or worsening visual field defects
- Deteriorating level of consciousness [1]
Conservative management is appropriate for patients with no visual compromise, or with pre-existing non-functioning adenoma and stable visual fields. Both approaches require ongoing endocrine follow-up — many patients develop permanent hypopituitarism. The decision is made in a multidisciplinary team with neurosurgery and endocrinology. [1]
DCE high-yield: "A 60-year-old man presents with sudden severe headache, left eye ptosis, and a bitemporal hemianopia. Blood pressure is 90/60. What is the immediate management?" The answer is emergency intravenous hydrocortisone. Do not order the MRI first — treat presumptive adrenal crisis. The cortisol-first rule applies even more urgently here than in chronic hypopituitarism. [1]
Craniopharyngioma
Craniopharyngioma is a benign epithelial tumour arising from remnants of Rathke's pouch along the pituitary stalk. It is the most common nonglial brain tumour in children but presents in a bimodal distribution — a childhood peak (5 to 15 years) and an adult peak (50 to 70 years). There are two histological types: adamantinomatous (cystic, calcified, childhood) and papillary (solid, adult). [1]
Clinical presentation:
- Mass effect — headache, visual field defect (chiasm compression from above, unlike pituitary adenoma which compresses from below), hydrocephalus (from third ventricle obstruction)
- Hypopituitarism — GH deficiency (growth failure in children is often the presenting feature), delayed puberty, hypothyroidism, adrenal insufficiency
- Diabetes insipidus — common and often early, because the tumour arises in or near the stalk and hypothalamus. DI at presentation distinguishes craniopharyngioma from pituitary adenoma, which rarely causes DI at diagnosis.
- Hypothalamic dysfunction — hyperphagia and obesity (from hypothalamic damage), temperature dysregulation, sleep disturbance, behavioural change [1]
Investigation:
- MRI pituitary and brain — shows a cystic suprasellar mass, often with calcification (adamantinomatous type). The cystic fluid is classically "machine oil" at surgery.
- Full pituitary panel and visual fields.
- DI is assessed clinically and biochemically (water deprivation test or copeptin — see below). [1]
Management:
- Surgery is the mainstay — the goal is maximal safe resection. Total resection is ideal but carries a high risk of hypothalamic injury (causing severe obesity, DI, cognitive impairment). Increasingly, limited surgery followed by radiotherapy is preferred to protect the hypothalamus.
- Radiotherapy — reduces recurrence. Proton beam therapy is increasingly used to spare surrounding structures.
- Intracystic therapy — for cystic craniopharyngioma, intracystic interferon-alpha or bleomycin may shrink the cyst and defer surgery.
- Hormone replacement — most patients require lifelong multiple-axis replacement, including desmopressin for permanent DI. Replace cortisol first. [1]
Prognosis: The 10-year survival is 80 to 95%, but recurrence is common and quality of life is often impaired from hypothalamic damage, visual deficits, and panhypopituitarism. [1]
Diabetes insipidus — central versus nephrogenic
Diabetes insipidus is a disorder of water homeostasis causing polyuria (typically above 3 litres per day of dilute urine) and polydipsia. It is caused by either deficient vasopressin (central DI) or renal resistance to vasopressin (nephrogenic DI). It must be distinguished from primary polydipsia (compulsive water drinking), which causes a similar clinical picture by a different mechanism. [1]
