Phys · gastrointestinal
Malabsorption and Small Bowel Disease
Also known as malabsorption · coeliac disease · tropical sprue · Whipple disease · small intestinal bacterial overgrowth · bile salt malabsorption · short bowel syndrome · refractory coeliac disease · enteropathy-associated T-cell lymphoma · protein-losing enteropathy
Consultant-physician-depth guide to malabsorption and small bowel disease — the three-level pathophysiology (luminal maldigestion, mucosal absorption, and transport), the mucosal causes (coeliac disease with anti-tTG IgA and anti-endomysial serology, duodenal Marsh classification, gluten-free diet, refractory coeliac, dermatitis herpetiformis, and selective IgA deficiency), tropical sprue, Crohn disease, eosinophilic gastroenteritis, amyloidosis, and Whipple disease (Tropheryma whipplei), the digestive causes (chronic pancreatic exocrine insufficiency, bile salt malabsorption, and SIBO), the structural causes (short bowel syndrome, blind loop, fistula, strictures), and the structured investigation pathway (FBC anaemia type, albumin and alpha-1-antitrypsin clearance, coeliac serology, faecal calprotectin and elastase, SeHCAT, hydrogen breath test, OGD with biopsy, capsule endoscopy, CT or MR enterography). Structured for FRACP DWE and DCE preparation.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Malabsorption and Small Bowel Disease

The answer first
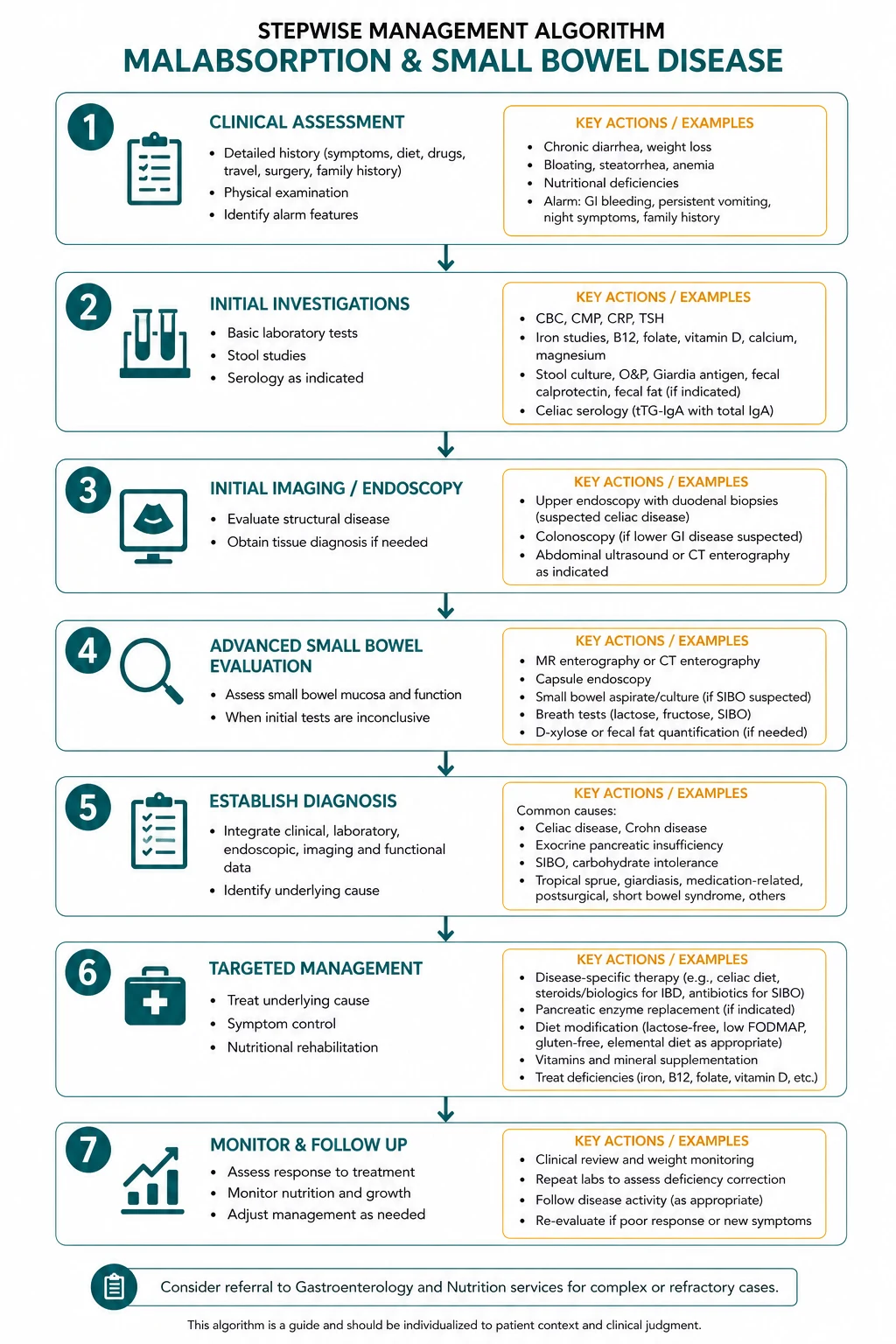
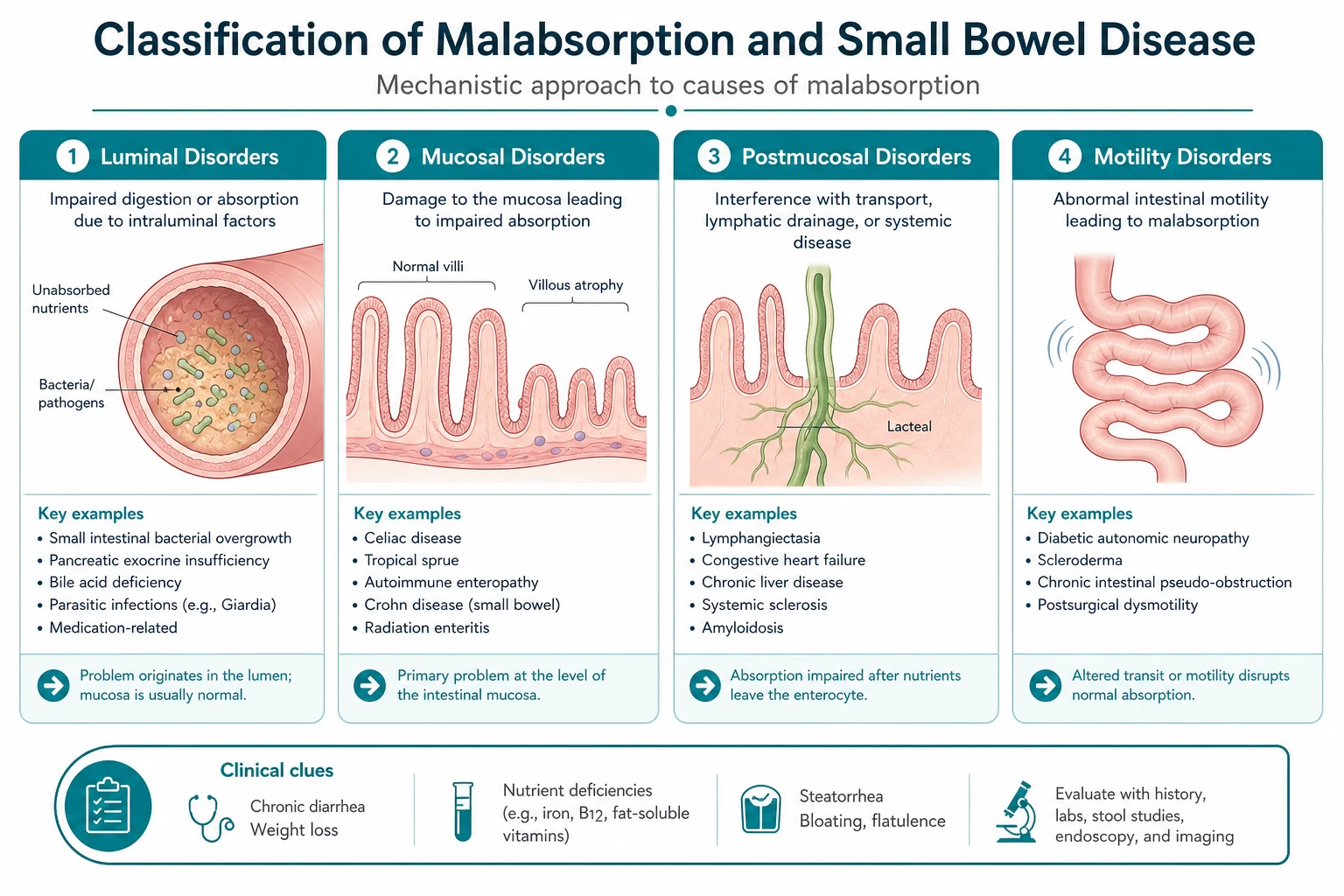
Malabsorption is the failure of the gut to absorb one or more dietary nutrients, and the physician's task is to localise the defect to one of three levels — luminal maldigestion (the food is not broken down, as in chronic pancreatitis or bile salt malabsorption), mucosal absorption (the enterocytes are damaged and cannot absorb, as in coeliac disease), or transport (the absorbed nutrients cannot be delivered to the body, as in short bowel syndrome or lymphatic obstruction). This three-level framework drives the entire investigation pathway and every management decision. [1]
