Phys · general-medicine
Primary Immunodeficiency in Adults
Also known as inborn errors of immunity · CVID · common variable immunodeficiency · selective IgA deficiency · specific antibody deficiency · terminal complement deficiency · hereditary angioedema · C1 inhibitor deficiency · chronic granulomatous disease · recurrent infection workup · Jeffrey Modell warning signs · humoral immunodeficiency · immunoglobulin replacement therapy
Consultant-physician guide to when to suspect primary immunodeficiency in adults (Jeffrey Modell Foundation warning signs), the four-arm classification (humoral, cellular, complement, phagocyte), the adult-presentation disorders in each arm (CVID as the most common symptomatic PID in adults; selective IgA deficiency; specific antibody deficiency; terminal complement and properdin deficiency with recurrent Neisseria; hereditary angioedema from C1 inhibitor deficiency; chronic granulomatous disease), and the structured investigation pathway (immunoglobulins, vaccine response, lymphocyte subsets, CH50 and AH50, and the mandatory HIV test to exclude secondary causes). Treatment principles for immunoglobulin replacement therapy, complement-deficiency prevention, and HAE-specific acute and prophylactic therapy are covered at consultant viva depth.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Primary Immunodeficiency in Adults
The one-minute consultant answer
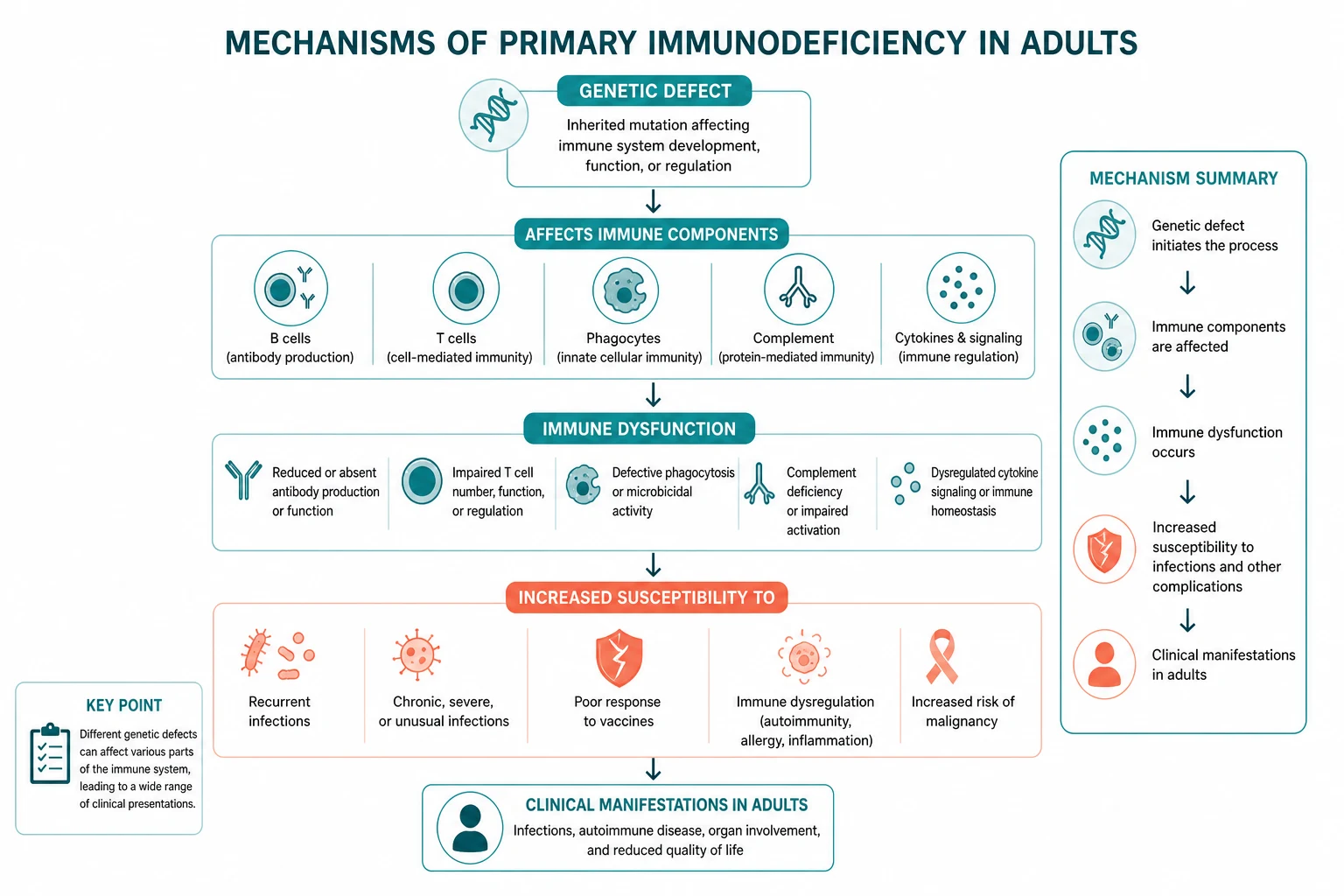
Primary immunodeficiency (PID) — now increasingly called inborn errors of immunity — is far more common than believed and the diagnosis is almost always delayed. The population prevalence of diagnosed PID is about 1 in 1,200 people, and the median diagnostic delay in common variable immunodeficiency (CVID) is 4 to 7 years from the first symptom to the correct diagnosis [1]. The reason is that most adults with PID present to general physicians, respiratory physicians, gastroenterologists, and haematologists — not to immunologists — with recurrent sinopulmonary infection, unexplained bronchiectasis, autoimmune cytopenia, chronic diarrhoea, or recurrent Neisseria infection, and the immunoglobulin screen that would make the diagnosis is never sent.
