Phys · haematological
Coagulation Disorders — Bleeding, Thrombosis and the Abnormal Coagulation Screen
Also known as coagulopathy · bleeding disorder · abnormal coagulation screen · prolonged aPTT · prolonged PT · elevated INR · haemophilia A · haemophilia B · Christmas disease · factor VIII deficiency · factor IX deficiency · von Willebrand disease · vWD · disseminated intravascular coagulation · DIC · antiphospholipid syndrome · APS · lupus anticoagulant · immune thrombocytopenia · ITP · heparin-induced thrombocytopenia · HIT · thrombotic thrombocytopenic purpura · TTP · mixing study · emicizumab · desmopressin · DDAVP
Consultant-physician-depth guide to coagulation disorders for FRACP DWE and DCE. Covers the coagulation cascade (intrinsic, extrinsic, common pathways), inherited bleeding disorders (haemophilia A, haemophilia B, von Willebrand disease), acquired coagulopathies (DIC, liver disease, warfarin, massive transfusion), antiphospholipid syndrome, thrombocytopenia (ITP, HIT, TTP/HUS), mixing study interpretation, and the systematic investigation of the abnormal coagulation screen.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Coagulation Disorders — Bleeding, Thrombosis and the Abnormal Coagulation Screen
The answer first
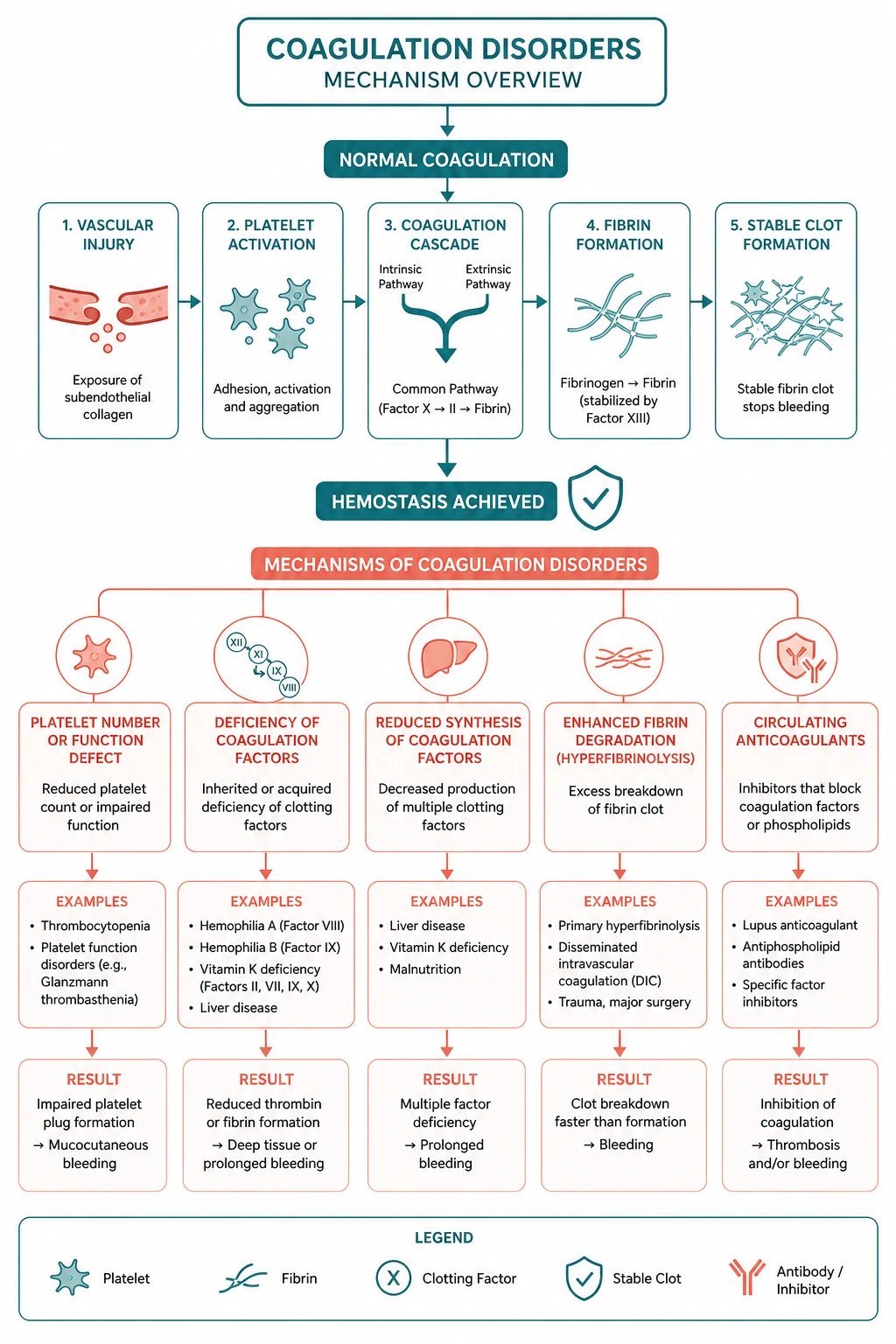
Coagulation disorders fall into three clinical questions the examiner asks: Is the patient bleeding, clotting, or does the lab look odd? The registrar who answers all three will pass this station. The one who chases a single prolonged clotting time without asking whether the patient is bleeding or thrombosing will fail it. [1]
The framework is built on three axes: [1]
- Platelets versus coagulation factors. Platelet bleeding is mucocutaneous — petechiae, epistaxis, menorrhagia, GI bleeding. Factor bleeding is deep — haemarthrosis, muscle bleeds, intracranial. The platelet count and the coagulation screen split these two worlds.