The single highest-yield concept is that the pattern of micronutrient deficiency localises the disease site: microcytic iron deficiency points to the proximal small bowel (coeliac disease), macrocytic vitamin B12 or folate deficiency points to the terminal ileum (Crohn disease, ileal resection), and fat-soluble vitamin deficiency with steatorrhoea points to pancreatic or diffuse mucosal disease. Coeliac disease is the dominant mucosal cause in adults and the one the examiner expects you to discuss in depth — serology (anti-tTG IgA with total IgA to exclude selective IgA deficiency), duodenal biopsy with the modified Marsh classification, a lifelong strict gluten-free diet, and vigilance for refractory disease and its malignant progression to enteropathy-associated T-cell lymphoma [1] [2].
DCE trap: The biggest pivot in the malabsorption long case is the diagnostic pathway. Do not jump to a diagnosis. State the three levels of pathophysiology, use the anaemia type and the specific micronutrient deficiency to localise the site, then use the targeted tests — coeliac serology, faecal elastase, faecal calprotectin, SeHCAT, hydrogen breath test, and endoscopy with biopsy — to confirm the cause. The two diagnoses you must not miss are refractory coeliac disease type 2 (premalignant for enteropathy-associated T-cell lymphoma) and Whipple disease (treatable but fatal if missed). [1]
Definition and pathophysiology — the three-level framework
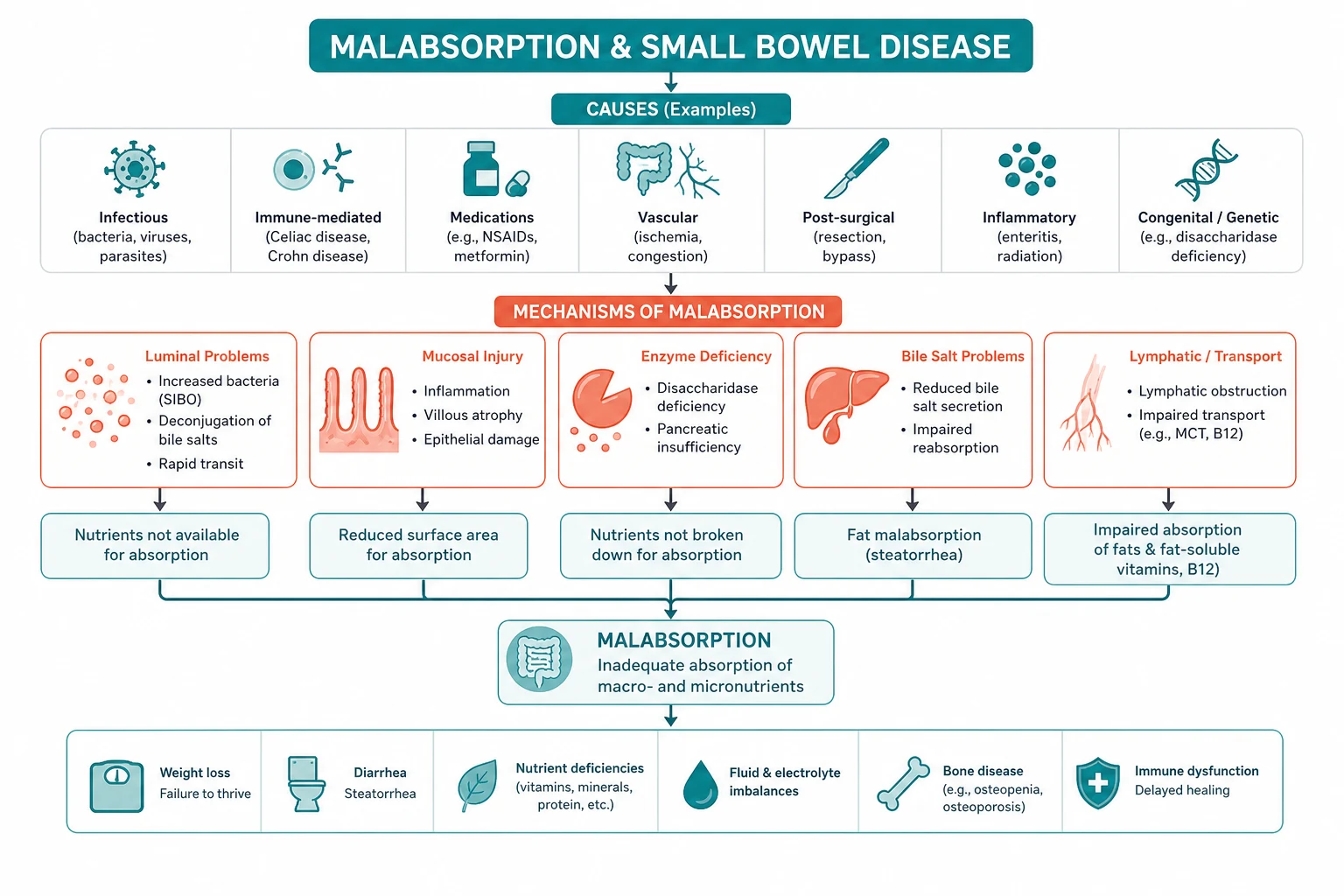
Malabsorption results from the disruption of normal digestion and absorption, a process that can be conceptually divided into three sequential stages. Disease at each stage produces a characteristic clinical and biochemical fingerprint, and the physician's diagnostic strategy is to identify which stage is failing [1].