The single most useful question at the bedside is does this pattern of infection suggest a defect in one of the four arms of the immune system? — because each arm has its own signature organism list, its own diagnostic test, and its own treatment. [1]
- Humoral (B-cell) defects (CVID, selective IgA deficiency, specific antibody deficiency) cause recurrent sinopulmonary and gastrointestinal infection with encapsulated organisms — Streptococcus pneumoniae, Haemophilus influenzae, Moraxella — and Giardia. CVID is the most common symptomatic PID in adults, presenting at age 20 to 40 with recurrent pneumonia, sinusitis, autoimmune disease in about 25 per cent, and a markedly increased lymphoma risk.
- T-cell defects cause opportunistic infection (PCP, Candida, mycobacteria, viruses) and usually present in childhood, but milder combined defects (ataxia-telangiectasia, Nijmegen break syndrome) can present in adulthood.
- Complement deficiencies cause recurrent Neisseria infection (terminal pathway, C5 to C9) or pyogenic infection (C3), and hereditary angioedema (C1 inhibitor deficiency).
- Phagocyte defects (chronic granulomatous disease) cause recurrent catalase-positive organisms (Staphylococcus aureus, Burkholderia, Serratia, Aspergillus) and granuloma formation. [1]
The non-negotiable principles: measure immunoglobulins in any adult with recurrent sinopulmonary infection, unexplained bronchiectasis, or autoimmune cytopenia; always rule out HIV before attributing hypogammaglobulinaemia to a primary defect; and remember that a normal immunoglobulin panel does not exclude PID — specific antibody deficiency and complement deficiency have normal immunoglobulins, so the next step is the pneumococcal vaccine response and the CH50 [2][3].
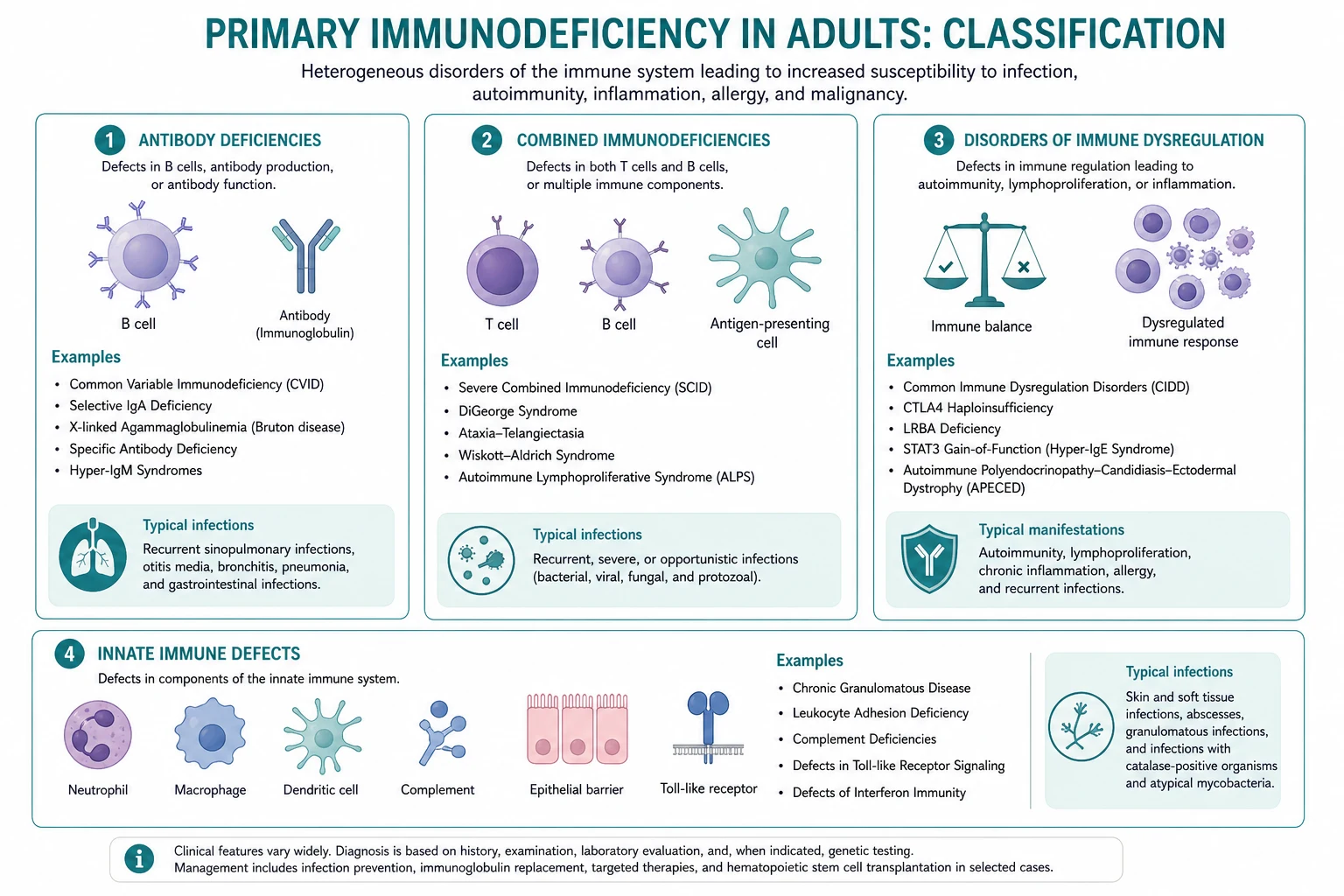
The central framework — the four arms of the immune system

The immune system has four functional arms. A defect in each arm produces a characteristic infection pattern. Memorise the arms and the organism list follows. [1]
| Arm | Function | Defect produces | Adult-presentation disorders | Characteristic organisms |
|---|---|---|---|---|
| Humoral (B-cell) | Antibody production, opsonisation | Recurrent sinopulmonary and gastrointestinal infection; autoimmunity; lymphoma | CVID, selective IgA deficiency, specific antibody deficiency | Encapsulated bacteria (S. pneumoniae, H. influenzae, Moraxella); Giardia; Campylobacter |
| Cellular (T-cell) | Cell-mediated immunity against intracellular pathogens | Opportunistic infection (childhood usually; milder forms in adults) | Ataxia-telangiectasia, Nijmegen break syndrome (mild combined) | PCP, Candida, mycobacteria, viruses (herpes family) |
| Complement | Serum opsonisation and lysis; MAC formation | Recurrent Neisseria (terminal pathway); pyogenic infection (C3); angioedema (C1 inhibitor) | Terminal complement (C5 to C9) deficiency; properdin deficiency; C1 inhibitor deficiency (HAE) | Neisseria meningitidis, N. gonorrhoeae; encapsulated bacteria (C3) |
| Phagocyte | Intracellular killing (respiratory burst) | Recurrent catalase-positive organisms; granulomas | Chronic granulomatous disease (X-linked carriers, autosomal recessive) | Staphylococcus aureus, Burkholderia cepacia, Serratia, Nocardia, Aspergillus |
The exam trap: the question stem will name the infection pattern (recurrent pneumonia, recurrent meningococcaemia, recurrent staphylococcal abscesses, recurrent non-urticarial angioedema) — and the correct answer is the diagnostic test (immunoglobulins for humoral, CH50 for complement, DHR for phagocyte) and the most likely disorder. A stem with "two episodes of meningococcaemia" expects terminal complement deficiency; a stem with "recurrent pneumonia and Giardia, low IgG" expects CVID; a stem with "recurrent facial swelling without urticaria, family history, low C4" expects hereditary angioedema. [1]