- Bleeding versus thrombosis. A prolonged aPTT from haemophilia causes bleeding. A prolonged aPTT from a lupus anticoagulant paradoxically causes thrombosis. The mixing study separates these. Never assume a prolonged clotting time means a bleeding tendency.
- Inherited versus acquired. A lifelong history of post-surgical or dental bleeding points to haemophilia or von Willebrand disease. A new coagulopathy in a sick patient points to DIC, liver disease, or warfarin. [1]
The single most tested principle for the exam: the mixing study is the gateway to the abnormal coagulation screen. Mix the patient's plasma with normal pooled plasma. If the prolonged clotting time corrects, a factor deficiency is present. If it does not correct, an inhibitor is present — and the next question is whether that inhibitor causes bleeding (a factor inhibitor, like a haemophilia inhibitor) or thrombosis (a lupus anticoagulant). [1]
Overview — the coagulation cascade in two minutes

The coagulation cascade is not a textbook schematic to memorise — it is a clinical map that tells you which test is abnormal and therefore where the problem lies. Learn it as two arms converging into one. [1]
The extrinsic pathway — tissue factor and factor VII
Vascular injury exposes tissue factor (a transmembrane protein on subendothelial cells). Tissue factor binds factor VII, activating it to VIIa. The tissue factor–VIIa complex directly activates factor X to Xa, the start of the common pathway. This is the primary initiator of coagulation in vivo — the extrinsic pathway is the "first responder" that gets the clot started [1].
The test that measures the extrinsic pathway is the prothrombin time (PT) and its standardised form the INR. If the PT is prolonged, the problem is in factor VII (the shortest half-life factor, so the first to fall in liver disease or warfarin therapy) or the common pathway (factors X, V, II, I). [1]
The intrinsic pathway — contact activation and factor VIII/IX
The intrinsic pathway begins with contact activation — factor XII binds to a negatively charged surface (in the test tube, glass or kaolin; in vivo, polyphosphates and collagen). Factor XII activates XI, XI activates IX, and IX (with its cofactor VIII) activates X. This is the "amplification" arm of the cascade — it sustains and amplifies the thrombin burst that the extrinsic pathway started [1].
The test that measures the intrinsic pathway is the activated partial thromboplastin time (aPTT). If the aPTT is prolonged, the problem is in factors XII, XI, IX, or VIII (or the common pathway). Factor XII deficiency prolongs the aPTT but causes no clinical bleeding — it is the classic "lab looks odd, patient is fine" scenario and a high-yield MCQ discriminator. [1]
The common pathway — thrombin and fibrin
Both arms converge at factor X. Factor Xa, with its cofactor Va, converts prothrombin (factor II) to thrombin. Thrombin is the master enzyme of coagulation: it converts fibrinogen to fibrin, activates platelets, activates factors V and VIII (positive feedback), and activates factor XIII (which cross-links the fibrin clot and stabilises it). Both the PT and the aPTT are prolonged by common pathway deficiencies (fibrinogen, prothrombin, factors V, X). [1]
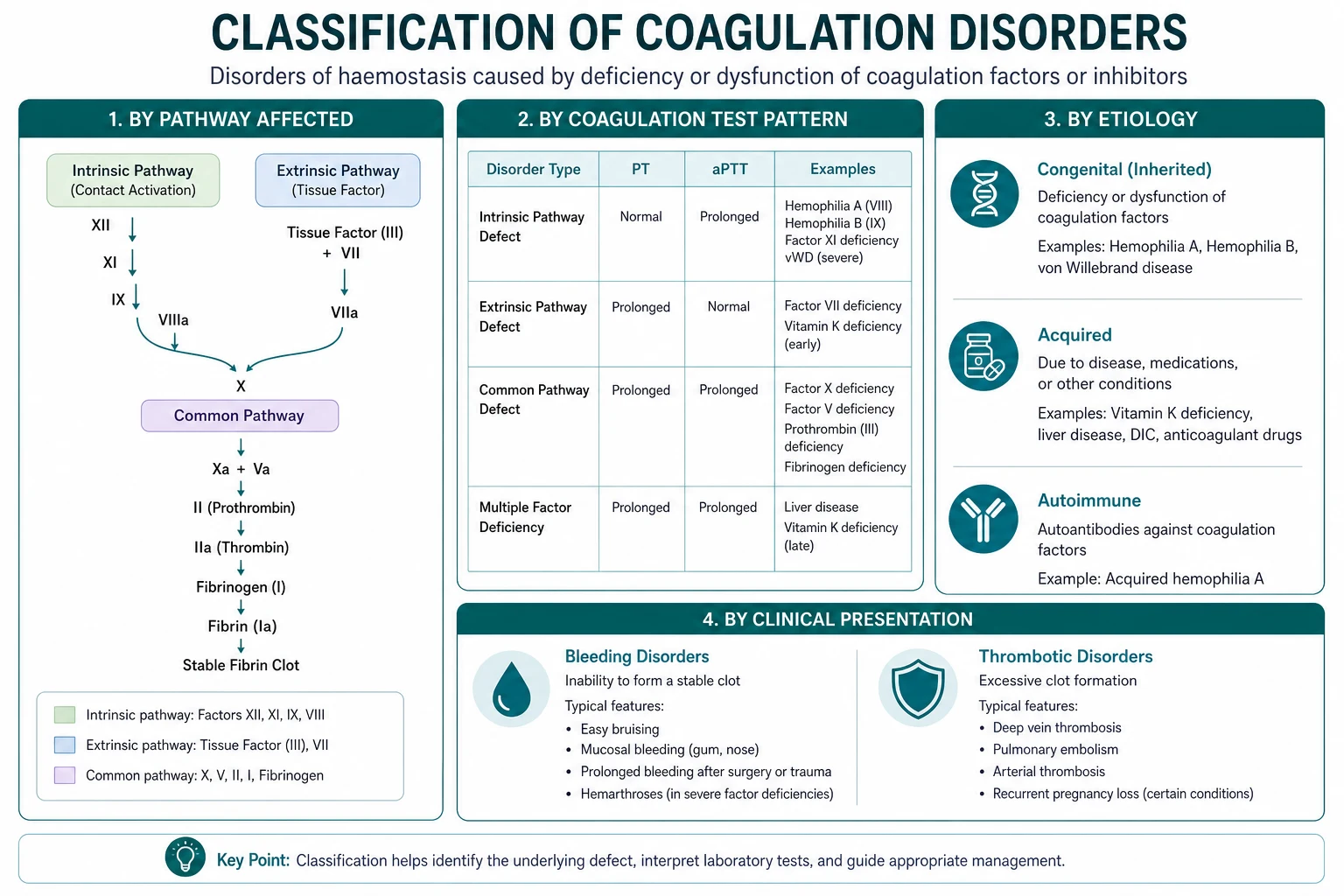
Coagulation screen: what each test measures