Level 1 — luminal maldigestion
The intestinal lumen must provide the right environment and the right enzymes to break food into absorbable units. Failure at this level occurs in three settings: [1]
- Pancreatic enzyme deficiency — chronic pancreatitis destroys exocrine acinar cells, reducing lipase, colipase, and protease secretion. Steatorrhoea (foul, bulky, greasy stools) and fat-soluble vitamin deficiency develop only after more than 90 per cent of secretory capacity is lost. Faecal elastase is the non-invasive marker, and pancreatic enzyme replacement is the treatment. This is covered in detail in the chronic pancreatitis topic.
- Bile salt deficiency — adequate micellar solubilisation of fat requires a critical concentration of conjugated bile salts in the duodenal lumen. This fails in terminal ileum disease (Crohn disease, ileal resection) where bile salt reabsorption is lost, depleting the enterohepatic circulation, and in cholestatic liver disease where bile delivery is impaired. The clinical fingerprint is steatorrhoea and fat-soluble vitamin deficiency.
- Bacterial overgrowth — when bacteria colonise the small bowel excessively (small intestinal bacterial overgrowth, SIBO), they deconjugate bile salts to free bile acids that cannot form micelles, directly consume vitamin B12, and damage the mucosa, producing a patchy villous atrophy. The result is steatorrhoea, weight loss, and B12 deficiency with a paradoxically normal or elevated folate (synthesised by the bacteria). [1]
Level 2 — mucosal absorption
The enterocyte, with its apical brush border enzymes and villous architecture, is responsible for the uptake of digested nutrients. Mucosal disease reduces the absorptive surface and impairs specific transport mechanisms. The dominant cause in adults is coeliac disease; others include tropical sprue, Crohn disease, eosinophilic gastroenteritis, amyloidosis, Whipple disease, and infectious enteritis (Giardia, HIV with cryptosporidiosis or microsporidiosis). The clinical fingerprint depends on the site and extent of damage — coeliac disease preferentially affects the proximal small bowel (iron and folate deficiency), while terminal ileum disease causes B12 and bile salt deficiency. [1]
Level 3 — transport
Once absorbed across the enterocyte, nutrients must be delivered to the body via the portal venous and lymphatic systems. Failure at this level occurs in short bowel syndrome (insufficient absorptive surface after resection), lymphatic obstruction (intestinal lymphangiectasia, primary or secondary), and rapid transit (post-vagotomy dumping, hyperthyroidism). The clinical fingerprint is global malabsorption with high-output diarrhoea or a stoma. [1]
DWE high-yield mechanism question: "Why does iron deficiency suggest proximal small bowel disease while B12 deficiency suggests terminal ileum disease?" Answer: Iron is absorbed predominantly in the duodenum and proximal jejunum, so iron deficiency (often the sole presentation of coeliac disease) implies disease there. Vitamin B12 binds intrinsic factor in the stomach and the complex is absorbed in the terminal ileum, so B12 deficiency implies terminal ileum disease (Crohn disease, ileal resection) or bacterial overgrowth (bacteria consume B12 before it reaches the ileum). [1]
Coeliac disease — the prototype mucosal malabsorption
Coeliac disease is an immune-mediated enteropathy triggered by dietary gluten (the prolamin fraction — gliadin in wheat, hordein in barley, secalin in rye) in genetically susceptible individuals. It affects approximately 1 per cent of the population, though the majority remain undiagnosed. It is the prototype mucosal malabsorption syndrome and the one the examiner expects in depth [1] [2].
Pathogenesis — the HLA-DQ2/DQ8 immune cascade
Coeliac disease occurs only in individuals carrying the HLA-DQ2 or HLA-DQ8 haplotype (present in over 95 per cent of patients), but the haplotype is necessary but not sufficient (present in 30 to 40 per cent of the general population). The pathogenic cascade is: [1]
- Gliadin peptides resist luminal digestion and cross the intestinal epithelium.
- Tissue transglutaminase (tTG) in the lamina propria deamidates specific glutamine residues in gliadin, creating negatively charged residues that bind with high affinity to the HLA-DQ2 or DQ8 antigen-binding groove.
- Deamidated gliadin is presented to CD4-positive T helper cells, driving a Th1 inflammatory response with interferon-gamma and cytokine release.
- The inflammatory cascade drives intraepithelial lymphocyte infiltration, crypt hyperplasia (to compensate for surface loss), and villous atrophy — the histological triad that defines the disease.
- B cells produce autoantibodies against tissue transglutaminase (anti-tTG) and endomysium (anti-endomysial), the serological markers of the disease. [1]
This autoimmune mechanism explains why tTG is both the autoantigen and the target of the diagnostic serology, and why the disease is strongly associated with other autoimmune conditions (type 1 diabetes, autoimmune thyroid disease, autoimmune hepatitis, and Addison disease). [1]
Clinical presentation
Coeliac disease now presents more often with atypical or silent features than with classic malabsorption [2]:
- Classic malabsorption — chronic diarrhoea with steatorrhoea, weight loss, abdominal bloating, and failure to thrive in children. This picture is now the minority of adult presentations.
- Iron deficiency anaemia — the single most common presentation in adults, often the sole finding. Iron is absorbed in the proximal small bowel, the site most affected by coeliac disease.