When to suspect primary immunodeficiency — the Jeffrey Modell Foundation warning signs

The Jeffrey Modell Foundation published warning signs for primary immunodeficiency as a public health screening tool. They are a trigger to investigate, not a diagnostic criterion. The adult-relevant signs are: [1]
| Warning sign | Clinical meaning |
|---|---|
| Two or more new pneumonias in one year | Failure of humoral defence against encapsulated organisms at the respiratory mucosa |
| Four or more new ear infections in one year | Recurrent mucosal infection beyond the normal range |
| Chronic sinusitis or bronchitis lasting more than 12 weeks (despite appropriate antibiotics) | Failure to clear mucosal infection; consider humoral defect or ciliary dysfunction |
| Two or more months of antibiotics with little effect | Poor response suggests impaired immune clearance rather than resistant organism alone |
| Two or more deep-seated infections (sepsis, cellulitis, osteomyelitis, meningitis) in one year | Failure of systemic humoral or complement defence |
| Recurrent deep abscesses (liver, brain, skin) | Consider phagocyte defect (CGD) or humoral defect |
| Family history of primary immunodeficiency | Heritable component in many PIDs (HAE autosomal dominant; CGD X-linked; familial CVID clustering) |
The exam principle: any adult meeting two or more of these criteria warrants a serum immunoglobulin measurement (IgG, IgA, IgM) and, depending on the pattern, a CH50 (for recurrent Neisseria) and a pneumococcal vaccine response (for suspected specific antibody deficiency). The single greatest source of diagnostic delay is that the immunoglobulin test is never ordered [1].
Humoral (B-cell) immunodeficiency — the adult presentation
Common variable immunodeficiency (CVID) — the most common symptomatic PID in adults
CVID is the most common symptomatic primary immunodeficiency in adults (selective IgA deficiency is the most common PID overall, but most patients are asymptomatic). It is a heterogeneous group of disorders characterised by defective B-cell differentiation into antibody-secreting plasma cells, producing hypogammaglobulinaemia and impaired specific antibody responses [2].
Presentation. The onset is typically between 20 and 40 years (a second peak occurs in adolescence). The clinical spectrum: [1]
- Recurrent sinopulmonary infection is the universal presentation — pneumonia, sinusitis, otitis media, bronchitis — with encapsulated organisms (Streptococcus pneumoniae, Haemophilus influenzae, Moraxella catarrhalis). Untreated, this progresses to bronchiectasis, the single most important preventable complication.
- Gastrointestinal infection and inflammation — Giardia lamblia (a classic clue), Campylobacter, Salmonella; CVID enteropathy (a sprue-like malabsorption syndrome); nodular lymphoid hyperplasia.
- Autoimmune disease in about 25 per cent — the dominant manifestations are immune thrombocytopenia (ITP) and autoimmune haemolytic anaemia (AIHA), alone or together (Evans syndrome); thyroiditis; rheumatoid arthritis; vitiligo; pernicious anaemia. Autoimmune cytopenia may be the presenting feature and precede the recognition of the humoral defect.
- Granulomatous disease mimicking sarcoidosis — non-caseating granulomas in lungs, liver, spleen, skin; this is a dysregulated immune response, not infection.
