Phys · haematological
Chronic Leukaemia and Myeloid Neoplasms
Also known as chronic lymphocytic leukaemia · CLL · chronic myeloid leukaemia · CML · BCR-ABL1 · Philadelphia chromosome · imatinib · dasatinib · nilotinib · ponatinib · ibrutinib · venetoclax · obinutuzumab · Richter transformation · myeloproliferative neoplasm · polycythaemia vera · essential thrombocythaemia · myelofibrosis · JAK2 · ruxolitinib · myelodysplastic syndrome · MDS · IPSS-R · azacitidine · luspatercept · lenalidomide
Consultant-physician guide to the chronic leukaemias and related myeloid neoplasms — the indolent clonal disorders that dominate outpatient haematology. Covers chronic lymphocytic leukaemia (the commonest leukaemia in the Western world; clonal CD5-positive, CD19-positive, CD23-positive B cells; Rai and Binet staging; watch and wait for early asymptomatic disease; chemoimmunotherapy with FCR or BR for fit patients; novel agents ibrutinib, venetoclax and obinutuzumab; autoimmune haemolytic anaemia, ITP, infection and Richter transformation), chronic myeloid leukaemia (the BCR-ABL1 t(9;22) Philadelphia chromosome; chronic, accelerated and blast phases; imatinib, dasatinib, nilotinib, bosutinib and ponatinib; BCR-ABL1 transcript monitoring by qPCR; the T315I gatekeeper mutation; treatment-free remission; pregnancy), and overviews of the myeloproliferative neoplasms (polycythaemia vera, essential thrombocythaemia, myelofibrosis) and myelodysplastic syndrome (IPSS-R risk stratification, azacitidine, luspatercept and lenalidomide for del(5q)). Structured for FRACP DWE and DCE, MRCP, and ABIM preparation.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Chronic Leukaemia and Myeloid Neoplasms

The answer first
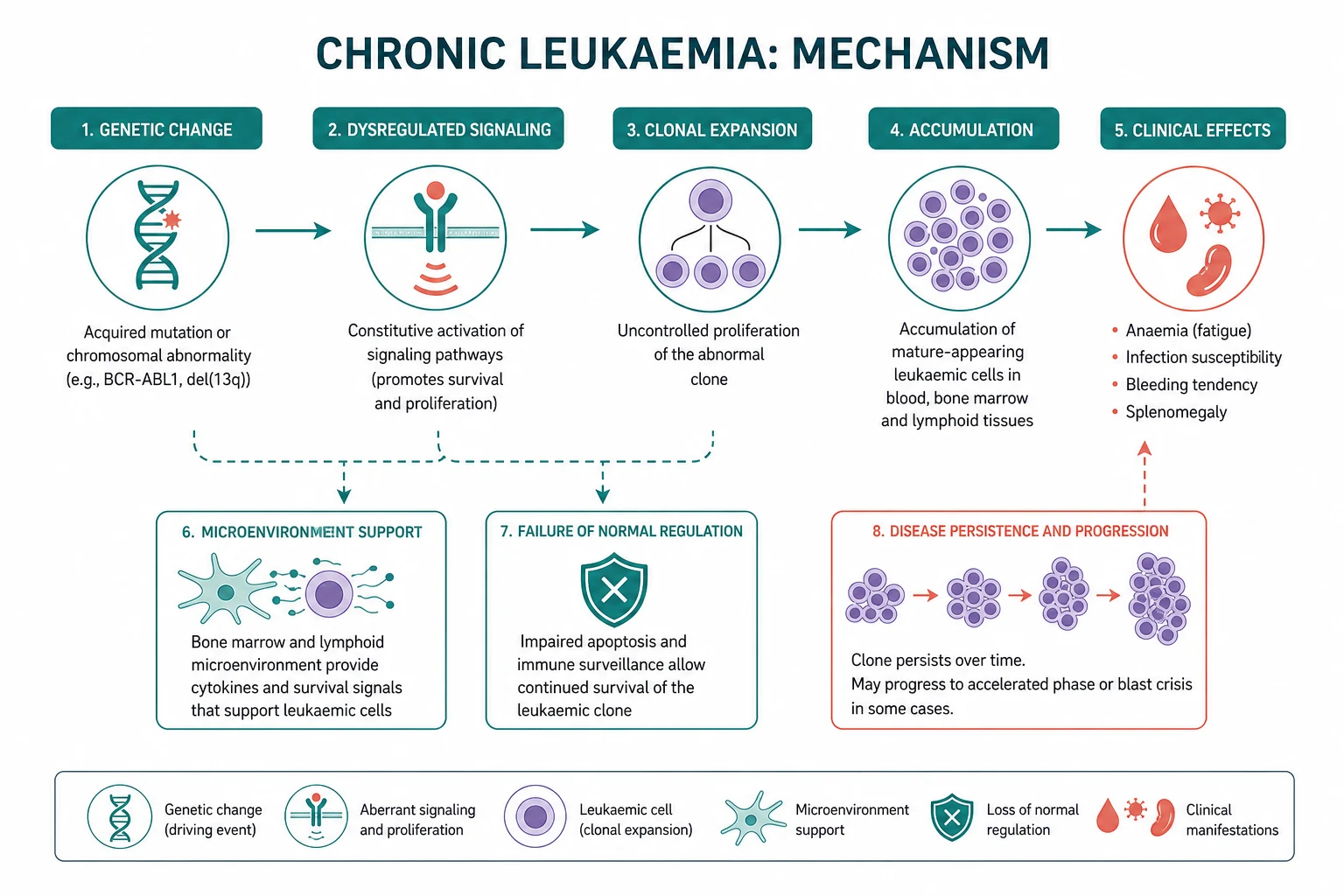