- Isolated osteoporosis or osteomalacia — from calcium and vitamin D malabsorption, often presenting with fractures.
- Dermatitis herpetiformis — the cutaneous manifestation: intensely pruritic symmetric vesicles and papules on extensor surfaces (elbows, knees, buttocks, scalp), diagnosed by skin biopsy showing granular IgA deposition at the dermoepidermal junction. Almost all patients have coeliac enteropathy on biopsy.
- Neurological — peripheral neuropathy, ataxia (gluten ataxia), and epilepsy with cerebral calcifications.
- Reproductive — infertility, recurrent miscarriage, and delayed puberty.
- Associated conditions — type 1 diabetes, autoimmune thyroid disease, IgA deficiency, Down syndrome, and Turner syndrome all warrant screening.
- Incidental — antibody positivity found on screening of first-degree relatives (who carry a 10 per cent risk). [1]
DCE trap: Iron deficiency anaemia is the single most common presentation of adult coeliac disease. Every adult with unexplained iron deficiency (especially in the absence of overt gastrointestinal blood loss) warrants coeliac serology before or alongside endoscopy. Missing coeliac disease in the iron-deficient patient is a common and avoidable error. [1]
Diagnosis — serology first, biopsy confirms
Diagnosis requires an integrated approach: serological screening followed by duodenal biopsy confirmation while the patient is on a gluten-containing diet (serology and histology both normalise on a gluten-free diet) [1] [2].
Serology: [1]
| Antibody | Class | Sensitivity | Specificity | Notes |
|---|---|---|---|---|
| Anti-tissue transglutaminase (anti-tTG) | IgA | Above 90 per cent | Above 95 per cent | First-line test. Uses human recombinant tTG; highly reproducible. |
| Anti-endomysial (EMA) | IgA | Above 85 per cent | Above 98 per cent | More specific, less sensitive, operator-dependent (monkey oesophagus or human umbilical cord substrate). Used as a confirmatory test. |
| Anti-deamidated gliadin peptide (anti-DGP) | IgG | Above 80 per cent | Above 90 per cent | The test of choice in IgA-deficient patients. |
| Total serum IgA | — | — | — | Must always be measured alongside anti-tTG IgA. |
The critical diagnostic trap is selective IgA deficiency, which is 10 to 40 times more common in coeliac disease (prevalence 1 in 40 to 1 in 500 in coeliac patients versus 1 in 500 to 1 in 2000 in the general population). Because anti-tTG IgA and anti-EMA IgA are IgA-class antibodies, an IgA-deficient patient produces a false-negative serological result. Always check total serum IgA alongside anti-tTG IgA. If the patient is IgA-deficient, use IgG-based alternatives — anti-tTG IgG or anti-DGP IgG [1].
High-titre anti-tTG IgA (above 10 times the upper limit of normal) with a positive anti-endomysial antibody has a very high positive predictive value for villous atrophy. In selected children, ESPGHAN criteria allow a no-biopsy pathway with these criteria; in adults, the duodenal biopsy remains the standard for confirmation [2].
Duodenal biopsy — the modified Marsh classification: [1]
At least four biopsies should be taken — from the duodenal bulb (where coeliac changes may be patchy and earliest) and the second part of the duodenum (D2). The modified Marsh classification grades the histological severity [1]:
| Grade | Histological features | Interpretation |
|---|---|---|
| Marsh 0 | Normal mucosa | No coeliac enteropathy |
| Marsh 1 | Increased intraepithelial lymphocytes (above 30 per 100 enterocytes), normal villi | Infiltrative; non-specific but may represent early coeliac in the right serological context |
| Marsh 2 | Increased IELs plus crypt hyperplasia | Hyperplastic; intermediate |
| Marsh 3a | Partial villous atrophy (villous height to crypt depth ratio below 1) | Destructive; consistent with coeliac disease with positive serology |
| Marsh 3b | Subtotal villous atrophy | Destructive; advanced |
| Marsh 3c | Total villous atrophy | Destructive; severe |
Marsh 3 (a, b, or c) with positive serology confirms coeliac disease. Marsh 1 is non-specific and requires serological correlation — it may be seen with Helicobacter pylori, non-steroidal anti-inflammatory drugs, or bacterial overgrowth. [1]
DWE trap: The most common serological error is ordering anti-tTG IgA without total serum IgA. A false-negative anti-tTG IgA in a selectively IgA-deficient patient with coeliac disease is a classic board-exam scenario. If total IgA is low, use IgG-based serology (anti-DGP IgG or anti-tTG IgG). [1]
Management — the lifelong gluten-free diet
The only treatment for coeliac disease is a lifelong strict gluten-free diet — complete exclusion of wheat, barley, and rye. Oats are tolerated by the majority (they contain avenin, a prolamin with lower toxicity) but should be introduced cautiously and sourced gluten-free to avoid cross-contamination [1].
Key elements of management:
- Dietitian referral — for education on label reading, hidden gluten sources, and cross-contamination, and to ensure nutritional adequacy.
- Screen for and correct deficiencies — iron, folate, vitamin B12, calcium, vitamin D, and the fat-soluble vitamins A, E, and K.
- Bone density (DEXA) at diagnosis and periodically — osteoporosis is common from chronic calcium and vitamin D malabsorption.
- Monitor serological response — anti-tTG IgA should fall to normal over 6 to 12 months on a strict diet; a persistently elevated titre suggests ongoing gluten exposure (intentional or inadvertent).
- Vaccinations — pneumococcal and influenza vaccination is recommended because of the association with hyposplenism and functional asplenia.
- Screen first-degree relatives — they carry a 10 per cent risk and should be tested with serology, even if asymptomatic. [1]
DCE integration point: In every coeliac long case, state the diagnosis by serology and Marsh grade, confirm adherence to the gluten-free diet, screen for deficiencies and osteoporosis, address associated autoimmune conditions, and outline the indications for repeat endoscopy (persistent or recurrent symptoms, suspicion of refractory disease). A candidate who covers all these domains passes comfortably. [1]
Refractory coeliac disease and its malignant progression
Refractory coeliac disease is defined as persistent or recurrent malabsorptive symptoms with villous atrophy despite a strict gluten-free diet for more than 12 months, after rigorous exclusion of inadvertent gluten ingestion (the most common cause of apparent non-response) and other causes of villous atrophy (tropical sprue, common variable immunodeficiency, autoimmune enteropathy, olmesartan enteropathy, and bacterial overgrowth) [3].