- Lymphoproliferation and malignancy — splenomegaly, lymphadenopathy, and a markedly increased risk of non-Hodgkin lymphoma (B-cell, often marginal zone or MALT, 30 to 200-fold increased risk) and gastric carcinoma. [1]
Diagnosis. The ICON consensus diagnostic criteria are: marked decrease in IgG (below 5 g/L) with marked decrease in IgA and/or IgM; impaired antibody response to vaccines (pneumococcal polysaccharide and tetanus); and exclusion of secondary causes of hypogammaglobulinaemia (protein loss, drug effects, malignancy) [3]. The onset is after age 4 (to exclude transient hypogammaglobulinaemia of infancy). Lymphocyte subsets show low or normal B-cell numbers (the defect is in differentiation, not necessarily number).
The diagnostic-delay trap: the median time from first symptom to diagnosis is 4 to 7 years, during which bronchiectasis develops, autoimmune cytopenia is treated as "idiopathic," and lymphoma may emerge. The rule is to measure immunoglobulins in any adult with recurrent sinopulmonary infection, unexplained bronchiectasis, or autoimmune cytopenia. [1]
Selective IgA deficiency — the most common PID overall
Selective IgA deficiency has a prevalence of about 1 in 500 in Caucasian populations, making it the most common primary immunodeficiency overall. It is defined by isolated IgA deficiency (IgA below 0.07 g/L) with normal IgG and IgM in a patient over 4 years, with normal vaccine responses and exclusion of secondary causes [4].
Presentation. Most patients are asymptomatic and never diagnosed. Symptomatic patients present with: [1]
- Recurrent mucosal sinopulmonary and gastrointestinal infection — because IgA is the predominant immunoglobulin at mucosal surfaces.
- Atopy — asthma, eczema, allergic rhinitis are over-represented.
- Autoimmune disease — coeliac disease (a strong association), systemic lupus erythematosus, thyroiditis, type 1 diabetes, rheumatoid arthritis.
- The transfusion risk — patients with selective IgA deficiency can develop IgG or IgE class anti-IgA antibodies after exposure to IgA in blood products, and re-exposure can trigger anaphylaxis. This is the classic exam point: a patient with a history of anaphylactic transfusion reaction may have selective IgA deficiency. In an emergency, do not delay transfusion to obtain IgA-deficient products, but for elective transfusion in a patient with a documented reaction, use washed red cells (removes plasma IgA) or IgA-deficient plasma products from identified donors. Universal IgA screening of all transfusion recipients is not recommended. [1]
Management. Selective IgA deficiency does not respond to immunoglobulin replacement therapy — commercial IgRT contains minimal IgA and does not correct the mucosal defect. Management is aggressive treatment and prevention of infection (standby antibiotics, vaccination with inactivated vaccines) and surveillance for the autoimmune associations. A small proportion of patients evolve into CVID over time, so unexplained worsening warrants re-evaluation. [1]
Specific antibody deficiency (SAD) — the diagnosis you miss with a normal immunoglobulin screen
Specific antibody deficiency is the diagnosis that is missed when you stop at a normal immunoglobulin screen. The patient has normal total immunoglobulins and normal IgG, IgA, IgM but a selective failure of antibody response to polysaccharide antigens, presenting as recurrent sinopulmonary infection with encapsulated organisms in an adult whose immunoglobulin panel looks "normal." [1]
Diagnosis. Measure specific IgG to pneumococcal polysaccharide antigens (PPSV23) before and 4 to 6 weeks after vaccination. A normal response is a two-fold rise or protective titres in the majority of serotypes. Failure of polysaccharide response with preserved protein (tetanus) response localises the defect to T-cell-independent B-cell function. [1]
Management. If recurrent infection is significant, immunoglobulin replacement may be considered despite normal total immunoglobulins — the functional defect justifies it. Aggressive prevention of sinopulmonary infection and surveillance for bronchiectasis are essential. [1]
Complement deficiencies — Neisseria and angioedema
Terminal complement deficiency (C5 to C9) — recurrent Neisseria
The terminal complement components (C5, C6, C7, C8, C9) assemble into the membrane attack complex (MAC), which forms the pore that lyses susceptible bacteria. Neisseria meningitidis and Neisseria gonorrhoeae are the organisms that critically depend on MAC-mediated lysis for clearance — they are the signature organisms of terminal complement deficiency [5][6].
Presentation. Recurrent invasive Neisseria infection — meningococcaemia, meningococcal meningitis, disseminated gonococcal infection — often with the first episode in adolescence or early adulthood (in contrast to immunocompetent hosts, in whom meningococcal disease peaks in infancy). The risk of meningococcal disease is 7,000 to 10,000 times that of the general population, though the mortality per episode is paradoxically lower (estimated 1 to 5 per cent versus about 10 per cent in immunocompetent hosts), possibly because of earlier recognition or antibody-opsonised clearance. [1]
The exam rule: any adult with two or more episodes of invasive meningococcal disease must have a CH50 measured. A low or absent CH50 mandates individual terminal component assay (C5, C6, C7, C8, C9). C9 deficiency is most common in Japanese populations; C6 and C8 deficiencies are common in Western populations. [1]
Properdin deficiency — the X-linked exception
Properdin is the only X-linked complement component. It stabilises the C3 convertase of the alternative pathway, amplifying the complement cascade. Its deficiency causes impaired opsonisation and lysis of Neisseria in males. Unlike terminal pathway deficiencies (autosomal recessive, recurrent but milder episodes), properdin deficiency produces fulminant single episodes with higher mortality because the alternative pathway amplification loop is disrupted. [1]
The discriminator: the pattern of CH50 and AH50. In terminal pathway deficiency, CH50 is low and AH50 is low (the terminal pathway is shared by both). In properdin deficiency, CH50 may be normal but AH50 is low (the alternative pathway is selectively affected). Suspect properdin deficiency in a male with fulminant meningococcal disease and a family history suggestive of X-linked inheritance. [1]
C3 deficiency — recurrent pyogenic infection
Deficiency of C3 (the central convergent component of all complement pathways) produces severe recurrent pyogenic infection with encapsulated organisms (S. pneumoniae, H. influenzae, N. meningitidis) and immune complex disease, because both opsonisation and lysis are impaired. Both CH50 and AH50 are low. C3 deficiency is rare and usually presents in childhood. [1]
Hereditary angioedema (HAE) — C1 inhibitor deficiency
Hereditary angioedema is an autosomal dominant disorder caused by deficiency (type 1, about 85 per cent of cases) or dysfunction (type 2) of C1 inhibitor. C1 inhibitor normally inhibits activated kallikrein (and hence bradykinin generation) and activated C1 (and hence classical pathway complement consumption). Its deficiency allows uncontrolled bradykinin-mediated vascular permeability, producing angioedema [7][8].