Why the cascade matters clinically
The cascade map lets you read a coagulation screen by inspection: [1]
| Pattern | PT | aPTT | Platelets | Think |
|---|---|---|---|---|
| Isolated PT prolongation | Prolonged | Normal | Normal | Factor VII deficiency — early liver disease, early warfarin, vitamin K deficiency |
| Isolated aPTT prolongation | Normal | Prolonged | Normal | Haemophilia (VIII or IX), vWD, factor XII deficiency, lupus anticoagulant, heparin contamination |
| Both prolonged | Prolonged | Prolonged | Normal or low | DIC, liver disease, massive transfusion, severe vitamin K deficiency, warfarin overdose |
| Both prolonged, platelets low | Prolonged | Prolonged | Low | DIC, massive transfusion with dilutional thrombocytopenia |
| Normal PT and aPTT, platelets low | Normal | Normal | Low | ITP, TTP/HUS, drug-induced thrombocytopenia, hypersplenism |
Inherited bleeding disorders
Haemophilia A — factor VIII deficiency
Haemophilia A is an X-linked recessive deficiency of factor VIII. It is the second most common inherited bleeding disorder after von Willebrand disease, with a prevalence of approximately 1 in 5,000 male births. One-third of cases arise from a new mutation — a family history is supportive but not required [1].
Severity is determined by the factor VIII level, and this is the single most important clinical fact: [1]
| Severity | Factor VIII level (IU/dL) | Clinical bleeding |
|---|---|---|
| Severe | Less than 1 | Spontaneous haemarthrosis, muscle bleeds, intracranial haemorrhage |
| Moderate | 1 to 5 | Bleeding after minor trauma or procedures; occasional spontaneous bleeds |
| Mild | 5 to 40 | Bleeding only after surgery, dental work, or significant trauma; may be diagnosed late |
The hallmark of severe haemophilia is the haemarthrosis — bleeding into joints (knees, ankles, elbows). Repeated haemarthroses cause a crippling arthropathy: chronic synovitis, cartilage destruction, joint deformity. Muscle bleeds (especially iliopsoas) cause pain, swelling, and compartment syndrome. Intracranial haemorrhage is the leading cause of death in severe haemophilia [1].
Investigation. The aPTT is prolonged (because factor VIII is in the intrinsic pathway). The PT is normal. The mixing study corrects (a factor deficiency, not an inhibitor). The definitive test is a factor VIII assay — the level determines severity and guides treatment. Always check for an inhibitor (a Betheshda assay) in patients who fail to respond to factor replacement. [1]
Treatment of haemophilia A: [1]
- Severe haemophilia A — prophylaxis. The standard of care has shifted from on-demand factor to prophylaxis. Emicizumab — a bispecific antibody that bridges factor IXa and factor X, effectively replacing the function of factor VIII — is given subcutaneously once weekly, every two weeks, or every four weeks. It transformed haemophilia A management by reducing annualised bleeding rates by over 80 per cent in the HAVEN trials, including in patients with inhibitors [2].
- Acute bleeds (if not on emicizumab or breakthrough). Recombinant factor VIII concentrate, dosed to achieve the target factor level: minor bleeds need 20-40 IU/dL, major bleeds (joint, muscle, intracranial) need 80-100 IU/dL. The rule of thumb: one unit per kilogram of factor VIII raises the level by approximately 2 IU/dL.