Refractory disease is classified into two types by intraepithelial lymphocyte (IEL) phenotyping and T-cell receptor clonality, a distinction that is prognostically critical: [1]
| Feature | Type 1 RCD | Type 2 RCD |
|---|---|---|
| IEL phenotype | Normal surface CD3, CD8 positive | Aberrant: surface CD3 and CD8 negative, cytoplasmic CD3 positive |
| T-cell clonality | Polyclonal | Monoclonal T-cell receptor rearrangement |
| Risk of EATL | Low | 30 to 50 per cent over 5 years |
| Treatment | Enteric-coated budesonide, steroids, azathioprine | Specialist centre; consider anti-IL-15, cladribine, autologous stem cell transplant |
| Prognosis | Good | Poor (5-year survival 40 to 60 per cent) |
Enteropathy-associated T-cell lymphoma (EATL) is the feared complication of type 2 refractory coeliac disease. The aberrant clonal intraepithelial lymphocytes are premalignant, and the lymphoma arises from them. EATL presents with new or worsening symptoms in a patient with known coeliac disease — fever, abdominal pain, worsening diarrhoea, weight loss, and sometimes perforation or obstruction from a small bowel mass. The prognosis is poor, with a 5-year survival of 20 to 60 per cent. The international peripheral T-cell lymphoma project confirmed the aggressive clinical course and poor outcomes of EATL [4].
DCE trap: Any patient with coeliac disease who develops new or worsening symptoms despite a strict gluten-free diet has refractory disease or enteropathy-associated T-cell lymphoma until proven otherwise. Type 2 refractory disease requires IEL phenotyping and T-cell clonality studies, specialist centre management, and surveillance for lymphoma. Never attribute a deterioration to dietary non-adherence without thorough investigation. [1]
Other mucosal causes
Tropical sprue
Tropical sprue is a chronic malabsorption syndrome occurring in residents of or travellers returning from tropical regions (the Caribbean, southern India, south-east Asia, and Central America). The cause is uncertain but presumed infectious (a persistent small bowel enterotoxigenic organism). It produces partial villous atrophy with a megaloblastic anaemia from folate and B12 deficiency. [1]
The key distinctions from coeliac disease:
- Negative coeliac serology — anti-tTG and anti-EMA are negative.
- No HLA-DQ2 or DQ8 association — the genetic predisposition is absent.
- Tropical exposure — a residence or travel history in an endemic area is essential.
- Response to folate and tetracycline — the disease responds dramatically to folate supplementation and a course of tetracycline (for 3 to 6 months), which is both diagnostic (response) and therapeutic. [1]
Tropical sprue is now uncommon but remains a high-yield exam entity because its distinction from coeliac disease (seronegative, tropical exposure, antibiotic response) is a classic board question. [1]
Crohn disease
Crohn disease causes malabsorption through multiple mechanisms: terminal ileitis (B12 and bile salt malabsorption), extensive small bowel involvement with villous atrophy, and post-resection short bowel syndrome. It is covered in detail in the inflammatory bowel disease topic, but the physician must always consider it in the malabsorption workup — faecal calprotectin and CT or MR enterography are the key investigations. Cross-reference: see the inflammatory bowel disease topic. [1]
Eosinophilic gastroenteritis
This rare condition is characterised by eosinophilic infiltration of the gastrointestinal wall, classified by the Klein classification into mucosal (diarrhoea, malabsorption, and protein-losing enteropathy), muscularis (obstruction), and serosal (eosinophilic ascites) forms. It presents with abdominal pain, diarrhoea, and malabsorption, often with peripheral eosinophilia and a history of atopy or food allergy. Diagnosis is by biopsy showing eosinophilic infiltration (greater than 30 per high-power field); peripheral eosinophilia supports but is not required. Treatment is with elimination diet, and for severe or refractory disease, oral corticosteroids. [1]
Amyloidosis
Systemic amyloidosis (AL or AA type) can deposit amyloid in the gastrointestinal tract, producing a stiff, poorly motile bowel with malabsorption, motility disturbance (pseudo-obstruction or diarrhoea), and protein-losing enteropathy. The diagnosis is suggested by other organ involvement (nephrotic syndrome, cardiomyopathy, macroglossia, and hepatomegaly) and confirmed by Congo red staining of a biopsy (rectal, abdominal fat, or duodenal). Treatment is of the underlying cause (myeloma in AL, chronic inflammation in AA). [1]
Whipple disease
Whipple disease is a rare chronic infection caused by Tropheryma whipplei that produces a characteristic multisystem syndrome. It predominantly affects middle-aged Caucasian men and classically presents with a tetrad of features [5]:
- Migratory arthralgia — typically preceding the gastrointestinal symptoms by months to years, and often the earliest manifestation. This is a critical diagnostic clue — a patient with chronic unexplained arthralgia who later develops malabsorption.
- Malabsorption — chronic diarrhoea, steatorrhoea, and profound weight loss, producing a malabsorption syndrome with hypoalbuminaemia and lymphadenopathy.
- Systemic features — low-grade fever, generalized lymphadenopathy, and hyperpigmentation.
- Neurological and cardiac involvement — the most serious features, including dementia, ophthalmoplegia, myoclonus, and the pathognomonic oculomasticatory myorhythmia (a pendular convergence nexion synchronous with rhythmic jaw contractions). Endocarditis and culture-negative valvular disease may also occur. [1]
Diagnosis rests on the duodenal biopsy, which shows PAS-positive foamy macrophages in the lamina propria (periodic acid-Schiff stain). However, PAS-positive macrophages are not entirely specific — they can also be seen in Mycobacterium avium-intracellulare infection in HIV patients. Modern diagnosis confirms Tropheryma whipplei by PCR on duodenal biopsy, synovial fluid, cerebrospinal fluid, or blood [5].
Treatment requires antibiotics that penetrate the central nervous system, because subclinical CNS infection is common and CNS relapse is devastating. The standard regimen is intravenous ceftriaxone (2 g daily) or penicillin G for 2 to 4 weeks, followed by oral trimethoprim-sulfamethoxazole for at least 12 months. Treatment that is too short risks relapse, particularly in the CNS. Tropheryma whipplei is intrinsically resistant to trimethoprim alone, which is why the combination with sulfamethoxazole is required [5].