Presentation. Recurrent episodes of non-pitting, non-urticarial, non-pruritic angioedema affecting: [1]
- The face, limbs, and genitalia (subcutaneous swelling).
- The gastrointestinal tract — bowel wall oedema produces severe cramping abdominal pain, nausea, vomiting, and can mimic a surgical abdomen (unnecessary laparotomy is a documented harm).
- The larynx — laryngeal oedema is the leading cause of death, with historical mortality from upper airway obstruction reported as high as 14 to 33 per cent before modern therapy. [1]
Attacks typically begin in childhood or adolescence, worsen at puberty, last 2 to 5 days, and are triggered by trauma, dental work, surgery, stress, oestrogen-containing medications, and angiotensin-converting enzyme inhibitors. A family history is present in most cases (autosomal dominant, about 75 per cent penetrance), but 25 per cent are de novo mutations. [1]
Diagnosis. The diagnostic triad during and between attacks: C4 is low (consumed by uncontrolled classical pathway activation — C4 is low even between attacks, making it the screening test); C1 inhibitor antigenic level is low in type 1 (about 85 per cent of cases) or normal to high with low function in type 2; and C1q is normal (distinguishing hereditary HAE from acquired C1 inhibitor deficiency, in which C1q is low). [1]
The "acquired C1 inhibitor deficiency in the older adult" trap: new-onset angioedema in an older adult with no family history, low C4, low C1 inhibitor, and low C1q is acquired, driven by an underlying B-cell lymphoproliferative disorder, monoclonal gammopathy, or autoantibody to C1 inhibitor. Investigate for the cause, because treating the angioedema alone misses the underlying malignancy. [1]
Management — acute attacks do NOT respond to adrenaline, antihistamines, or corticosteroids. This is the single most important management point. Acute attacks are treated with: [1]
- C1 inhibitor concentrate — plasma-derived (Berinert) 20 units per kg intravenously, or recombinant (conestat alfa, Ruconest) 50 units per kg (maximum 4,200 units).
- Icatibant — a bradykinin B2 receptor antagonist, 30 mg subcutaneously, with one repeat dose if symptoms recur.
- Ecallantide — a kallikrein inhibitor (where available). [1]
For laryngeal attacks, give C1 inhibitor concentrate or icatibant immediately and secure the airway. Failing to recognise HAE and giving repeated ineffective doses of adrenaline is a documented source of fatal laryngeal oedema. [1]
Long-term prophylaxis. The WAO guideline recommends lanadelumab (anti-kallikrein monoclonal antibody, 300 mg subcutaneously every 2 weeks, reducing attack frequency by 80 to 90 per cent), berotralstat (oral kallikrein inhibitor, 150 mg daily), C1 inhibitor concentrate (regular intravenous or subcutaneous dosing), or attenuated androgens (danazol, 50 to 200 mg daily, used as second-line because of androgenic side effects) [7]. Short-term prophylaxis with C1 inhibitor concentrate 1 to 6 hours before dental or surgical procedures is essential because trauma is a classic trigger.
Cellular (T-cell) defects presenting in adulthood
Severe combined immunodeficiency and most T-cell defects present in infancy and are fatal without haematopoietic stem cell transplantation. However, milder combined immunodeficiencies can present in adulthood: [1]
- Ataxia-telangiectasia (ATM gene) — progressive cerebellar ataxia, oculocutaneous telangiectasia, combined immunodeficiency (often IgA deficiency with low IgG and IgM), chromosomal instability, and markedly increased cancer risk (lymphoma, leukaemia). Milder phenotypes survive into adulthood.
- Nijmegen break syndrome (NBS1 gene) — microcephaly, bird-like facies, combined immunodeficiency, chromosomal instability, and lymphoid malignancy. Also a DNA repair disorder. [1]
These are rare, but the general physician should recognise the syndrome of sinopulmonary infection with ataxia, telangiectasia, or lymphoid malignancy in a young adult as a possible combined immunodeficiency and refer for immunological and genetic evaluation. DiGeorge syndrome (22q11 deletion) with partial T-cell deficiency can also present in adulthood with subtle hypocalcaemia, cardiac history, and recurrent infection. [1]
Phagocyte defects — chronic granulomatous disease
Chronic granulomatous disease (CGD) is caused by defects in the NADPH oxidase complex (the enzyme that generates the respiratory burst in phagocytes). The most common form is X-linked (gp91phox defect); autosomal recessive forms (p47phox most commonly) also occur. Phagocytes cannot generate superoxide and hydrogen peroxide, so they cannot kill catalase-positive organisms (which destroy their own hydrogen peroxide, removing the compensatory killing mechanism) [6].