- Mild haemophilia A. Desmopressin (DDAVP) 0.3 micrograms per kilogram IV or subcutaneously releases stored factor VIII and vWF from endothelial Weibel-Palade bodies, roughly doubling the factor VIII level. A DDAVP challenge test confirms responsiveness before relying on it. Avoid in young children (hyponatraemia) and cardiovascular disease.
- Inhibitor development. Approximately 30 per cent of severe haemophilia A patients develop alloantibodies against factor VIII. The inhibitor neutralises infused factor, so bleeding is uncontrolled by standard doses. Treatment of acute bleeding with an inhibitor: bypassing agents (recombinant activated factor VII, or activated prothrombin complex concentrate). Emicizumab prophylaxis has largely solved the day-to-day problem of the inhibitor patient [2].
DCE examiner trap: A young man presents with a swollen painful knee and a prolonged aPTT. The examiner asks: what is the most likely diagnosis? The answer is haemophilia A (factor VIII deficiency) — the most common inherited factor deficiency that causes haemarthrosis. Factor IX deficiency (haemophilia B) is clinically identical. The discriminator is the factor assay, not the clinical picture. [1]
Haemophilia B — Christmas disease (factor IX deficiency)
Haemophilia B is an X-linked recessive deficiency of factor IX. It is approximately five times less common than haemophilia A (prevalence 1 in 25,000 to 30,000 male births). The clinical presentation is identical to haemophilia A — the same severity classification by factor level, the same pattern of haemarthrosis and deep tissue bleeding, the same X-linked inheritance [1].
Investigation. Prolonged aPTT, normal PT, mixing study corrects, and a factor IX assay confirms the diagnosis. Factor IX deficiency must be distinguished from factor VIII deficiency before treatment because the treatments differ — giving factor VIII to a haemophilia B patient does nothing. [1]
Treatment. Recombinant factor IX concentrate for acute bleeds and prophylaxis. Factor IX has a longer half-life (18-24 hours) than factor VIII (12 hours), so dosing is less frequent. Extended half-life factor IX products (PEGylated or Fc-fusion) allow once-weekly prophylaxis. DDAVP is not effective in haemophilia B (it releases factor VIII and vWF, not factor IX). [1]
Von Willebrand disease (vWD) — the most common inherited bleeding disorder
Von Willebrand disease is the most common inherited bleeding disorder, with a prevalence of approximately 1 per cent (though many cases are mild and undiagnosed). It is caused by a deficiency or dysfunction of von Willebrand factor (vWF), a large multimeric glycoprotein with two functions: it mediates platelet adhesion to the subendothelium at sites of vascular injury (primary haemostasis), and it is the carrier protein for factor VIII in the circulation, protecting it from premature clearance [3].
Clinical presentation. Unlike haemophilia, which causes deep tissue bleeding (haemarthrosis), vWD causes mucocutaneous bleeding: epistaxis, gum bleeding, menorrhagia, easy bruising, and prolonged bleeding after dental work or surgery. Heavy menstrual bleeding is often the presenting complaint in women. Gastrointestinal bleeding from angiodysplasia occurs in some subtypes. Bleeding severity varies enormously — many patients are asymptomatic until challenged by surgery or menstruation. [1]
Classification (three types): [1]
| Type | Mechanism | Inheritance | Severity | Treatment |
|---|---|---|---|---|
| Type 1 (75 per cent) | Partial quantitative deficiency of vWF | Autosomal dominant | Mild to moderate | DDAVP; vWF concentrate if unresponsive |
| Type 2 (20-25 per cent) | Qualitative defect (dysfunctional vWF) | Usually autosomal dominant | Variable | vWF concentrate (DDAVP may worsen type 2B) |
| Type 3 (rare) | Complete absence of vWF | Autosomal recessive | Severe | vWF concentrate |
Type 2 has four subtypes. Type 2B is the exam favourite: the vWF has enhanced binding to platelets, causing spontaneous platelet aggregation and thrombocytopenia. Giving DDAVP to a type 2B patient worsens the thrombocytopenia — a dangerous and easily missed trap [3].
Investigation. The coagulation screen is often normal or shows a mildly prolonged aPTT (from reduced factor VIII, since vWF is its carrier). The diagnostic tests are: [1]
- vWF antigen — measures the quantity of vWF.
- vWF activity (ristocetin cofactor assay, vWF:RCo) — measures the function of vWF. A low ratio of activity to antigen indicates type 2 (dysfunctional vWF).
- Factor VIII level — may be low because vWF is not there to carry it.
- Blood group effect — vWF levels are 25 per cent lower in blood group O individuals; this is a physiological variant, not a disease. [1]
Treatment:
- Type 1 (and some type 2): DDAVP 0.3 micrograms per kilgram IV or subcutaneously, releasing stored vWF and factor VIII from endothelium. A DDAVP trial confirms responsiveness before relying on it for surgery.
- Type 3, type 2B, and non-responsive type 1/2: vWF-containing concentrate (plasma-derived vWF plus factor VIII). Recombinant vWF is now available.