DCE trap: Whipple disease is one of the great mimics. The migratory arthralgia that precedes malabsorption by years is the cardinal early clue. PAS-positive macrophages on duodenal biopsy with Tropheryma whipplei PCR confirm the diagnosis. Treatment must include a CSF-penetrating induction regimen (ceftriaxone) and prolonged maintenance (trimethoprim-sulfamethoxazole for at least a year) to prevent CNS relapse. Do not use trimethoprim alone — the organism is resistant. [1]
Digestive (luminal) causes
Chronic pancreatic exocrine insufficiency
Chronic pancreatitis destroys exocrine acinar cells, reducing lipase, colipase, and protease secretion. Steatorrhoea and fat-soluble vitamin deficiency develop only after more than 90 per cent of secretory capacity is lost. Faecal elastase (below 200 micrograms per gram indicates insufficiency, below 100 severe) is the non-invasive marker. Treatment is pancreatic enzyme replacement therapy (PERT) — enteric-coated porcine pancreatin 25,000 to 40,000 units of lipase with meals, with a proton pump inhibitor to protect the enzymes from gastric acid degradation. This is covered in detail in the chronic pancreatitis topic. [1]
Bile salt malabsorption
The terminal ileum is the site of active reabsorption of bile acids via the apical sodium-dependent bile acid transporter. Disease or resection of the terminal ileum disrupts the enterohepatic circulation, and the consequences depend on the extent of loss [7]:
- Mild ileal resection or disease (less than 100 cm) — the liver compensates for the increased bile acid loss by upregulating synthesis from cholesterol, maintaining an adequate bile salt pool for fat digestion. The excess bile acids spilling into the colon irritate the colonic mucosa, stimulating water and electrolyte secretion — cholerrhoeic diarrhoea. This responds well to a bile acid sequestrant (cholestyramine, colesevelam, or colestipol).
- Extensive ileal resection (more than 100 cm) — the bile acid loss exceeds the liver's synthetic capacity, depleting the bile salt pool below the critical micellar concentration. Fat digestion fails and steatorrhoea develops. In this situation, a bile acid sequestrant worsens the steatorrhoea by further depleting the pool. The correct management is a low-fat diet with medium-chain triglycerides (which do not require micellar solubilisation for absorption). [1]
Diagnosis is by the SeHCAT test (selenium-75-homotaurocholic acid retention scan). The radiolabelled bile acid analogue is administered orally and whole-body retention is measured at 7 days. Retention above 15 per cent is normal; 10 to 15 per cent mild; 5 to 10 per cent moderate; below 5 per cent severe bile salt malabsorption — the lower the retention, the better the response to a sequestrant [7].
Bile salt malabsorption is common after ileal resection for Crohn disease, and it is also increasingly recognised as a cause of chronic diarrhoea (idiopathic or primary bile acid diarrhoea) in patients without ileal disease — the proposed mechanism is impaired feedback inhibition of bile acid synthesis by fibroblast growth factor 19 (FGF19) from the ileum. [1]
Small intestinal bacterial overgrowth (SIBO)
SIBO is defined as an abnormally high bacterial population in the small bowel (above 10 to the 5 colony-forming units per mL on jejunal aspirate). It causes malabsorption through bile salt deconjugation (free bile acids cannot form micelles), direct mucosal damage producing patchy villous atrophy, and consumption of vitamin B12 (with paradoxically normal or elevated folate from bacterial synthesis) [6].
Risk factors are the key to the diagnosis and fall into four groups:
- Impaired motility — the most important protective mechanism is the migrating motor complex that sweeps bacteria distally. Its failure in diabetes with autonomic neuropathy, systemic sclerosis, and chronic intestinal pseudo-obstruction allows stasis and bacterial colonisation.
- Anatomical abnormalities — surgical blind loops (Billroth II, Roux-en-Y), small bowel diverticula, strictures (Crohn disease), and fistulae create regions of stasis.
- Reduced gastric acid — gastric acid is a barrier to bacterial colonisation. Chronic proton pump inhibitor use, achlorhydria (pernicious anaemia), and vagotomy remove this barrier.
- Immune deficiency — IgA deficiency, common variable immunodeficiency, and HIV impair mucosal defence. [1]
Diagnosis:
- Hydrogen breath test — the non-invasive standard. The patient ingests glucose or lactulose, and exhaled hydrogen (and methane) is measured. A rise in hydrogen of more than 20 ppm within 90 minutes (glucose) or a double peak (lactulose, from early small bowel fermentation) indicates SIBO. False positives (rapid transit) and false negatives (non-hydrogen-producing bacteria) occur.
- Jejunal aspirate culture — the gold standard but invasive. A colony count above 10 to the 5 CFU per mL is diagnostic. [1]
Treatment:
- Rifaximin is the preferred antibiotic — a non-absorbable rifamycin that acts locally in the gut lumen with broad-spectrum activity, minimal systemic absorption, and low resistance. It is also effective in irritable bowel syndrome with diarrhoea, where SIBO may play a role [10].
- Alternatives — metronidazole, ciprofloxacin, doxycycline, and amoxicillin-clavulanate, often used in rotating cycles (one week in four) for recurrent disease.
- Correct the underlying cause where possible — discontinue unnecessary proton pump inhibitors, treat strictures and fistulae, and manage motility disorders with prokinetics.
- Nutritional supplementation — vitamin B12 (often deficient from bacterial consumption) and fat-soluble vitamins.
DWE trap: Recurrent SIBO is the norm when the underlying cause is irreversible (scleroderma, blind loop, diabetes). A single antibiotic course rarely cures it. Expect to use rotating antibiotic cycles (rifaximin or metronidazole, one week per month) and to address the underlying cause where possible. Do not forget to check and replace vitamin B12. [1]
Structural causes
Short bowel syndrome
Short bowel syndrome (SBS) results from extensive intestinal resection that leaves insufficient absorptive surface to maintain nutrition and hydration. The most common causes in adults are mesenteric ischaemia (arterial or venous thrombosis, or volvulus), Crohn disease (cumulative resections), surgical adhesions with obstruction and resection, radiation enteritis, and trauma. [1]
The clinical severity and the potential for intestinal adaptation depend on four factors:
- Remaining bowel length — normal adult small bowel is 600 to 800 cm. SBS is generally defined as less than 200 cm remaining. Below 100 cm, parenteral nutrition is almost always required; above 150 cm, oral nutrition may suffice.
- Site of resection — ileal resection is worse tolerated than jejunal because the ileum is the only site of bile acid and vitamin B12 absorption and has slower transit. Jejunal resection is better tolerated because the ileum can compensate.
- Colon preservation — the colon salvages fluid and electrolytes, absorbs short-chain fatty acids (produced by bacterial fermentation of unabsorbed carbohydrate) as an energy source, and slows transit. An intact colon dramatically improves the outcome of SBS. When the colon is in continuity, however, increased oxalate absorption predisposes to calcium oxalate kidney stones.
- Ileocaecal valve preservation — the valve slows transit and prevents colonic bacterial reflux into the small bowel (reducing SIBO). [1]
Intestinal adaptation — over the first 1 to 2 years after resection, the remaining bowel undergoes structural and functional adaptation: villous lengthening, increased crypt depth, dilatation, and enhanced absorptive capacity. This process is driven by luminal nutrients (enteral feeding stimulates adaptation), trophic hormones, and pancreaticobiliary secretions. Maximising adaptation is the central therapeutic goal. [1]
Management — the therapeutic ladder:
- Oral intake and oral rehydration — enteral feeding is the most powerful stimulus for adaptation; it should be introduced and increased as early as possible. A high-energy, high-protein diet with oral rehydration solution (hypotonic fluids worsen high-output diarrhoea by drawing water into the gut).