Central DI (arginine vasopressin deficiency) is caused by damage to the hypothalamic vasopressin neurons or the pituitary stalk. Causes include pituitary surgery (transient or permanent), trauma, tumours (craniopharyngioma, metastasis, germinoma), infiltrative disease (Langerhans cell histiocytosis, sarcoidosis, neurosarcoidosis), autoimmune hypophysitis, and idiopathic. The 2018 nomenclature change from "central DI" to "arginine vasopressin deficiency" reflects the pathophysiology — the posterior pituitary is a storage site, and the defect is in the hypothalamic neurons. [1]
Nephrogenic DI (arginine vasopressin resistance) is caused by renal resistance to vasopressin. Causes include lithium (the most common acquired cause), electrolyte disturbances (hypercalcaemia, hypokalaemia), congenital (V2 receptor or aquaporin-2 mutations), and drugs (demeclocycline — now rarely used). [1]
Diagnosis — the water deprivation test with desmopressin trial: [1]
The water deprivation test (Miller-Moses) is the classic diagnostic approach. It is performed under supervision with hourly monitoring of weight, urine output, urine osmolality, and plasma osmolality and sodium. Fluids are withheld until the patient concentrates urine or reaches an endpoint (weight loss above 3%, plasma osmolality above 295 mOsm per kg, or sodium above 145 mmol per litre). Desmopressin (1 to 2 microgram intramuscularly or intravenously, or 4 microgram subcutaneously) is then administered. [1]
| Condition | Urine osmolality after deprivation | Response to desmopressin |
|---|---|---|
| Normal | Concentrates above 800 mOsm per kg | Minimal further increase |
| Primary polydipsia | Concentrates above 500 to 600 mOsm per kg (partial washout effect) | Minimal further increase |
| Complete central DI | Remains dilute, below 300 mOsm per kg | Dramatic increase above 800 mOsm per kg |
| Partial central DI | Partial concentration, 300 to 800 mOsm per kg | Further increase, above 50% |
| Nephrogenic DI | Remains dilute, below 300 mOsm per kg | No significant response |
Modern approach — copeptin: Copeptin is the C-terminal fragment of provasopressin, released stoichiometrically with vasopressin. It is stable in serum and does not require the cumbersome water deprivation test. The Refardt study (PMID 30067922) showed that the hypertonic saline-stimulated copeptin test (copeptin cutoff above 4.9 pmol per litre after hypertonic saline) has a diagnostic accuracy of 96.5% for central DI, superior to the indirect water deprivation test (76.6%). This is now the preferred diagnostic approach in specialist centres. A baseline copeptin above 21 pmol per litre excludes central DI and suggests nephrogenic DI or primary polydipsia. [1]
Management of central DI:
- Desmopressin (DDAVP) — a synthetic vasopressin analogue with potent V2 (antidiuretic) and minimal V1 (pressor) activity. Given orally (100 to 200 microgram two to three times daily), intranasally (10 to 40 microgram daily), or sublingually. Titrate to control polyuria without causing hyponatraemia. The goal is to allow a mild diuresis each day to prevent water intoxication.
- Treat the underlying cause — for infiltrative disease, treat the primary condition. [1]
Management of nephrogenic DI:
- Desmopressin is ineffective — the kidney is resistant.
- Maintain adequate water intake — the patient must drink to thirst.
- Thiazide diuretics (paradoxical effect — mild volume depletion enhances proximal tubular sodium and water reabsorption, reducing polyuria).
- Low-sodium diet and NSAIDs (indomethacin — enhances concentrating ability, but use with caution).
- Remove the offending drug (lithium if possible). [1]
The three-phase pattern of DI after pituitary surgery: A triphasic response occurs in some patients after pituitary stalk injury:
- Phase 1 (days 1 to 5): Transient DI from oedema of the stalk and posterior pituitary, impairing vasopressin release.
- Phase 2 (days 5 to 14): SIADH from uncontrolled vasopressin release from the degenerating posterior pituitary.