The chronic leukaemias and their related myeloid neoplasms are the indolent clonal disorders of haematopoiesis. Two diseases dominate: chronic lymphocytic leukaemia (CLL) — the commonest leukaemia in the Western world, a clonal B-cell disorder whose management turns on a single principle — and chronic myeloid leukaemia (CML) — the archetypal molecular cancer, defined by the BCR-ABL1 fusion of the Philadelphia chromosome t(9;22) and transformed by tyrosine kinase inhibitors. The myeloproliferative neoplasms (polycythaemia vera, essential thrombocythaemia, myelofibrosis) and the myelodysplastic syndromes complete the family. [1]
Two organising principles carry the whole topic: [1]
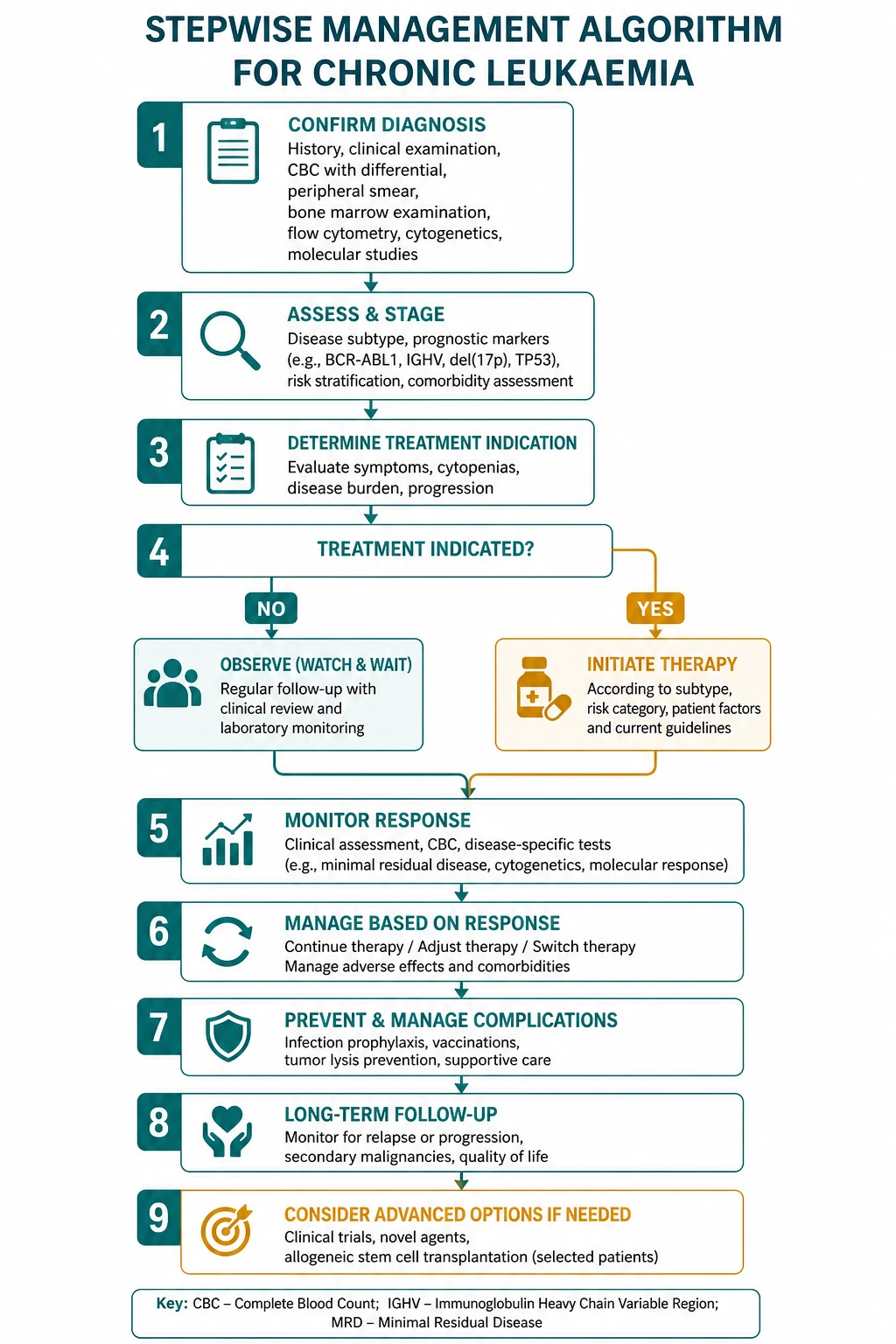
- CLL turns on active versus inactive disease, not on the lymphocyte count. Asymptomatic early-stage CLL is observed (watch and wait) because early treatment confers no survival advantage; therapy is reserved for the iwCLL-defined active disease criteria [10].
- CML turns on the BCR-ABL1 transcript level. Diagnosis, response assessment, resistance detection and the decision to stop therapy are all read off the quantitative PCR for BCR-ABL1 on the International Scale.
The treatment ladders: [1]
- CLL — watch and wait if asymptomatic; for active disease, chemoimmunotherapy (FCR for fit patients with favourable biology, BR for less fit) or a novel agent (ibrutinib, venetoclax with obinutuzumab) for adverse biology or frailty [7][8][9].
- CML — first-line tyrosine kinase inhibitor (imatinib, dasatinib, nilotinib, bosutinib), monitored by serial BCR-ABL1 transcript levels; ponatinib for the T315I mutation; allogeneic transplant only for TKI failure [1][4][5].
- MPN — risk-stratified cytoreduction (phlebotomy and aspirin for polycythaemia vera, hydroxycarbamide or anagrelide for essential thrombocythaemia, ruxolitinib for myelofibrosis).
- MDS — risk-stratified by the IPSS-R: supportive care, erythropoiesis-stimulating agents, luspatercept and lenalidomide for lower-risk; azacitidine and transplant for higher-risk [14][15][16][17].