- Parenteral nutrition and intravenous fluids — required initially and potentially long-term, tailored to fluid, electrolyte, and nutritional needs. Complications include line sepsis, loss of venous access, metabolic bone disease, and parenteral nutrition-associated liver disease.
- Pharmacological intestinal rehabilitation — teduglutide. A recombinant analogue of glucagon-like peptide-2 (GLP-2), teduglutide stimulates intestinal mucosal growth, increases villous height and crypt depth, and enhances fluid and nutrient absorption. The Jeppesen randomised trial showed it reduces parenteral nutrition and intravenous fluid requirements by at least 20 per cent in a significant proportion of patients with SBS and intestinal failure [8]. Other pharmacological options include octreotide (to reduce output, though it may inhibit adaptation), loperamide and codeine to slow transit, and PPIs to reduce gastric hypersecretion (which is transiently excessive after resection) [9].
- Surgical rehabilitation — intestinal lengthening procedures (Bianchi, serial transverse enteroplasty or STEP) and reversal of a segment to slow transit, in specialist centres.
- Intestinal transplantation — reserved for life-threatening complications of parenteral nutrition (recurrent line sepsis, loss of venous access, progressive liver disease) after pharmacological and surgical rehabilitation has been exhausted.
Metabolic complications beyond malnutrition:
- Dehydration and electrolyte disturbance — hypokalaemia, hypomagnesaemia, and metabolic acidosis from high-output diarrhoea or stoma.
- D-lactic acidosis — bacterial fermentation of unabsorbed carbohydrate in the colon produces D-lactic acid, absorbed and causing an anion-gap metabolic acidosis with neurological symptoms (confusion, ataxia, slurred speech).
- Cholelithiasis — loss of the ileal enterohepatic bile acid circulation depletes the bile acid pool, predisposing to cholesterol gallstones.
- Calcium oxalate kidney stones — when the colon is in continuity, unabsorbed fatty acids bind calcium (normally it binds oxalate), increasing free oxalate absorption from the colon.
- Parenteral nutrition-associated liver disease — cholestasis and steatosis from long-term parenteral nutrition. [1]
DCE integration point: In the short bowel syndrome long case, state the four factors determining outcome (remaining length, site, colon, ileocaecal valve), the goals of intestinal adaptation over the first 1 to 2 years, the therapeutic ladder from oral intake through teduglutide to transplantation, and the metabolic complications (dehydration, D-lactic acidosis, gallstones, oxalate stones). The teduglutide evidence (Jeppesen 2011) is the high-yield pharmacology point. [1]
Blind loop syndrome
A surgical blind loop (after Billroth II gastrectomy, Roux-en-Y, or end-to-side anastomosis), small bowel diverticulosis, or a stricture creates a region of stasis that is colonised by bacteria, producing SIBO and its consequences (bile salt deconjugation, B12 deficiency, and malabsorption). The treatment combines antibiotics for SIBO with surgical correction of the anatomical abnormality where feasible. [1]
Fistulae and strictures
Crohn disease fistulae (entero-enteric, enterocolic) bypass absorptive mucosa and create blind loops. Strictures from Crohn disease, radiation, or ischaemia produce stasis upstream, predisposing to SIBO. Diagnosis is by CT or MR enterography and capsule endoscopy; treatment combines medical management of the underlying disease, antibiotics for overgrowth, and endoscopic (balloon dilatation) or surgical correction of the structural lesion. [1]
The structured investigation pathway
The investigation of malabsorption should follow a logical sequence that the examiner expects you to articulate: first, establish the malabsorption and its pattern; second, localise the site using the micronutrient deficiencies; third, confirm the specific cause with targeted tests; and fourth, assess for complications. [1]
Step 1 — Establish malabsorption and nutritional status
- Full blood count — anaemia is the most common haematological abnormality. The type of anaemia localises the disease: microcytic (low MCV) iron deficiency points to the proximal small bowel (coeliac disease, where iron absorption is impaired); macrocytic (high MCV) from B12 or folate deficiency points to the terminal ileum (B12, from Crohn disease or ileal resection) or diffuse mucosal disease (folate, from coeliac disease or SIBO).
- Albumin — hypoalbuminaemia reflects protein-losing enteropathy, chronic inflammation (Crohn disease), malnutrition, or impaired hepatic synthesis. For suspected protein-losing enteropathy, measure alpha-1-antitrypsin clearance (a serum protein that is normally not degraded in the gut; its presence in stool indicates protein loss through the intestinal mucosa).
- Coagulation (INR) — vitamin K malabsorption prolongs the INR in cholestatic or diffuse mucosal disease.
- Bone profile — calcium and vitamin D for osteomalacia and osteoporosis.
- Vitamin levels — iron studies, vitamin B12, folate, and fat-soluble vitamins (A, D, E, K) as guided by the clinical picture. [1]
Step 2 — Localise with targeted tests
| Test | What it measures | When to use it |
|---|---|---|
| Coeliac serology (anti-tTG IgA + total IgA) | IgA-class antibodies against tissue transglutaminase, with total IgA to exclude IgA deficiency | Every patient with suspected malabsorption, iron deficiency, osteoporosis, or associated conditions. If IgA-deficient, use IgG-based alternatives. |
| Faecal calprotectin | Neutrophil-derived protein in stool, a marker of intestinal inflammation | To distinguish IBD (elevated) from irritable bowel syndrome or coeliac disease (normal), and to monitor IBD activity. |
| Faecal elastase | Human pancreatic elastase in stool, a marker of exocrine pancreatic function | To diagnose pancreatic exocrine insufficiency (below 200 indicates insufficiency). Not affected by enzyme replacement. |
| Stool culture, microscopy, and ova/cysts/parasites | Infective organisms including Giardia, Cryptosporidium, and Microsporidium | To exclude infection, especially in immunocompromised or returning travellers. |
| SeHCAT | Bile acid retention at 7 days | To diagnose bile salt malabsorption. Below 15 per cent retention is abnormal. |
| Hydrogen breath test (glucose or lactulose) | Exhaled hydrogen after a substrate load | To diagnose SIBO (rise above 20 ppm within 90 minutes) or lactose intolerance. |
| OGD with duodenal biopsies | Macroscopic and histological (Marsh classification) assessment of the duodenum | To confirm coeliac disease (Marsh 3), and to diagnose Whipple disease (PAS-positive macrophages), eosinophilic gastroenteritis, and lymphangiectasia. |
| Capsule endoscopy | Direct visualisation of the entire small bowel mucosa | For small bowel Crohn disease, tumours (lymphoma, adenocarcinoma), and obscure GI bleeding; not suitable for known or suspected strictures. |
| CT or MR enterography | Cross-sectional imaging of the bowel wall and lumen with contrast | For Crohn disease (wall thickening, strictures, fistulae), small bowel masses, and structural abnormalities (blind loops, adhesions). |
Step 3 — Confirm the specific cause
The choice of confirmatory test depends on the pattern established in steps 1 and 2. A few principles: [1]
- Microcytic iron deficiency with positive serology — coeliac disease; confirm with duodenal biopsy (Marsh classification).