Presentation. CGD usually presents in childhood, but X-linked carrier females (because of lyonisation — random X-inactivation, which can produce a skewed population of deficient neutrophils) and autosomal recessive forms can present in adulthood with: [1]
- Recurrent catalase-positive infection — Staphylococcus aureus (abscesses, osteomyelitis), Burkholderia cepacia complex (particularly dangerous, a classic cause of fatal pneumonia and sepsis in CGD), Serratia marcescens (liver abscess, osteomyelitis), Nocardia (pulmonary, brain abscess), and Aspergillus (invasive pulmonary aspergillosis, often with osteomyelitis of the chest wall).
- Granulomatous complications — obstructive granulomas (gastric outlet obstruction, genitourinary obstruction), CGD-related colitis mimicking Crohn disease, and disseminated granulomatous inflammation.
- The infection history is the most powerful discriminator — a patient with recurrent staphylococcal abscesses, Burkholderia or Serratia infection, Nocardia, or invasive Aspergillus warrants a DHR test. [1]
Diagnosis. The dihydrorhodamine 123 (DHR) flow cytometry test is the modern standard, measuring the oxidative burst of stimulated neutrophils (absent or reduced fluorescence in CGD). The older nitroblue tetrazolium (NBT) slide test is an alternative. Genetic testing confirms the specific NADPH oxidase defect and allows carrier testing of females and genetic counselling. [1]
Management. Lifelong antibacterial prophylaxis (co-trimoxazole, trimethoprim 5 mg per kg per day) and antifungal prophylaxis (itraconazole 200 mg daily) are the standard; infections are treated aggressively with prolonged targeted therapy; corticosteroids manage granulomatous complications (obstructive granulomas, colitis); and haematopoietic stem cell transplantation or gene therapy offers definitive treatment in selected patients. [1]
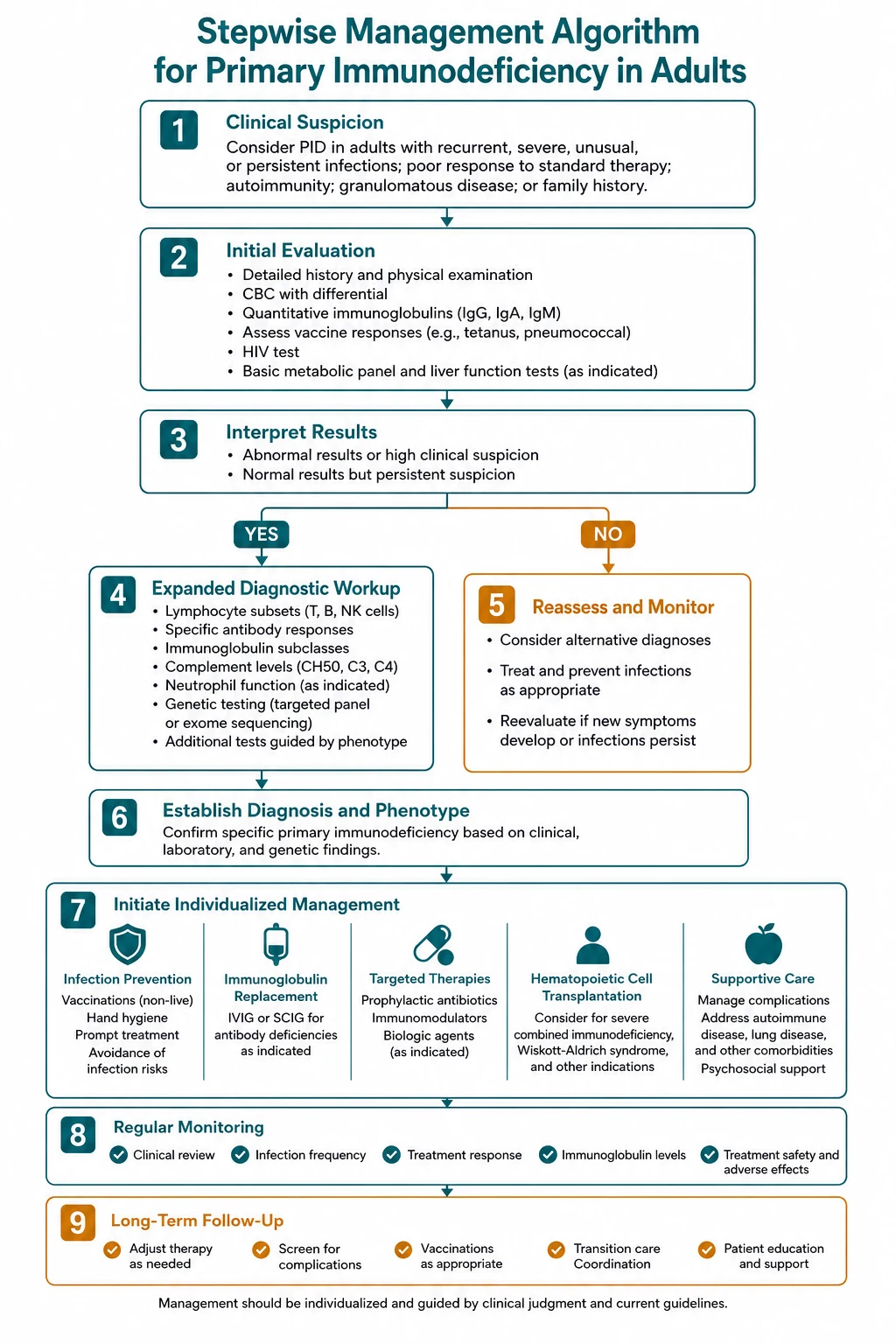
The investigation pathway — a structured approach

The workup is staged. The first-line panel is available in any hospital; the second-line tests are guided by the pattern and by the first-line results. [1]
First-line panel (every adult with suspected PID)
| Test | What it finds |
|---|---|
| Serum immunoglobulins (IgG, IgA, IgM, IgE) | Hypogammaglobulinaemia (CVID); isolated IgA deficiency; IgM elevation (hyper-IgM syndromes); IgE elevation (atopy, hyper-IgE syndrome) |
| Full blood count with differential | Lymphopenia (T-cell defect, HIV); neutropenia; eosinophilia (atopy); thrombocytoneutropenia (autoimmune cytopenia in CVID) |
| HIV test | Excludes the most common secondary immunodeficiency — mandatory before attributing hypogammaglobulinaemia to a primary defect |
The rule: a normal immunoglobulin panel does not exclude PID. Specific antibody deficiency and complement deficiency have normal immunoglobulins — go to the second-line tests if the clinical pattern warrants. [1]
Second-line tests (guided by the clinical pattern and first-line results)
| Clinical pattern | Second-line test | Finding and diagnosis |
|---|---|---|
| Recurrent sinopulmonary infection with normal or low immunoglobulins | Pneumococcal vaccine response (PPSV23, measure specific IgG before and 4 to 6 weeks after) | Poor polysaccharide response with normal immunoglobulins = specific antibody deficiency; poor response with low IgG = CVID |
| Recurrent Neisseria infection | CH50, then AH50, then individual components (C3, C4, C5 to C9) | Low or absent CH50 = terminal pathway deficiency; low AH50 with normal CH50 = properdin deficiency |
| Recurrent non-urticarial angioedema | C4, C1 inhibitor antigen and function, C1q | Low C4 with low C1 inhibitor (antigen or function) and normal C1q = hereditary HAE; low C1q = acquired C1 inhibitor deficiency |