DWE high-yield: The answer to almost every chronic leukaemia question is "stage or risk-stratify the patient, then treat by fitness and molecular biology." Asymptomatic early CLL is watched. Active CLL gets FCR or BR (fit) or ibrutinib/venetoclax (adverse biology or frail). CML gets a TKI monitored by BCR-ABL1 qPCR. The T315I mutation needs ponatinib. Never treat an asymptomatic patient with early CLL. [1]
Chronic lymphocytic leukaemia (CLL)
Epidemiology and the indolent B-cell clone
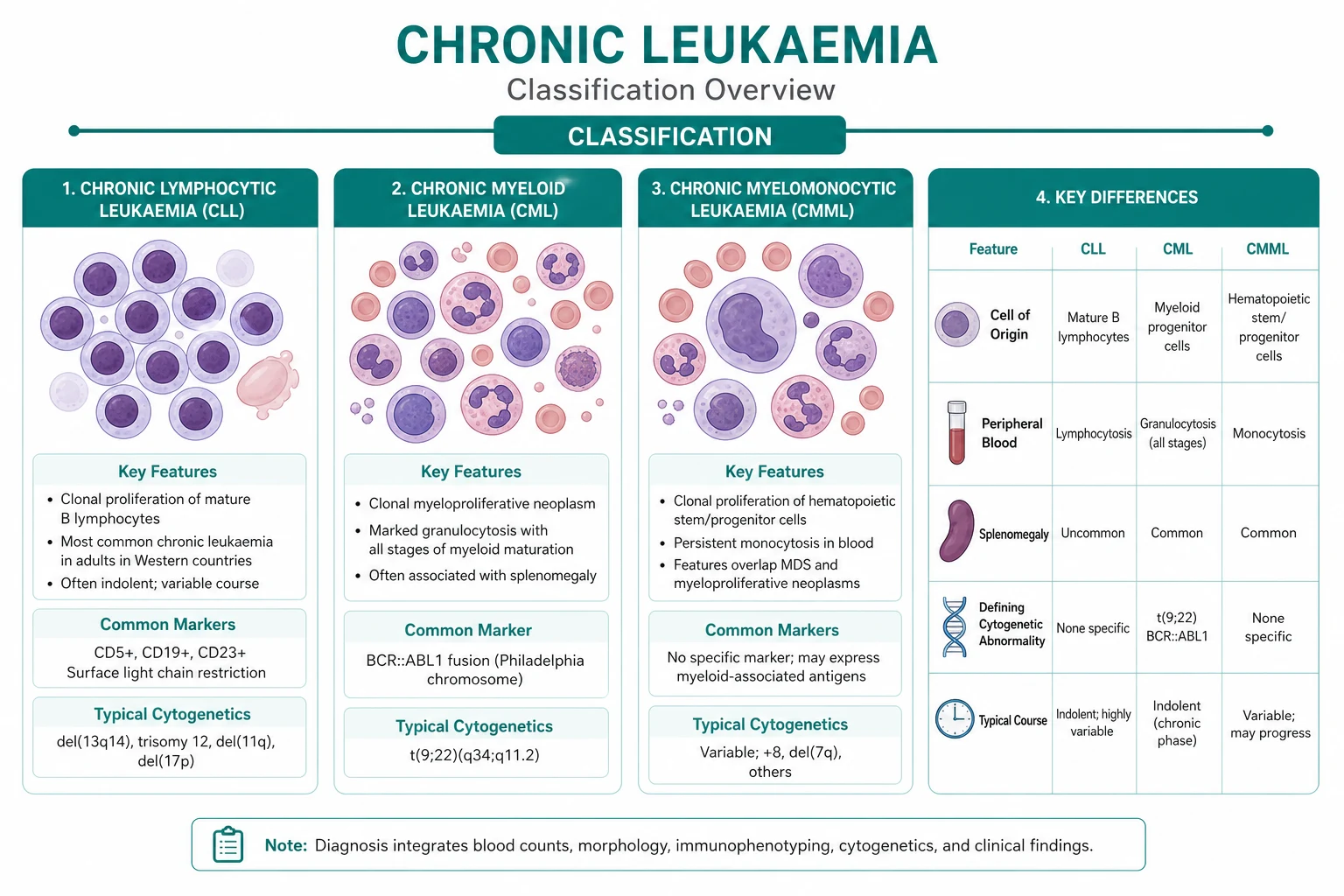
CLL is the most common leukaemia in the Western world, with a median age at diagnosis around 70 years and a slight male predominance. It is a clonal proliferation of mature-appearing but functionally incompetent B lymphocytes that accumulate in the blood, bone marrow, spleen and lymph nodes. The disease runs a highly variable course — some patients live for decades without ever needing therapy, while others follow an aggressive path that resists treatment. [1]
The immunophenotype is the diagnostic fingerprint: CD5-positive, CD19-positive, CD23-positive, with dim surface immunoglobulin and restricted (clonal) kappa or lambda light chain. The CD5 positivity (shared only with mantle cell lymphoma, which is CD23-negative and cyclin-D1-positive) is the key discriminator. Flow cytometry on peripheral blood is usually sufficient for diagnosis — bone marrow biopsy is reserved for unexplained cytopenia, a rising lymphocyte count, or before treatment. [1]
Staging — Rai and Binet
Two systems are in routine use, both based on disease burden and marrow failure rather than on molecular biology: [1]
| Stage | Rai system | Binet system | Median survival |
|---|---|---|---|
| Low risk | Lymphocytosis only (stage 0) | Fewer than 3 involved lymphoid areas, no anaemia or thrombocytopenia (stage A) | Over 10 years |
| Intermediate risk | Lymphadenopathy (I), or hepatosplenomegaly (II) | 3 or more involved areas, no anaemia or thrombocytopenia (stage B) | 5 to 7 years |
| High risk | Anaemia (Hb under 110) (III), or thrombocytopenia (platelets under 100) (IV) | Anaemia (Hb under 100) or thrombocytopenia (under 100) regardless of areas (stage C) | 2 to 3 years |
The stage answers two questions: is there marrow failure (cytopenia), and is the disease disseminated? But the stage does not by itself trigger treatment — a patient with Binet stage A disease and a slowly rising lymphocyte count may be observed for years. [1]
DWE trap: Staging does not determine when to treat. A patient with Binet stage A (early) CLL who is asymptomatic is observed regardless of the absolute lymphocyte count. The decision to treat rests on the iwCLL active-disease criteria, not on a number. [1]
The molecular prognostic markers — biology over stage
What separates an indolent from an aggressive CLL is the molecular biology, captured at diagnosis: [1]
- IGHV mutation status. CLL arising from a post-germinal-centre B cell (with somatic hypermutation of the immunoglobulin heavy-chain variable region — mutated IGHV) behaves indolently; CLL from a naive B cell (unmutated IGHV) is aggressive. This is the single most powerful baseline prognostic marker.
- TP53 disruption — either deletion of 17p (del(17p)) or a TP53 mutation — confers chemoresistance and a poor prognosis. It is present in about 5 to 10 percent at diagnosis and rises with each line of therapy. These patients must not receive chemoimmunotherapy, which fails them.
- FISH cytogenetics stratifies risk: del(17p) and del(11q) (ATM) are adverse; trisomy 12 is intermediate; del(6q) and a normal karyotype or isolated del(13q) are favourable. [1]
Every patient with newly diagnosed CLL should have IGHV mutation status and TP53 (FISH del(17p) plus sequencing) performed, because these direct the first treatment decision [10].
Watch and wait — the principle of no early treatment
The defining management principle of early CLL is that treatment is withheld until the disease becomes active. Multiple randomised trials established that treating asymptomatic early-stage disease (Rai 0 or Binet A) with chlorambucil or chemoimmunotherapy confers no overall survival advantage and only adds toxicity. The patient is monitored — blood counts every 3 to 6 months — and therapy begins only when an iwCLL criterion for active disease is met. [1]
The iwCLL 2018 active-disease criteria are [10]:
- Progressive marrow failure — worsening anaemia or thrombocytopenia (Hb under 100, or platelets under 100).