- Macrocytic anaemia with negative coeliac serology — terminal ileum disease (Crohn disease) or SIBO; use faecal calprotectin, enterography, and hydrogen breath test.
- Steatorrhoea with normal coeliac serology — pancreatic insufficiency (faecal elastase) or bile salt malabsorption (SeHCAT).
- Chronic diarrhoea after ileal resection — bile salt malabsorption (SeHCAT) or SIBO (hydrogen breath test); both can coexist.
- Travel history with seronegative partial villous atrophy — tropical sprue; trial of folate and tetracycline. [1]
DWE trap: The anaemia type is the single most powerful bedside localiser in malabsorption. Microcytic (iron) means proximal small bowel; macrocytic (B12 or folate) means terminal ileum or diffuse mucosal disease. State this explicitly in the DWE and DCE — it demonstrates diagnostic reasoning and frames the investigation pathway. [1]
High-yield DWE points and exam traps
- Always check total serum IgA with anti-tTG IgA. Selective IgA deficiency is 10 to 40 times more common in coeliac disease and causes false-negative serology. If IgA-deficient, use IgG-based alternatives (anti-tTG IgG or anti-DGP IgG).
- All coeliac testing must be on a gluten-containing diet. Serology and villous atrophy both normalise on a gluten-free diet. A gluten challenge (10 g of gluten per day for 2 to 6 weeks) is needed if the patient has already started the diet. [1]- Iron deficiency anaemia is the single most common adult presentation of coeliac disease. Test coeliac serology in every adult with unexplained iron deficiency.
- Tropical sprue is seronegative, occurs in tropical residents or travellers, and responds to folate and tetracycline. Do not confuse it with coeliac disease.
- Whipple disease: migratory arthralgia preceding malabsorption, PAS-positive macrophages, Tropheryma whipplei PCR. Treatment is ceftriaxone induction followed by prolonged trimethoprim-sulfamethoxazole.
- SIBO risk factors: PPIs, diabetes, scleroderma, blind loops, post-surgical. Rifaximin is the preferred antibiotic; recurrent disease requires rotating cycles.
- Bile salt malabsorption: SeHCAT retention below 15 per cent. Cholestyramine for mild ileal loss; low-fat diet with MCT for resections over 100 cm (bile salt pool too depleted for sequestrant to work).
- Refractory coeliac disease type 2 has aberrant clonal IELs and a 30 to 50 per cent risk of EATL over 5 years. Needs specialist centre management and lymphoma surveillance.
- Short bowel syndrome: the four factors are remaining length, site (ileum worse), colon, and ileocaecal valve. Teduglutide (GLP-2 analogue) promotes adaptation and reduces parenteral nutrition requirements.
- The anaemia type localises the site: microcytic iron (proximal small bowel), macrocytic B12 or folate (terminal ileum or diffuse mucosa). State this in every DWE and DCE answer.
- Dermatitis herpetiformis: intensely pruritic vesicles on extensor surfaces, granular IgA on skin biopsy. Treated with gluten-free diet and dapsone (check G6PD first). [1]
References and further reading
The British Society of Gastroenterology guidelines for adult coeliac disease (Ludvigsson 2014) provide the current UK diagnostic and management framework [1]. The American College of Gastroenterology guideline (Rubio-Tapia 2013) is the parallel US standard [2]. Refractory coeliac disease is reviewed by Malamut and Cellier, emphasising the type 1 and type 2 distinction and the lymphoma risk [3]. Enteropathy-associated T-cell lymphoma is characterised by the international peripheral T-cell lymphoma project (Delabie 2011) [4]. Whipple disease pathogenesis and treatment are reviewed by Schneider (2008), with emphasis on the CSF-penetrating antibiotic strategy [5]. SIBO is defined by the Bures consensus (World J Gastroenterol 2010) [6]. Bile acid diarrhoea management is reviewed by Walters (2010), including the SeHCAT grading [7]. Teduglutide in short bowel syndrome is established by the Jeppesen randomised trial (2011) [8], and pharmacological intestinal rehabilitation options are reviewed by Jeppesen (2014) [9]. Rifaximin for IBS and SIBO is established by the Pimentel TARGET trial (NEJM 2011) [10].
References
- [1]Ludvigsson JF, Bai JC, Biagi F, et al. Diagnosis and management of adult coeliac disease: guidelines from the British Society of Gastroenterology Gut, 2014.PMID 24917550
- [2]Rubio-Tapia A, Hill ID, Kelly CP, et al. ACG clinical guidelines: diagnosis and management of celiac disease Am J Gastroenterol, 2013.PMID 23609613
- [3]Malamut G, Cellier C Refractory Celiac Disease Gastroenterol Clin North Am, 2019.PMID 30711206
- [4]Delabie J, Holte H, Vose JM, et al. Enteropathy-associated T-cell lymphoma: clinical and histological findings from the international peripheral T-cell lymphoma project Blood, 2011.PMID 21566094
- [5]Schneider T, Moos V, Loddenkemper C, et al. Whipple's disease: new aspects of pathogenesis and treatment Lancet Infect Dis, 2008.PMID 18291339
- [6]Bures J, Cyrany J, Kohoutova D, et al. Small intestinal bacterial overgrowth syndrome World J Gastroenterol, 2010.PMID 20572300
- [7]Walters JRF, Johnston IM, Nolan JD, et al. Managing bile acid diarrhoea Ther Adv Gastroenterol, 2010.PMID 21180614
- [8]Jeppesen PB, Gilroy R, Pertkiewicz M, et al. Randomised placebo-controlled trial of teduglutide in reducing parenteral nutrition and/or intravenous fluid requirements in patients with short bowel syndrome Gut, 2011.PMID 21317170
- [9]Jeppesen PB Pharmacologic options for intestinal rehabilitation in patients with short bowel syndrome JPEN J Parenter Enteral Nutr, 2014.PMID 24615689
- [10]Pimentel M, Lembo A, Chey WD, et al. Rifaximin therapy for patients with irritable bowel syndrome without constipation N Engl J Med, 2011.PMID 21208106