- Adjuncts: tranexamic acid (antifibrinolytic) for dental procedures and menorrhagia; hormonal therapy (oral contraceptive pill or Mirena IUD) for menorrhagia. [1]
Exam pearl: The three-part discriminator between the inherited bleeding disorders: haemophilia A and B cause deep bleeds (haemarthrosis) with an isolated prolonged aPTT; vWD causes mucocutaneous bleeds with a normal or mildly prolonged aPTT and abnormal vWF studies; and the inheritance is X-linked in haemophilia but autosomal in vWD. [1]

Acquired coagulopathy
Disseminated intravascular coagulation (DIC)
DIC is not a disease — it is a syndrome of pathological, systemic activation of coagulation triggered by an underlying condition. Tissue factor is released into the circulation, generating widespread thrombin production, which deposits fibrin throughout the microvasculature. This consumes platelets and coagulation factors faster than the liver and marrow can replace them. The simultaneous activation of fibrinolysis (via plasmin) produces fibrin degradation products (D-dimer). The result is a paradox of microvascular thrombosis and consumption coagulopathy — the patient bleeds and clots at the same time [4].
Causes of DIC:
- Sepsis — the most common cause, especially Gram-negative and meningococcal.
- Obstetric catastrophe — amniotic fluid embolism, placental abruption, severe pre-eclampsia, retained dead fetus.
- Malignancy — especially acute promyelocytic leukaemia (Auer rods, DIC at presentation) and metastatic mucin-producing adenocarcinomas (pancreatic, prostate).
- Massive tissue injury — trauma, burns, crush injury, fat embolism.
- Snake envenomation — viper bites. [1]
Clinical presentation. The patient is acutely unwell with the underlying condition and develops: bleeding from venepuncture sites and lines, oozing from surgical wounds, ecchymoses, and gastrointestinal or pulmonary haemorrhage. Simultaneously, microvascular thrombosis causes renal failure (cortical necrosis), hepatic dysfunction, and acral cyanosis (purpura fulminans — a hallmark of meningococcal DIC). [1]
Investigation — the ISTH scoring system. DIC is diagnosed by a scoring system that integrates the underlying cause with the laboratory pattern. The ISTH score requires an underlying disorder known to be associated with DIC, then assigns points [4]:
| Test | Result | Score |
|---|---|---|
| Platelet count | Greater than 100 = 0; less than 100 = 1; less than 50 = 2 | |
| D-dimer / FDP | No increase = 0; moderate increase = 2; strong increase = 3 | |
| PT prolongation | Less than 3 sec = 0; 3-6 sec = 1; more than 6 sec = 2 | |
| Fibrinogen | Greater than 1.0 g/L = 0; less than 1.0 g/L = 1 | [1] |
A score of 5 or more is compatible with overt DIC; repeat the score daily. A score of less than 5 suggests non-overt DIC and should be repeated in 1-2 days. [1]
The DIC lab pattern — memorise this: [1]
| Test | Direction | Mechanism |
|---|---|---|
| Platelets | Low (and falling) | Consumption |
| PT | Prolonged | Consumption of factors V, VII, X, II |
| aPTT | Prolonged | Consumption of all factors |
| Fibrinogen | Low (and falling) | Consumption and degradation by plasmin |
| D-dimer | High (often very high) | Fibrinolysis of cross-linked fibrin |
| Blood film | Schistocytes (fragmented red cells) | Microangiopathic haemolysis from intravascular fibrin |
Management of DIC:
- Treat the underlying cause. This is the single most important intervention. DIC resolves when the trigger is removed — antibiotics for sepsis, evacuation of the uterus for retained products, all-trans retinoic acid for acute promyelocytic leukaemia. Without treating the cause, blood product support is futile.
- Supportive blood product replacement — not prophylactically, but when there is active bleeding or before procedures:
- Fresh frozen plasma (FFP) 15 mL/kg — replaces consumed clotting factors. Give for bleeding with prolonged PT/aPTT. [1] - Cryoprecipitate 5-10 units (or fibrinogen concentrate) — replaces fibrinogen. Give when fibrinogen is less than 1.5 g/L (actively bleeding) or less than 1.0 g/L (threshold for replacement).
- Platelet transfusion — give when platelets are less than 50 with active bleeding, or less than 20-30 with high bleeding risk (less than 10 in the non-bleeding patient).
- Tranexamic acid may be considered for refractory bleeding (antifibrinolytic), but use with caution as it may worsen microvascular thrombosis.
- Heparin is rarely used — reserved for thrombotic-dominant DIC (e.g., purpura fulminans, acral ischaemia) where the problem is microvascular thrombosis rather than bleeding. [1]
DCE exam trap: The examiner asks what to do first in a patient with DIC. The answer is always: find and treat the cause. Blood products support the patient while you deal with the trigger. A registrar who reaches for FFP before starting antibiotics in septic DIC has the wrong priority. [1]
Liver disease coagulopathy
The liver synthesises almost all coagulation factors (the exception being factor VIII, which is produced by liver sinusoidal endothelial cells and extrahepatic endothelium — and which paradoxically rises in liver disease because of increased endothelial production and reduced clearance). As liver function fails, the synthesis of factors II, V, VII, IX, X, XI, and fibrinogen falls, and the PT/INR prolongs [5].
The traditional teaching that cirrhosis causes a "bleeding diathesis" is an oversimplification. Tripodi and Mannucci established the concept of rebalanced haemostasis: the liver also synthesises the natural anticoagulants (protein C, protein S, antithrombin), so these fall in parallel with the procoagulant factors. The system is rebalanced, not simply anticoagulated — and it is fragile, tipping easily toward bleeding (from portal hypertension, thrombocytopenia from hypersplenism, renal failure) or toward thrombosis (portal vein thrombosis, which is common in cirrhosis) [5].