- Massive (over 6 cm below the costal margin) or progressive splenomegaly.
- Massive or progressive lymphadenopathy (over 10 cm, or rapidly enlarging).
- Progressive lymphocytosis with a rise of over 50 percent over 2 months, or a lymphocyte doubling time under 6 months (after excluding other causes).
- Autoimmune cytopenias (anaemia or thrombocytopenia) poorly responsive to corticosteroids.
- Symptomatic or functional extranodal involvement, or disease-related B-symptoms (fevers, drenching night sweats, weight loss over 10 percent). [1]
The lymphocyte count alone is not an indication — a patient with a lymphocyte count of 200 who is well, with stable counts and no cytopenia or symptoms, is still observed. [1]
DWE high-yield — do not treat early asymptomatic CLL. Watch and wait is the standard for Binet A or Rai 0 disease. Early treatment does not improve survival and causes harm. This is among the most frequently tested facts in haematology. [1]
First-line therapy — chemoimmunotherapy or a novel agent
For fit patients with active CLL and favourable biology (mutated IGHV, intact TP53), the gold-standard chemoimmunotherapy is FCR — fludarabine, cyclophosphamide and rituximab. The CLL8 trial established that adding rituximab to fludarabine and cyclophosphamide improved both progression-free and overall survival (3-year PFS 65 percent with FCR versus 45 percent with FC), and long-term follow-up shows a subset of mutated-IGHV patients achieving durable remission consistent with cure [7].
For less fit patients or those with comorbidity, BR — bendamustine and rituximab is preferred (less myelosuppression and infection than FCR). For the frail elderly, chlorambucil plus obinutuzumab remains a reasonable low-intensity option. [1]
The modern alternative, increasingly first-line for adverse biology, is a novel-agent regimen: [1]
- Ibrutinib — an irreversible Bruton tyrosine kinase (BTK) inhibitor that blocks B-cell receptor signalling. The RESONATE-2 trial showed first-line ibrutinib (with or without obinutuzumab) superior to chlorambucil in older patients, with durable single-agent activity even in TP53-disrupted and unmutated-IGHV disease [8]. It is given continuously until progression. Characteristic toxicities are bleeding (antiplatelet effect), atrial fibrillation, hypertension, infection and rash.
- Venetoclax — a selective BCL-2 inhibitor that restores the intrinsic apoptotic pathway in CLL cells. Combined with rituximab (MURANO, relapsed disease) or obinutuzumab (first-line), it produces high rates of measurable residual disease (MRD) negativity and is given as a time-limited course (typically 12 to 24 months) [9]. Its life-threatening risk is tumour lysis syndrome, managed by the mandatory 5-week ramp-up dosing schedule, TLS risk stratification, hydration, allopurinol or rasburicase, and frequent electrolyte monitoring.
The treatment choice is therefore personalised by fitness and biology: [1]
| Clinical scenario | First-line choice |
|---|---|
| Fit, young, mutated IGHV, intact TP53 | FCR (curative intent in a subset) |
| Less fit, comorbidity | Bendamustine plus rituximab (BR) |
| Frail elderly | Obinutuzumab plus chlorambucil, or venetoclax plus obinutuzumab |
| TP53-disrupted (del(17p)/mutation) or unmutated IGHV | Ibrutinib (plus obinutuzumab) or venetoclax plus obinutuzumab |
Measurable residual disease (MRD)
A deep response — particularly MRD negativity (fewer than 1 CLL cell per 10 000 leucocytes in blood or marrow) — predicts durable remission. The MURANO trial (venetoclax-rituximab) showed that MRD negativity at end of treatment predicted longer progression-free survival [9]. MRD is now an integral response and research endpoint, though it is not yet used routinely to decide when to stop therapy outside trials.
Complications of CLL
CLL is complicated by a predictable set of problems the physician must anticipate: [1]
- Infection — the leading cause of morbidity and mortality. Mechanisms include hypogammaglobulinaemia, impaired T-cell function, and treatment-related immunosuppression (particularly with BTK and BCL-2 inhibitors). Management is vaccination (preferably before immunosuppression), prompt antibiotics, and intravenous immunoglobulin replacement for the patient with recurrent bacterial infection and a low IgG.
- Autoimmune cytopenias — autoimmune haemolytic anaemia (warm IgG, positive direct antiglobulin test) and immune thrombocytopenia occur in 5 to 10 percent, more often in advanced disease. Treat with corticosteroids first-line, rituximab for steroid-refractory disease, and directed CLL therapy if the underlying disease is active.
- Richter transformation — transformation to an aggressive diffuse large B-cell lymphoma (rarely Hodgkin lymphoma) in 2 to 10 percent. Suspect it with rapid lymph-node enlargement, new B-symptoms, a rising LDH and a discordant PET-avid node. Diagnosis requires node biopsy. Prognosis is poor; treatment is with aggressive chemoimmunotherapy (R-CHOP or similar), with a consideration of transplant in responders. [1]
DWE trap: Rapid lymph-node enlargement and a rising LDH in known CLL is Richter transformation until biopsy proves otherwise — do not assume disease progression and re-challenge with the same therapy. [1]
Chronic myeloid leukaemia (CML)
The Philadelphia chromosome and BCR-ABL1
CML is defined by a single molecular lesion: the reciprocal translocation t(9;22)(q34;q11), which fuses the BCR gene on chromosome 22 with the ABL1 tyrosine kinase gene on chromosome 9. The resulting derivative chromosome 22 is the Philadelphia chromosome, and the BCR-ABL1 fusion gene encodes a constitutively active tyrosine kinase that drives uncontrolled proliferation of the myeloid lineage. CML was the first cancer defined by a molecular lesion and the first to be treated with a rationally designed targeted therapy (imatinib) — it is the paradigm of precision oncology. [1]
The disease accounts for about 15 percent of adult leukaemias, with a median age around 60. Most patients (over 90 percent) present in chronic phase; untreated, it progresses through an accelerated phase to a uniformly fatal blast crisis within 3 to 5 years. With tyrosine kinase inhibitor therapy, the median survival now exceeds that of the general population. [1]
The three phases
| Phase | Defining features | Significance |
|---|---|---|
| Chronic | Under 10 percent blasts; controlled disease | The treatable phase; excellent TKI outcomes |
| Accelerated | 10 to 19 percent blasts, or basophilia over 20 percent, or clonal cytogenetic evolution, or treatment resistance | Adverse; signals TKI resistance or progression |
| Blast crisis | 20 percent or more blasts, or extramedullary blast infiltrate (myeloid or lymphoid) | Treated as acute leukaemia; poor prognosis |
Diagnosis