- Phase 3 (after day 14): Permanent DI if the hypothalamic neurons are destroyed. [1]
This pattern explains why post-operative sodium must be monitored closely for 2 weeks. [1]
The DCE long case — acromegaly and hypopituitarism
Long case archetype — acromegaly with complications: [1]
A typical long-case patient is a 52-year-old man referred for assessment of enlarged hands, headaches, and recently diagnosed type 2 diabetes and hypertension. He snores loudly and is sleepy during the day. On examination he has coarse facial features, frontal bossing, prognathism with interdental spaces, large hands and feet, a carpal tunnel release scar, skin tags, and a blood pressure of 156/94. IGF-1 is 3 times the upper limit of normal. OGTT GH nadir is 8 microgram per litre. MRI shows a 16 mm pituitary macroadenoma with suprasellar extension abutting but not elevating the chiasm. [1]
Problem list:
- Acromegaly from a somatotroph macroadenoma — biochemical activity and mass effect
- Obstructive sleep apnoea (macroglossia) — requires sleep study and CPAP
- Cardiomyopathy screen — echocardiogram for LVH and diastolic dysfunction
- Hypertension and type 2 diabetes — metabolic consequences of GH excess
- Colonic polyp and cancer risk — colonoscopy at diagnosis
- Carpal tunnel syndrome (bilateral) — may improve with biochemical control
- Visual field assessment — chiasm abutment
- Full anterior pituitary panel — exclude hypopituitarism [1]
Management plan:
- Transsphenoidal surgery first-line (experienced pituitary surgeon)
- If not cured (likely for macroadenoma), medical therapy with first-generation somatostatin analogue (octreotide LAR or lanreotide autogel); escalate to pasireotide or pegvisomant if IGF-1 not controlled
- Treat OSA with CPAP pre-operatively
- Colonoscopy, echocardiogram, visual fields, DEXA
- Aggressive cardiovascular risk factor management
- Lifelong surveillance — IGF-1 and GH every 3 to 6 months, annual MRI for first 3 years [1]
The cortisol-first rule in the long case: If this patient has post-operative hypopituitarism, replace hydrocortisone first, then levothyroxine (monitor free T4 not TSH), then testosterone, then GH if indicated. [1]
DCE short case — visual fields and the acromegalic face
Visual field examination routine: [1]
Examination instruction: "Examine this patient's visual fields." [1]
- General inspection — glasses, ptosis, proptosis, facial asymmetry
- Visual acuity — Snellen chart each eye with and without correction; pinhole if reduced
- Visual fields (confrontation) — test each eye individually for the four quadrants (or use a red target for subtle defects). Ask the patient to fixate on your nose and compare their field to yours. Look specifically for a temporal defect in each eye — bitemporal hemianopia is the classic finding of chiasm compression.
- Eye movements — for cranial nerve III, IV, VI palsy from cavernous sinus involvement
- Fundoscopy — look for optic atrophy (pale disc from chronic chiasm compression)
- Complete the examination — check cranial nerves, skin for acromegalic or Cushingoid features, hands for acromegaly, blood pressure, and signs of hypopituitarism [1]
Presentation template for a bitemporal hemianopia: [1]
"I examined this patient's visual fields. Visual acuity is 6/9 in the right eye and 6/12 in the left. Confrontation testing reveals a bitemporal hemianopia, with loss of the temporal field in each eye, sparing the nasal fields. The defect respects the vertical midline. Eye movements are full. Fundoscopy shows mild temporal pallor of the right optic disc. There are no cranial nerve palsies. [1]
This bitemporal hemianopia is caused by compression of the central decussating fibres of the optic chiasm, most commonly by a pituitary macroadenoma with suprasellar extension. I would confirm with formal perimetry (Humphrey), perform an MRI of the pituitary, check a full anterior pituitary panel including prolactin (to exclude a macroprolactinoma, which is treated medically), and refer urgently to a pituitary multidisciplinary team for consideration of transsphenoidal decompression." [1]
Discussion questions:
- Why does the chiasm compress the temporal fields first? The central decussating fibres (from the nasal retina, representing the temporal visual field) are in the inferior and central chiasm — directly in the path of a superiorly expanding pituitary mass.
- What hormone must you check before surgery? Prolactin — a macroprolactinoma is treated with cabergoline, not surgery.
- What is the order of hormone loss in chiasm compression? GH, then LH/FSH, then TSH, then ACTH. [1]
Communication and shared decision-making
- Prolactinoma and dopamine agonist: Explain that cabergoline is highly effective, tumours usually shrink, and medical therapy is the first choice even for large tumours. Discuss the small risk of impulse control disorders (gambling, hypersexuality) and the need to stop before driving if drowsiness occurs. For pregnancy planning, stop at confirmation and monitor for symptomatic enlargement.
- Acromegaly and treatment options: Explain that surgery is first-line, that biochemical control restores life expectancy to normal, and that medical therapy is effective if surgery is not curative. Discuss the colonoscopy and sleep study as essential, not optional.
- Hypopituitarism and the cortisol-first rule: Give the patient a steroid card, MedicAlert bracelet, and emergency hydrocortisone injection kit. Teach sick day rules (double oral dose for fever or minor illness; emergency injection for vomiting, severe trauma, or collapse). Explain that levothyroxine is monitored by free T4, not TSH.