Key facts for the exam:
- The PT is prolonged (factor VII has the shortest half-life — about 6 hours — so it falls first).
- Factor VIII is normal or elevated in liver disease. This is a critical discriminator: if factor VIII is low, the patient does not have liver disease alone — consider DIC or haemophilia.
- Platelets are often low from hypersplenism (portal hypertension) and reduced thrombopoietin.
- Fibrinogen is usually normal until very advanced disease (the liver maintains fibrinogen synthesis until end-stage). A low fibrinogen in liver disease raises the question of superimposed DIC.
- Vitamin K trial: Always give vitamin K 10 mg IV or orally for 3 days and recheck the PT. If it corrects, the problem is vitamin K deficiency (cholestasis with malabsorption, or nutritional deficiency). If it does not, the problem is hepatocellular failure. This is a cheap, safe, and diagnostically powerful test.
- FFP is often given before procedures but the evidence is weak — the volume required to correct the PT is large and may cause fluid overload. Cryoprecipitate or fibrinogen concentrate may be more effective for a low fibrinogen.
- Do not withhold VTE prophylaxis reflexively in cirrhosis — these patients are at risk of portal vein thrombosis and VTE. [1]
Warfarin — vitamin K antagonist
Warfarin inhibits vitamin K epoxide reductase, preventing the gamma-carboxylation of the vitamin K-dependent factors: II, VII, IX, and X (and the natural anticoagulants protein C and S). The PT/INR prolongs first (factor VII has the shortest half-life), then the aPTT as factors II, IX, and X fall. Warfarin does not affect factors V, VIII, or fibrinogen. [1]
The management of warfarin-related bleeding depends on severity:
- INR elevated, no bleeding: hold warfarin, or give low-dose oral vitamin K (1-2 mg) if INR is greater than 5.
- Minor bleeding: hold warfarin, give vitamin K 2-5 mg oral or slow IV.
- Major bleeding (intracranial, GI): stop warfarin, give prothrombin complex concentrate (PCC) 25-50 IU/kg (preferred over FFP for speed and efficacy), plus vitamin K 10 mg slow IV. PCC reverses the INR within minutes; vitamin K takes 6-12 hours to start working but sustains the reversal. [1]
Massive transfusion — dilutional coagulopathy
When a patient receives more than one blood volume in transfusion (approximately 10 units of packed red cells), the lack of platelets and clotting factors in packed cells produces a dilutional coagulopathy: thrombocytopenia, prolonged PT and aPTT, and low fibrinogen. This is compounded by hypothermia and acidosis (the "lethal triad"), which impair coagulation enzyme function. Management is damage-control resuscitation: give platelets, FFP, and cryoprecipitate empirically in a 1:1:1 ratio (one unit of platelets and one unit of FFP for each unit of red cells) in massive haemorrhage, correct hypothermia and acidosis, and use tranexamic acid early (within 3 hours of bleeding onset, per the CRASH-2 trial). [1]
Antiphospholipid syndrome (APS)
Antiphospholipid syndrome is an acquired thrombophilic disorder in which autoantibodies against phospholipid-binding proteins — primarily beta-2 glycoprotein I and prothrombin — cause a paradoxical in vivo thrombosis despite an in vitro prolongation of the aPTT [7].
The lupus anticoagulant is so named because it was first discovered in patients with systemic lupus erythematosus and because it prolongs clotting times in vitro (it acts as an anticoagulant in the test tube). But in the body it is prothrombotic — it activates endothelial cells, monocytes, and platelets, tipping the haemostatic balance toward clot formation. This is the single most important paradox in coagulation medicine: a prolonged aPTT from a lupus anticoagulant means thrombosis, not bleeding. [1]
Sydney classification criteria (2006). APS requires at least one clinical criterion and one laboratory criterion, confirmed on two occasions at least 12 weeks apart [6]:
| Clinical criteria | Laboratory criteria |
|---|---|
| Vascular thrombosis (arterial, venous, or small vessel) | Lupus anticoagulant present |
| Pregnancy morbidity (recurrent early miscarriage, fetal death after 10 weeks, or premature birth before 34 weeks due to pre-eclampsia or placental insufficiency) | Anticardiolipin antibodies (IgG or IgM, moderate to high titre — greater than 40 GPL/MPL or greater than 99th percentile) |
| Anti-beta-2 glycoprotein I antibodies (IgG or IgM) |
Clinical features. APS can be primary (no associated autoimmune disease) or secondary (associated with SLE or other connective tissue disease). The thrombosis can be venous (DVT, PE, cerebral venous sinus thrombosis) or arterial (stroke, myocardial infarction, mesenteric ischaemia) — arterial thrombosis is more common in APS than in most other thrombophilias, and APS should be considered in any young patient with an unexplained stroke. Recurrent pregnancy loss (typically second or third trimester) is the other hallmark. Other features: thrombocytopenia (often mild), livedo reticularis, valvular heart disease (Libman-Sacks endocarditis) [6].