The presentation is often insidious — fatigue, weight loss, early satiety from splenomegaly, or incidental finding on a routine blood count. The blood film shows a markedly elevated white cell count (often over 100) with the full spectrum of myeloid maturation (myelocytes, metamyelocytes, promyelocytes), basophilia (a hallmark), eosinophilia, and often thrombocytosis. The leucocyte alkaline phosphatase (LAP) score is low, distinguishing CML from a leukaemoid reaction (high LAP). [1]
Confirmation requires demonstrating BCR-ABL1 — by conventional cytogenetics (the Philadelphia chromosome on bone marrow karyotype), FISH, or quantitative RT-PCR for the BCR-ABL1 transcript. A baseline marrow establishes the phase (blast percentage) and provides a baseline karyotype. The baseline BCR-ABL1 transcript level sets the reference for monitoring. [1]
First-line therapy — the tyrosine kinase inhibitors
The five TKIs in routine use target the ATP-binding site of BCR-ABL1: [1]
| TKI | Generation | Key toxicity | Note |
|---|---|---|---|
| Imatinib 400 mg daily | First | Oedema, fluid retention, nausea, cytopenias, periorbital oedema | The original; well tolerated, inexpensive |
| Nilotinib 300 mg twice daily | Second | QT prolongation, pancreatitis, hyperglycaemia, vascular events | Fasting requirement; cardiovascular caution |
| Bosutinib 500 mg daily | Second | Diarrhoea, transaminitis | An alternative second-line option |
| Ponatinib 45 mg daily | Third | Arterial and venous thrombosis (major safety concern) | The only TKI active against T315I |
The IRIS trial established imatinib as first-line therapy for chronic-phase CML, with a complete cytogenetic response rate of 87 percent at 5 years and an overall survival of 89 percent [1][2]. The second-generation TKIs achieve faster, deeper molecular responses — DASISION (dasatinib) and ENESTnd (nilotinib) both showed higher rates of early molecular response and major molecular response than imatinib [3][4] — but without a clear overall survival advantage, and at the cost of different toxicities. The choice is individualised: a younger, fitter patient with high-risk disease may prefer a second-generation TKI for deeper response (and the prospect of treatment-free remission); an older patient with cardiovascular comorbidity may be better served by imatinib.
Monitoring by BCR-ABL1 transcript level — the milestones
The cornerstone of CML management is serial quantitative RT-PCR for BCR-ABL1 on the International Scale (IS). Transcript levels are measured at 3, 6, 12 and 18 months, and the response is graded against milestones: [1]
- Early molecular response (EMR) — BCR-ABL1 under 10 percent at 3 months. Failure to reach this predicts poorer outcomes and prompts a review of adherence, interactions, and a TKI switch consideration.
- Major molecular response (MMR) — BCR-ABL1 under 0.1 percent (MR3), typically by 12 months. The goal of first-line therapy.
- Deep molecular response (DMR) — MR4 (under 0.01 percent) or MR4.5 (under 0.0032 percent), sustained for over 2 years. The prerequisite for considering treatment-free remission. [1]
Loss of a previously achieved response (a confirmed rise in BCR-ABL1, or loss of MMR) signals treatment failure and triggers: (1) a check of adherence and drug interactions (the commonest causes), and (2) BCR-ABL1 kinase domain mutation testing to guide a TKI switch [5].
Resistance — the T315I gatekeeper mutation
Acquired resistance to imatinib, dasatinib, nilotinib and bosutinib arises from BCR-ABL1 kinase domain mutations. The most important is the T315I "gatekeeper" mutation (threonine to isoleucine at position 315), which sterically blocks all first- and second-generation TKIs. Only ponatinib (and the newer asciminib) inhibits T315I-positive disease [5]. The PACE trial demonstrated ponatinib's activity in heavily pre-treated and T315I-positive CML, though at the cost of a significant risk of arterial and venous thromboembolism that demands careful cardiovascular risk assessment and dose optimisation.
DWE high-yield — T315I needs ponatinib. When asked which TKI treats a CML patient with the T315I mutation, the answer is ponatinib (or asciminib). Imatinib, dasatinib, nilotinib and bosutinib all fail. This is one of the highest-yield single facts in the chronic leukaemia topic. [1]
Blast crisis
Blast crisis is treated as de novo acute leukaemia (AML or ALL, depending on the blast lineage) — induction chemotherapy plus a TKI, with urgent assessment for allogeneic stem cell transplant in any responder. The prognosis remains poor, with a median survival measured in months, which is why preventing progression through effective chronic-phase TKI therapy is the central goal. [1]
Pregnancy — interferon is safe, TKIs are not
CML may be diagnosed in pregnancy (a minority of patients are young women). The key principle: TKIs are teratogenic and are stopped before conception and throughout pregnancy and breastfeeding. Imatinib, dasatinib and nilotinib are all avoided. The agent that can be used safely in pregnancy is interferon-alpha, which does not cross the placenta. Leukapheresis is reserved for symptomatic leucostasis. Management requires a joint haematology and obstetric plan. [1]
Treatment-free remission (TFR)
A landmark development is the realisation that a carefully selected subset of patients in stable deep molecular response can safely stop the TKI without immediate relapse. The STIM trial (imatinib discontinuation) was the proof of concept: around 40 to 60 percent of well-selected patients maintained a molecular remission off therapy, with relapse (when it occurred) usually within 6 months and reliably regained on restarting the TKI [6]. Eligibility requires: chronic-phase disease, at least 3 years of TKI therapy, and a sustained deep molecular response (MR4 or deeper) for over 2 years. After stopping, BCR-ABL1 is monitored monthly for 6 months, then less frequently, with re-initiation if loss of MMR. TFR is a shared decision weighing the benefit (freedom from lifelong therapy, toxicity and cost) against the monitoring burden and relapse risk.
Allogeneic stem cell transplant — a diminishing role
In the pre-TKI era, allogeneic stem cell transplant was the only curative treatment for CML. With TKIs achieving near-normal life expectancy, transplant is now reserved for TKI failure — T315I-positive disease without ponatinib response, progression to accelerated or blast phase, or intolerance of all available TKIs. Transplant carries substantial treatment-related mortality and chronic graft-versus-host disease, so it is never first-line. [1]
Myeloproliferative neoplasms — overview
The myeloproliferative neoplasms are clonal stem cell disorders producing excess mature blood cells. The three classic "BCR-ABL1-negative" MPNs share a common molecular driver — the JAK2 V617F mutation (a valine-to-phenylalanine substitution at position 617 in the JH2 pseudo-kinase domain of JAK2), identified by James and colleagues in 2005 [11]. JAK2 V617F is found in nearly all (over 95 percent) of polycythaemia vera and in about half to two-thirds of essential thrombocythaemia and primary myelofibrosis.