| Recurrent catalase-positive organisms, granulomas | Dihydrorhodamine (DHR) flow cytometry | Absent or reduced oxidative burst = chronic granulomatous disease |
| Suspected combined immunodeficiency | Lymphocyte subsets (CD3, CD4, CD8, CD19/CD20, CD16 and CD56) | Low CD19 = B-cell defect (CVID, agammaglobulinaemia); low CD4 = T-cell defect or HIV; low CD16 and CD56 = NK-cell defect |
How to interpret the vaccine response
Measure specific IgG to pneumococcal polysaccharide antigens before and 4 to 6 weeks after PPSV23 vaccination. A normal response is a two-fold rise or protective titres in the majority of serotypes. The response is interpreted alongside the protein antigen response (tetanus): a preserved protein response with failed polysaccharide response localises the defect to T-cell-independent B-cell function (CVID, specific antibody deficiency). A failed response to both suggests a more global B-cell defect. [1]
How to interpret CH50 and AH50 together
The combination of CH50 (classical and terminal pathway) and AH50 (alternative pathway) localises the defect: [1]
| CH50 | AH50 | Localisation |
|---|---|---|
| Low | Normal | Classical pathway (C1, C2, C4) — consider C1 inhibitor deficiency (HAE) |
| Normal | Low | Alternative pathway or properdin deficiency |
| Low | Low | Terminal pathway (C5 to C9) deficiency, or C3 deficiency |
Individual component testing follows the pattern. A low C4 with normal C3 between attacks is the signature of hereditary angioedema. [1]
Management principles
Immunoglobulin replacement therapy (IgRT)
IgRT is the standard of care for significant antibody deficiency — CVID and other combined defects with documented low IgG and impaired vaccine response [9]. It is not indicated for isolated IgA deficiency (it contains minimal IgA and does not correct the mucosal defect).
| Route | Dose | Frequency | Pros | Cons |
|---|---|---|---|---|
| Intravenous (IVIG) | 400 to 600 mg per kg | Every 3 to 4 weeks | Clinic-administered; suitable for poor venous access for SCIG; reliable | Peaks and troughs; systemic reactions; clinic attendance |
| Subcutaneous (SCIG) | 100 to 200 mg per kg | Weekly (self-administered at home) | Steady IgG levels; fewer systemic reactions; patient autonomy | Local infusion-site reactions; requires training and self-management |
The trough IgG target is above 5 to 7 g/L and above the level at which the patient had breakthrough infection. The dose is individualised by clinical response (infection frequency), not by weight alone. Both routes are therapeutically equivalent for preventing serious bacterial infection; the choice is patient-centred. [1]
Adverse effects of IgRT: infusion reactions (headache, fever, chills, myalgia), aseptic meningitis (especially high-dose IVIG, rapid infusion), thromboembolic events (rare), haemolysis from isohaemagglutinins, and (extremely rarely with modern products) viral or prion transmission. SCIG has fewer systemic reactions but more local site reactions. [1]
Prevention for complement deficiency
- Meningococcal vaccination — quadrivalent ACWY (MenACWY, two doses 8 weeks apart then booster every 3 to 5 years) and serogroup B (two or three doses).
- Pneumococcal and Haemophilus influenzae type b vaccination — overlapping pyogenic risk.
- Standby antibiotic plan — a patient-held dose (e.g., ceftriaxone) to take at the first sign of fever, with instruction to attend hospital immediately.
- Patient education and medical alert — the patient and family must understand the risk and the plan.
- Long-term penicillin prophylaxis — considered on a case-by-case basis, particularly for properdin deficiency (higher mortality per episode).