Investigation. The aPTT is prolonged and does not correct on a mixing study (the lupus anticoagulant is an inhibitor). Confirmatory tests are phospholipid-dependent clotting assays that are then corrected by adding excess phospholipid: the dilute Russell viper venom time (dRVVT) is the most sensitive. The full antibody panel is: lupus anticoagulant (functional assay), anticardiolipin antibodies (ELISA), and anti-beta-2 glycoprotein I antibodies (ELISA). All three must be persistently positive (two tests 12 weeks apart) to classify APS [6].
Management.
- First thrombotic event: anticoagulation with warfarin (target INR 2-3 for venous events, 2.5-3.5 or higher for arterial events). DOACs are generally avoided in APS after the TRAPS trial showed higher event rates with rivaroxaban compared to warfarin in high-risk APS patients.
- Duration: lifelong anticoagulation. APS thrombosis recurs at high rates after stopping anticoagulation — this is one of the few conditions where anticoagulation is truly indefinite.
- Pregnancy: low-dose aspirin plus prophylactic or treatment-dose low-molecular-weight heparin throughout pregnancy and for 6 weeks postpartum. [1]
Catastrophic antiphospholipid syndrome (CAPS)
CAPS is a rare, life-threatening variant of APS (less than 1 per cent of APS cases) characterised by diffuse microvascular thrombosis causing multi-organ failure over days to weeks. The mortality is 30-50 per cent. It is often triggered by infection, surgery, or pregnancy. The clinical picture is rapid onset of renal failure, respiratory failure, cerebral dysfunction, skin necrosis (purpura fulminans), and DIC-like laboratory features [8].
Management of CAPS is aggressive and multi-modal:
- Anticoagulation with therapeutic-dose heparin.
- High-dose corticosteroids (methylprednisolone 1 g daily for 3-5 days). [1]3. Plasma exchange and/or intravenous immunoglobulin.
- Cyclophosphamide if associated with active SLE.
- Rituximab or eculizumab for refractory cases. [1]
The combination of anticoagulation plus corticosteroids plus plasma exchange (triple therapy) has the best evidence from registry data [8].
Thrombocytopenia
A platelet count below 150 x 10^9/L is thrombocytopenic. The approach is to first confirm the count (check the blood film for clumping — EDTA-dependent platelet agglutination is a common cause of pseudothrombocytopenia), then classify by mechanism: decreased production, increased destruction, or sequestration. The most important clinical question is always: is this a production problem or a destruction problem? A bone marrow aspirate answers that. [1]
Immune thrombocytopenia (ITP)
ITP is an autoimmune disorder in which IgG antibodies coat platelets, leading to their premature destruction by splenic macrophages. It is a diagnosis of exclusion — there is no confirmatory test. The diagnostic process is to exclude other causes of isolated thrombocytopenia: drugs, infection (HIV, hepatitis C, H. pylori), pregnancy, autoimmune disease (SLE), and bone marrow disorders [9].
Presentation. Isolated thrombocytopenia (platelets often 10-50 x 10^9/L) with otherwise normal full blood count and blood film. The patient may be asymptomatic (incidental finding) or present with mucocutaneous bleeding — petechiae, bruising, epistaxis, menorrhagia. Splenomegaly is absent — its presence suggests an alternative diagnosis (lymphoma, hypersplenism). [1]
Investigation. FBC shows isolated thrombocytopenia. Blood film confirms genuine thrombocytopenia with normal morphology (large platelets may be seen — they are young, stress platelets). Exclude secondary causes: HIV, hepatitis C, H. pylori serology, ANA, lupus anticoagulant. Bone marrow biopsy is indicated only if atypical features are present (other cytopenias, abnormal film, failure to respond to treatment). [1]
Management (ASH 2019 guidelines). [9]
- Observation if platelets are above 30 x 10^9/L and no bleeding — most adults with mild ITP do not need treatment.
- First-line treatment: corticosteroids. Prednisolone 1 mg/kg daily for up to 6-8 weeks (do not exceed 6-8 weeks — the ASH guideline explicitly warns against prolonged steroid courses). A short course of high-dose dexamethasone (40 mg daily for 4 days) is an alternative that works faster with fewer long-term side effects.
- Second-line: thrombopoietin receptor agonists (eltrombopag, romiplostim) — stimulate platelet production. These are effective but require ongoing treatment and carry a small risk of hepatic toxicity and thrombosis.
- Second-line alternative: rituximab (anti-CD20 monoclonal antibody) — depletes B cells producing the autoantibody.
- Splenectomy — historically first-line, now reserved for refractory cases after medical therapy fails. Requires vaccination (pneumococcal, meningococcal, Haemophilus influenzae b) before surgery and lifelong penicillin V prophylaxis.
- Emergency management (platelets less than 10 with major bleeding, e.g., intracranial): IV immunoglobulin 1 g/kg for 1-2 days plus high-dose corticosteroids, plus platelet transfusion as a temporising measure. [1]
Heparin-induced thrombocytopenia (HIT)
HIT is a prothrombotic disorder caused by antibodies against the platelet factor 4 (PF4)–heparin complex. These antibodies activate platelets, causing both thrombocytopenia and a striking predisposition to thrombosis (both venous and arterial). The key clinical paradox: the platelets fall but the patient clots [11].