Polycythaemia vera (PV)
PV is an erythrocytosis driven by JAK2-mutant erythroid precursors that proliferate independently of erythropoietin. It presents with a high haematocrit, thrombosis (cerebral, splanchnic, peripheral), headache, aquagenic pruritus (itching after a warm shower), erythromelalgia, and splenomegaly. The diagnosis requires JAK2 V617F (or the exon 12 variant), a raised red cell mass, and a low serum erythropoietin. [1]
Management is risk-stratified. The single most important intervention is maintaining the haematocrit below 0.45 by phlebotomy — the CYTO-PV trial showed that tighter haematocrit control reduced thrombosis and cardiovascular death. Low-dose aspirin is given unless contraindicated. High-risk disease (age over 60 or prior thrombosis) adds cytoreduction — hydroxycarbamide first-line, switching to ruxolitinib (a JAK1/JAK2 inhibitor) if resistant or intolerant. The RESPONSE trial established ruxolitinib for hydroxycarbamide-resistant or intolerant PV, achieving haematocrit control and spleen reduction [12]. Cardiovascular risk factor control is essential.
Essential thrombocythaemia (ET)
ET presents with a sustained platelet count over 450, with thrombosis (arterial and venous, including microvascular erythromelalgia) or, paradoxically, bleeding (from acquired von Willebrand disease at very high counts). It must be distinguished from reactive thrombocytosis (iron deficiency, inflammation, infection, post-splenectomy, rebound) — a sustained clonal thrombocytosis with the JAK2, CALR or MPL mutation, and exclusion of other causes, secures the diagnosis. [1]
Management is risk-stratified: [1]
- Very low and low risk (under 60, no prior thrombosis) — observation or low-dose aspirin only (especially if JAK2-positive, which raises thrombotic risk).
- Intermediate risk (age, platelet count, cardiovascular risk factors) — aspirin if symptomatic.
- High risk (over 60 or prior thrombosis) — cytoreduction with hydroxycarbamide first-line. In younger patients (to avoid the theoretical leukaemogenicity of hydroxycarbamide) or in pregnancy, anagrelide or interferon are alternatives. Anagrelide acts on megakaryocyte maturation but causes headache, palpitations and fluid retention. [1]
Primary myelofibrosis (MF)
Myelofibrosis is the most aggressive MPN, characterised by bone marrow fibrosis (from cytokine-driven megakaryocyte hyperplasia), extramedullary haematopoiesis with massive splenomegaly, cytopenias, and constitutional symptoms. The blood film is leucoerythroblastic — teardrop cells (dacrocytes), nucleated red cells and immature myeloid precursors. Diagnosis is confirmed by a fibrotic marrow trephine and JAK2 (or CALR/MPL) testing. [1]
Management is risk-stratified by the Dynamic International Prognostic Scoring System (DIPSS)-Plus. Ruxolitinib (COMFORT-I and COMFORT-II) reduces splenomegaly and constitutional symptoms and improves survival, though it does not eliminate the clone [13]. Fedratinib is an alternative JAK inhibitor. Allogeneic stem cell transplant is the only curative option, reserved for selected fit higher-risk patients because of its substantial morbidity and mortality. Supportive care — transfusion, infection management, and managing splenomegaly — is central for the unfit.
DWE high-yield — splanchnic vein thrombosis needs JAK2 testing. Budd-Chiari syndrome and portal or mesenteric vein thrombosis frequently herald an occult myeloproliferative neoplasm. Test JAK2 V617F even when the blood count is normal — a masked PV or early myelofibrosis may present with thrombosis before the count rises. [1]
Myelodysplastic syndrome (MDS) — overview
A clonal disorder of ineffective haematopoiesis
MDS is a clonal stem cell disorder of ineffective, dysplastic haematopoiesis, producing one or more cytopenias (anaemia most often) in an older adult, with a variable risk of transformation to AML. The marrow is typically hypercellular but ineffective — cells are produced but die before release, hence cytopenia with a busy marrow. Dysplastic features on the blood film (macrocytosis, dimorphic red cells, hypogranular or hyposegmented neutrophils) and on the marrow (dysplastic erythroid, granulocytic and megakaryocytic lineages, ring sideroblasts on iron stain) raise the diagnosis, which is confirmed by marrow morphology and cytogenetics. [1]
DWE trap — exclude B12 and folate deficiency first. Megaloblastic anaemia can mimic dysplasia morphologically. Always check B12 and folate before diagnosing MDS on morphology alone; a deficiency is a treatable mimic. [1]
IPSS-R risk stratification
The Revised International Prognostic Scoring System (IPSS-R) is the standard risk tool, combining five cytogenetic categories, marrow blast percentage, haemoglobin, platelet count and neutrophil count into five risk groups — very low, low, intermediate, high, and very high — with median survivals from 8.8 years (very low) to 0.8 years (very high) [14]. The IPSS-R drives the intensity of therapy: lower-risk disease is managed supportively, higher-risk disease escalates to hypomethylating therapy and transplant consideration.
Lower-risk MDS — supportive and targeted
For lower-risk disease (IPSS-R very low, low, and selected intermediate), the goal is symptom control and transfusion independence: [1]
- Supportive care — red cell transfusion for symptomatic anaemia, platelet transfusion for bleeding or prophylactically, and infection management.
- Erythropoiesis-stimulating agents (ESAs) — for symptomatic anaemia with a low endogenous serum erythropoietin (under 500), ESA therapy achieves transfusion independence in a meaningful subset.
- Luspatercept — a recombinant fusion protein (a TGF-beta superfamily ligand trap) that enhances late-stage erythroid maturation. The MEDALIST trial showed that luspatercept achieved transfusion independence in 38 percent versus 13 percent with placebo in transfusion-dependent lower-risk MDS with ring sideroblasts [17].
- Lenalidomide for del(5q) disease — an isolated interstitial deletion of chromosome 5q defines a distinct MDS subtype with macrocytic anaemia, a normal or high platelet count and hypolobulated megakaryocytes. Lenalidomide produces transfusion independence in around two-thirds and cytogenetic response in around three-quarters, a remarkable targeted effect [16].