- Family screening — first-degree relatives tested (CH50) because of the autosomal recessive inheritance. [1]
Surveillance for CVID complications
- Annual lung function and biennial chest CT for bronchiectasis.
- Regular monitoring for autoimmune cytopenia (full blood count) and thyroid disease.
- Vigilance for lymphoma — any new lymphadenopathy, B-symptoms, or unexplained cytopenia warrants urgent investigation (imaging, biopsy).
- Gastric carcinoma surveillance — increased risk; investigate upper gastrointestinal symptoms promptly.
- Annual influenza vaccination and age-appropriate inactivated vaccines. [1]
High-yield exam traps
- The diagnostic-delay trap. CVID is missed for 4 to 7 years because recurrent sinopulmonary infection is attributed to asthma or chronic sinusitis. Measure immunoglobulins in any adult with recurrent sinopulmonary infection, unexplained bronchiectasis, or autoimmune cytopenia.
- The "normal immunoglobulins do not exclude PID" trap. Specific antibody deficiency and complement deficiency have normal immunoglobulins. Measure pneumococcal vaccine response (for SAD) and CH50 (for complement deficiency) when the pattern warrants.
- The "two meningococcal infections means test complement" trap. Any adult with two or more episodes of invasive meningococcal disease must have a CH50. Terminal complement deficiency (C5 to C9) is the cause in a significant proportion.
- The HAE treatment trap. Acute attacks of hereditary angioedema do not respond to adrenaline, antihistamines, or corticosteroids. Give C1 inhibitor concentrate or icatibant. Failing to recognise HAE in the emergency department is a documented source of fatal laryngeal oedema.
- The IgA deficiency transfusion trap. Selective IgA deficiency with anti-IgA antibodies causes anaphylaxis to blood products. In an emergency do not delay transfusion, but for elective work in a patient with a known reaction use washed or IgA-deficient products. Universal IgA screening is not recommended.
- The "isolated IgA deficiency does not need IgRT" trap. IgRT is not indicated for isolated IgA deficiency (it contains minimal IgA and does not correct the mucosal defect). It is indicated for CVID and significant combined antibody deficiency.
- The "CVID is not just infection" trap. About 25 per cent of CVID patients have autoimmune disease; the lymphoma risk is markedly increased. The non-infectious complications drive much of the morbidity and mortality.
- The "rule out HIV first" trap. In any adult with unexplained immunodeficiency, secondary causes (HIV, CLL, myeloma, immunosuppressive drugs, protein-losing states) are far more common than primary defects. An HIV test is mandatory.
- The "CH50 versus AH50" trap. Low CH50 with normal AH50 = classical pathway (consider HAE); low AH50 with normal CH50 = properdin deficiency; both low = terminal pathway (C5 to C9) deficiency or C3 deficiency.
- The "acquired C1 inhibitor deficiency in the older adult" trap. New-onset angioedema in an older adult with low C4, low C1 inhibitor, and low C1q is acquired, driven by a B-cell lymphoproliferative disorder or autoantibody. Investigate for the cause. [1]
Key points for the viva
- Primary immunodeficiency in adults is common (prevalence about 1 in 1,200) and the diagnosis is delayed — measure immunoglobulins in any adult with recurrent sinopulmonary infection, unexplained bronchiectasis, or autoimmune cytopenia.
- The four arms of the immune system predict the organism: humoral (encapsulated bacteria, Giardia), cellular (opportunistic), complement (Neisseria, angioedema), phagocyte (catalase-positive organisms, granulomas).
- CVID is the most common symptomatic PID in adults — low IgG with low IgA and/or IgM, poor vaccine response, recurrent sinopulmonary infection, autoimmune disease in 25 per cent, increased lymphoma risk; treated with immunoglobulin replacement.
- Any adult with two or more meningococcal infections must have a CH50 — terminal complement deficiency (C5 to C9) is the cause in a significant proportion.
- Hereditary angioedema presents with recurrent non-urticarial angioedema and low C4 — acute attacks do NOT respond to adrenaline; give C1 inhibitor concentrate or icatibant.
- Always rule out HIV before attributing hypogammaglobulinaemia to a primary defect — secondary causes are far more common in adults. [1]
References
- [1]Boyle JM, Buckley RH Population prevalence of diagnosed primary immunodeficiency diseases in the United States J Clin Immunol, 2007.PMID 17577648
- [2]Bonilla FA Common variable immunodeficiency Pediatr Res, 2009.PMID 19190529
- [3]Bonilla FA, Barlan I, Chapel H, et al. International Consensus Document (ICON): Common Variable Immunodeficiency Disorders J Allergy Clin Immunol Pract, 2016.PMID 26563668
- [4]Yel L Selective IgA deficiency J Clin Immunol, 2010.PMID 20101521
- [5]Figueroa JE, Densen P. Infectious diseases associated with complement deficiencies Clin Microbiol Rev, 1991.PMID 1889047
- [6]Grumach AS, Kirschfink M. Are complement deficiencies really rare? Overview on prevalence, clinical importance and modern diagnostic approach Mol Immunol, 2014.PMID 25037634
- [7]Craig T, Aygoren-Pursun E, Bork K, et al. WAO Guideline for the Management of Hereditary Angioedema World Allergy Organ J, 2012.PMID 23282420
- [8]Busse PJ, Christiansen SC. Hereditary Angioedema N Engl J Med, 2020.PMID 32187470
- [9]Patel SY, Carbone J, Jolles S. The Expanding Field of Secondary Antibody Deficiency: Causes, Diagnosis, and Management Front Immunol, 2019.PMID 30800120