- Pituitary apoplexy: In the acute setting, explain to the patient and family that this is an emergency, that immediate steroid treatment is life-saving, and that surgery may be needed depending on visual assessment. [1]
Common exam traps and high-yield discriminators
- The stalk effect. A macroadenoma with a prolactin of 200 mU/L is a non-functioning adenoma with stalk effect — treat with surgery, not cabergoline. A true macroprolactinoma has a prolactin above 4000 mU/L proportional to tumour size.
- Macroprolactin. Always request PEG precipitation if the prolactin is elevated in an asymptomatic patient with normal gonadal function. Treating macroprolactin with cabergoline is wrong.
- Cortisol before thyroid. The single most dangerous pituitary prescribing error. Giving levothyroxine without cortisol precipitates adrenal crisis.
- Pituitary apoplexy — give hydrocortisone first. Do not wait for the MRI or cortisol result. Empirical hydrocortisone 100 to 200 mg IV is life-saving. [1]5. Monitor free T4, not TSH, in hypopituitarism. TSH is unreliable in central hypothyroidism. The target is mid-normal free T4.
- IGF-1 is the screening test for acromegaly. A normal age-matched IGF-1 excludes it. Confirm with OGTT GH nadir below 1.0 microgram per litre (0.4 on modern assays).
- Cabergoline is first-line for prolactinoma even with visual compromise. It shrinks tumours within weeks. Surgery is for resistance or CSF rhinorrhoea.
- DI at diagnosis suggests craniopharyngioma, not pituitary adenoma. Pituitary adenomas rarely cause DI at presentation because the posterior pituitary is anatomically separate.
- Post-pituitary surgery sodium monitoring. Watch for the triphasic response — DI, then SIADH, then permanent DI. Monitor sodium daily for 2 weeks.
- Acromegaly mortality is from cardiovascular disease. Control GH/IGF-1, screen for cardiomyopathy, treat sleep apnoea and cardiovascular risk factors aggressively. [1]
References
- [1]Katznelson L, Laws ER, Melmed S, et al. Acromegaly: an endocrine society clinical practice guideline J Clin Endocrinol Metab, 2014.PMID 25356808
- [2]Melmed S, Casanueva FF, Hoffman AR, et al. Diagnosis and treatment of hyperprolactinemia: an Endocrine Society clinical practice guideline J Clin Endocrinol Metab, 2011.PMID 21296991
- [3]Rajasekaran S, Vanderpump M, Baldeweg S, et al. UK guidelines for the management of pituitary apoplexy Clin Endocrinol (Oxf), 2011.PMID 21044119
- [4]Nieman LK, Biller BMK, Findling JW, et al. The diagnosis of Cushing's syndrome: an Endocrine Society Clinical Practice Guideline J Clin Endocrinol Metab, 2008.PMID 18334580
- [5]Colao A, Bronstein M, Freda P, et al. Pasireotide versus continued treatment with octreotide or lanreotide in patients with inadequately controlled acromegaly (PAOLA): a randomised, phase 3 trial Lancet Diabetes Endocrinol, 2014.PMID 25260838
- [6]Trainer PJ, Drake WM, Katznelson L, et al. Treatment of acromegaly with the growth hormone-receptor antagonist pegvisomant N Engl J Med, 2000.PMID 10770982
- [7]Tomlinson JW, Holden N, Hills RK, et al. Association between premature mortality and hypopituitarism. West Midlands Prospective Hypopituitary Study Group Lancet, 2001.PMID 11273062
- [8]Pappachan JM, Raskauskiene D, Kutty VR, Clayton RN Excess mortality associated with hypopituitarism in adults: a meta-analysis of observational studies J Clin Endocrinol Metab, 2015.PMID 25658016
- [9]Refardt J, Winzeler B, Christ-Crain M, et al. A Copeptin-Based Approach in the Diagnosis of Diabetes Insipidus N Engl J Med, 2018.PMID 30067922
- [10]Jasim S, Alahdab F, Ahmed AT, et al. Mortality in adults with hypopituitarism: a systematic review and meta-analysis Endocrine, 2017.PMID 27817141