- Iron chelation — for the chronically transfused patient with a rising ferritin, to prevent iron-overload end-organ damage (liver, cardiac, endocrine).
Higher-risk MDS — hypomethylating therapy and transplant
For higher-risk disease (IPSS-R high and very high, and selected intermediate-2), therapy aims to modify the disease course: [1]
- Azacitidine (or decitabine) — a hypomethylating agent. The AZA-001 trial established that azacitidine improves overall survival (median 24.5 versus 15.0 months) compared with conventional care in higher-risk MDS [15]. It is the standard first-line for patients not proceeding directly to transplant.
- Allogeneic stem cell transplant — the only curative modality, reserved for fit patients with higher-risk disease. Selection balances the patient's age, comorbidity (often by a transplant comorbidity index) and the IPSS-R against the substantial treatment-related mortality and graft-versus-host disease. Many higher-risk MDS patients are not transplant candidates, in whom azacitidine is the mainstay.
- Supportive and palliative care — for the frail or transplant-ineligible patient, transfusion support, infection management, and honest prognostic discussion are central.
The common exam traps
- Treating asymptomatic early CLL. Watch and wait is the standard for Binet A or Rai 0 disease. Early treatment confers no survival benefit and only adds toxicity — the lymphocyte count alone is never an indication.
- Choosing the wrong TKI for T315I. Only ponatinib (or asciminib) inhibits the T315I gatekeeper mutation. Imatinib, dasatinib, nilotinib and bosutinib all fail.
- Missing TP53 disruption in CLL. Patients with del(17p) or a TP53 mutation must not receive chemoimmunotherapy (which fails) — they go straight to a novel agent (ibrutinib or venetoclax-based).
- Treating a rising BCR-ABL1 as simple relapse. Before escalating, check adherence and drug interactions (CYP3A4 inhibitors), then test for a kinase domain mutation. Non-adherence is the commonest cause of loss of response.
- Using a TKI in pregnancy. TKIs are teratogenic — the safe agent is interferon-alpha. Stop the TKI before conception and throughout pregnancy.
- Diagnosing MDS on morphology alone. Always exclude B12 and folate deficiency first — megaloblastic change mimics dysplasia.
- Missing an occult MPN in splanchnic vein thrombosis. Test JAK2 V617F even with a normal blood count in Budd-Chiari or portal vein thrombosis.
- Stopping venetoclax without TLS precautions. The 5-week ramp-up and aggressive TLS prophylaxis are mandatory, particularly in bulky or high-lymphocyte-count CLL. [1]
Regional guideline anchoring
- ANZ (HSANZ, eviQ) — broadly aligned with ELN and NCCN, with local drug access and protocol variations documented in the eviQ chronic leukaemia protocols.
- ELN (European LeukemiaNet) — the international authority on CML response milestones and resistance management.
- NCCN / ESMO — the standard for MPN and MDS risk-stratified management.
- BSH (British Society for Haematology) — UK-aligned guidance frequently referenced in MRCP contexts. [1]
Drug dosing is verified against current guidelines. Imatinib 400 mg once daily; dasatinib 100 mg once daily; nilotinib 300 mg twice daily (fasting); bosutinib 500 mg once daily; ponatinib 45 mg once daily (with dose reduction once response achieved); ibrutinib 420 mg once daily; venetoclax ramp-up to 400 mg daily (with rituximab) or 400 mg with obinutuzumab; azacitidine 75 mg per square metre subcutaneously for 7 days every 28 days; lenalidomide 10 mg daily for 21 days per 28-day cycle in del(5q) MDS; ruxolitinib 15 to 20 mg twice daily for myelofibrosis and 10 mg twice daily for PV. [1]
Summary
Chronic leukaemia and the related myeloid neoplasms are the indolent clonal disorders of haematopoiesis, each anchored to a defining molecular lesion. Chronic lymphocytic leukaemia is the commonest leukaemia in the Western world — a clonal CD5-positive, CD19-positive, CD23-positive B-cell disorder in which the central principle is that asymptomatic early disease (Binet A, Rai 0) is observed, because early treatment confers no survival advantage; therapy is reserved for iwCLL-defined active disease, and is chosen by fitness and biology — FCR for fit patients with favourable biology, BR for the less fit, and a novel agent (ibrutinib or venetoclax with obinutuzumab) for TP53-disrupted or frail disease. Complications include infection, autoimmune haemolytic anaemia and ITP, and Richter transformation to diffuse large B-cell lymphoma. Chronic myeloid leukaemia is defined by the BCR-ABL1 t(9;22) Philadelphia chromosome, managed with a tyrosine kinase inhibitor (imatinib, dasatinib, nilotinib, bosutinib) monitored by serial BCR-ABL1 transcript levels at 3, 6, 12 and 18 months, with ponatinib for the T315I mutation, interferon as the safe agent in pregnancy, and treatment-free remission attempted in sustained deep molecular response. The myeloproliferative neoplasms are risk-stratified — polycythaemia vera (phlebotomy, aspirin, hydroxycarbamide or ruxolitinib), essential thrombocythaemia (aspirin and hydroxycarbamide or anagrelide for high risk), and myelofibrosis (ruxolitinib or fedratinib, transplant for selected fit patients). Myelodysplastic syndrome is risk-stratified by the IPSS-R — lower-risk disease receives supportive care, ESAs, luspatercept and lenalidomide for del(5q); higher-risk disease receives azacitidine and consideration of allogeneic stem cell transplant. [1]
One-line answer for the viva: "I manage chronic leukaemia by the molecular biology and the patient's fitness. In CLL, asymptomatic early disease is watched — early treatment adds no survival benefit. For active CLL I use FCR for fit patients with favourable biology, BR for the less fit, and a novel agent — ibrutinib or venetoclax with obinutuzumab — for TP53-disrupted or frail disease, watching for infection, autoimmune cytopenia and Richter transformation. In CML, I start a tyrosine kinase inhibitor and monitor the BCR-ABL1 transcript at 3, 6, 12 and 18 months; I use ponatinib for the T315I mutation, interferon in pregnancy, and consider treatment-free remission in sustained deep molecular response. I risk-stratify the myeloproliferative neoplasms and MDS, treating PV with phlebotomy and aspirin, ET and high-risk PV with cytoreduction, myelofibrosis with ruxolitinib, and MDS by the IPSS-R — supportive care, ESAs, luspatercept and lenalidomide for lower-risk, and azacitidine with transplant consideration for higher-risk disease." [1]
Sources
O'Brien SG, Guilhot F, Larson RA, et al. Imatinib compared with interferon and low-dose cytarabine for newly diagnosed chronic-phase CML (IRIS). N Engl J Med 2003;348:994-1004 [1]; Druker BJ, Guilhot F, O'Brien SG, et al. Five-year follow-up of patients receiving imatinib for CML. N Engl J Med 2006;355:2408-2417 [2]; Saglio G, Kim DW, Issaragrisil S, et al. Nilotinib versus imatinib for newly diagnosed CML (ENESTnd). N Engl J Med 2010;362:2251-2259 [3]; Kantarjian H, Shah NP, Hochhaus A, et al. Dasatinib versus imatinib in newly diagnosed chronic-phase CML (DASISION). N Engl J Med 2010;362:2260-2270 [4]; Cortes JE, Kim DW, Pinilla-Ibarz J, et al. Ponatinib in Philadelphia chromosome-positive leukemias (PACE). N Engl J Med 2013;369:1783-1796 [5]; Mahon FX, Rea D, Guilhot J, et al. Discontinuation of imatinib in CML (STIM). Lancet Oncol 2010;11:1029-1035 [6]; Hallek M, Fingerle-Rowson G, Fink AM, et al. Addition of rituximab to fludarabine and cyclophosphamide in CLL (CLL8). Lancet 2010;376:1164-1174 [7]; Burger JA, Tedeschi A, Barr PM, et al. Ibrutinib as initial therapy for CLL (RESONATE-2). N Engl J Med 2015;373:2425-2437 [8]; Seymour JF, Kipps TJ, Eichhorst B, et al. Venetoclax-rituximab in relapsed/refractory CLL (MURANO). N Engl J Med 2018;378:1107-1120 [9]; Hallek M, Cheson BD, Catovsky D, et al. iwCLL guidelines for diagnosis, treatment and response assessment of CLL. Blood 2018;131:2745-2760 [10]; James C, Ugo V, Le Couedic JP, et al. A unique clonal JAK2 mutation causes polycythaemia vera. Nature 2005;434:1144-1148 [11]; Vannucchi AM, Kiladjian JJ, Griesshammer M, et al. Ruxolitinib versus standard therapy for polycythemia vera (RESPONSE). N Engl J Med 2015;372:426-435 [12]; Verstovsek S, Mesa RA, Gotlib J, et al. Ruxolitinib for myelofibrosis (COMFORT-I). N Engl J Med 2012;366:799-807 [13]; Greenberg PL, Tuechler H, Schanz J, et al. Revised IPSS for MDS (IPSS-R). Blood 2012;120:2454-2465 [14]; Fenaux P, Mufti GJ, Hellstrom-Lindberg E, et al. Azacitidine in higher-risk MDS (AZA-001). Lancet Oncol 2009;10:223-232 [15]; List A, Dewald G, Bennett J, et al. Lenalidomide in the MDS with chromosome 5q deletion. N Engl J Med 2006;355:1456-1465 [16]; Fenaux P, Platzbecker U, Mufti GJ, et al. Luspatercept in lower-risk MDS (MEDALIST). N Engl J Med 2020;382:140-151 [17].
European LeukemiaNet recommendations for CML; NCCN Guidelines for CML and CLL; British Society for Haematology guidelines; eviQ chronic leukaemia protocols. [1]
References
- [1]O'Brien SG, Guilhot F, Larson RA, et al. Imatinib compared with interferon and low-dose cytarabine for newly diagnosed chronic-phase chronic myeloid leukemia N Engl J Med, 2003.PMID 12637609
- [2]Druker BJ, Guilhot F, O'Brien SG, et al. Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia N Engl J Med, 2006.PMID 17151364
- [3]Saglio G, Kim DW, Issaragrisil S, et al. Nilotinib versus imatinib for newly diagnosed chronic myeloid leukemia N Engl J Med, 2010.PMID 20525993
- [4]Kantarjian H, Shah NP, Hochhaus A, et al. Dasatinib versus imatinib in newly diagnosed chronic-phase chronic myeloid leukemia N Engl J Med, 2010.PMID 20525995
- [5]Cortes JE, Kim DW, Pinilla-Ibarz J, et al. A phase 2 trial of ponatinib in Philadelphia chromosome-positive leukemias N Engl J Med, 2013.PMID 24180494
- [6]Mahon FX, Rea D, Guilhot J, et al. Discontinuation of imatinib in patients with chronic myeloid leukaemia who have maintained complete molecular remission for at least 2 years: the prospective, multicentre Stop Imatinib (STIM) trial Lancet Oncol, 2010.PMID 20965785
- [7]Hallek M, Fingerle-Rowson G, Fink AM, et al. Addition of rituximab to fludarabine and cyclophosphamide in patients with chronic lymphocytic leukaemia: a randomised, open-label, phase 3 trial Lancet, 2010.PMID 20888994
- [8]Burger JA, Tedeschi A, Barr PM, et al. Ibrutinib as Initial Therapy for Patients with Chronic Lymphocytic Leukemia N Engl J Med, 2015.PMID 26639149
- [9]Seymour JF, Kipps TJ, Eichhorst B, et al. Venetoclax-Rituximab in Relapsed or Refractory Chronic Lymphocytic Leukemia N Engl J Med, 2018.PMID 29562156
- [10]Hallek M, Cheson BD, Catovsky D, et al. iwCLL guidelines for diagnosis, indications for treatment, response assessment, and supportive management of CLL Blood, 2018.PMID 29540348
- [11]James C, Ugo V, Le Couedic JP, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera Nature, 2005.PMID 15793561
- [12]Vannucchi AM, Kiladjian JJ, Griesshammer M, et al. Ruxolitinib versus standard therapy for the treatment of polycythemia vera N Engl J Med, 2015.PMID 25629741
- [13]Verstovsek S, Mesa RA, Gotlib J, et al. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis N Engl J Med, 2012.PMID 22375971
- [14]Greenberg PL, Tuechler H, Schanz J, et al. Revised international prognostic scoring system for myelodysplastic syndromes Blood, 2012.PMID 22740453
- [15]Fenaux P, Mufti GJ, Hellstrom-Lindberg E, et al. Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher-risk myelodysplastic syndromes: a randomised, open-label, phase III study Lancet Oncol, 2009.PMID 19230772
- [16]List A, Dewald G, Bennett J, et al. Lenalidomide in the myelodysplastic syndrome with chromosome 5q deletion N Engl J Med, 2006.PMID 17021321
- [17]Fenaux P, Platzbecker U, Mufti GJ, et al. Luspatercept in Patients with Lower-Risk Myelodysplastic Syndromes N Engl J Med, 2020.PMID 31914241