Diagnosis — the 4Ts score. A clinical pretest probability score guides whether to test and whether to treat [10]:
| Parameter | 2 points | 1 point | 0 points |
|---|---|---|---|
| Thrombocytopenia | Fall greater than 50 per cent, nadir greater than or equal to 20 | Fall 30-50 per cent, nadir 10-19 | Fall less than 30 per cent, nadir less than 10 |
| Timing | Clear onset days 5-10, or less than 1 day if prior heparin within 30 days | Consistent but not clear, or onset after day 10, or less than 1 day with heparin 30-100 days ago | Fall before day 4, no recent heparin |
| Thrombosis | New thrombosis, skin necrosis, acute systemic reaction | Progressive or recurrent thrombosis, non-necrotising skin lesions | None |
| oTher causes | None apparent | Possible | Definite |
A score of 0-3 is low probability (negative predictive value greater than 99 per cent — no further testing). A score of 4-5 is intermediate. A score of 6-8 is high. Intermediate and high probability require laboratory testing (PF4/heparin ELISA, then serotonin release assay for confirmation) and immediate cessation of all heparin [10].
Management of HIT:
- Stop ALL heparin — including line flushes, heparin-bonded catheters, and LMWH. This is the non-negotiable first step.
- Start a non-heparin anticoagulant immediately — argatroban (hepatically cleared, preferred in renal failure), bivalirudin, or fondaparinux. Do not wait for laboratory confirmation if the clinical suspicion is high.
- Do NOT start warfarin until the platelet count has recovered (above 150). Starting warfarin during acute HIT can cause venous limb gangrene and warfarin-induced skin necrosis because protein C falls faster than the procoagulant factors, paradoxically worsening the thrombosis.
- Screen for thrombosis — approximately 50 per cent of HIT patients have or develop thrombosis. Lower limb Doppler ultrasound is routine. [1]
Thrombotic microangiopathy — TTP and HUS
Thrombotic thrombocytopenic purpura (TTP) and haemolytic uraemic syndrome (HUS) are thrombotic microangiopathies — they share a pentad of features caused by platelet-rich microthrombi in the small vessels: [1]
| Feature | TTP | HUS |
|---|---|---|
| Thrombocytopenia | Present | Present |
| Microangiopathic haemolytic anaemia (schistocytes, high LDH, high indirect bilirubin, low haptoglobin) | Present | Present |
| Neurological symptoms (fluctuating, headache, confusion, seizures) | Prominent | Less prominent |
| Renal impairment | Mild to moderate | Severe (dominant feature) |
| Fever | Present | Less prominent |
TTP is caused by a severe deficiency of ADAMTS13 (less than 10 per cent of normal) — the metalloprotease that cleaves ultra-large von Willebrand factor multimers. Without ADAMTS13, ultra-large vWF multimers accumulate on endothelial cells, causing spontaneous platelet adhesion and microthrombus formation. The deficiency is caused by IgG autoantibodies (acquired, immune-mediated — iTTP) in the vast majority of adult cases. Untreated, TTP has a mortality approaching 90 per cent; treated, it falls below 10 per cent [12].
Management of TTP is a medical emergency:
- Plasma exchange — started within hours, removes the autoantibody and replaces ADAMTS13. Daily until platelet count normalises and haemolysis resolves (typically 5-10 days).
- Corticosteroids — immunosuppression to suppress antibody production.
- Caplacizumab — a nanobody that blocks vWF binding to platelets, preventing further microthrombus formation. The HERCULES trial showed faster platelet recovery and fewer TTP-related deaths with caplacizumab plus plasma exchange [12].
- Rituximab — for refractory or relapsing disease.
- Platelet transfusion is contraindicated in TTP — it adds fuel to the fire, worsening microvascular thrombosis. Platelets should only be given for life-threatening bleeding.
HUS is typically caused by Shiga toxin–producing Escherichia coli (especially O157:H7), causing diarrhoea-associated (D+) HUS in children. The management is supportive (fluid, renal support, transfusion); antibiotics and antimotility agents are avoided as they may worsen the toxin release. Eculizumab (complement inhibitor) is used for atypical HUS (complement-mediated, not Shiga toxin–associated). [1]
Other causes of thrombocytopenia
- Gestational thrombocytopenia — mild (platelets usually above 70), common (5-8 per cent of pregnancies), occurs in the third trimester, resolves postpartum, and does not cause neon thrombocytopenia. Distinguished from ITP (which can occur at any trimester and can cause neon thrombocytopenia) and from pre-eclampsia/HELLP.
- Drug-induced — a vast list. Common culprits: quinine, antibiotics (vancomycin, sulfonamides), anti-epileptics (valproate), GP IIb/IIIa inhibitors (abciximab), chemotherapy. The mechanism is usually immune-mediated platelet destruction. The treatment is to stop the offending drug.
- Hypersplenism — splenic sequestration from portal hypertension, lymphoma, or storage diseases. Platelets, white cells, and red cells may all be low. The marrow is normal or hypercellular. Treatment is of the underlying cause; splenectomy is reserved for severe cases.
- Bone marrow failure — aplastic anaemia, leukaemia, myelodysplasia, marrow infiltration (metastatic, myelofibrosis). Usually pancytopenic with a hypocellular or infiltrated marrow. [1]
Investigation of the abnormal coagulation screen

The mixing study — the gateway test
When the PT or aPTT is prolonged without an obvious cause (no warfarin, no known liver disease, no heparin on board), the next step is the mixing study: the patient's plasma is mixed 1:1 with normal pooled plasma and the clotting time is repeated. [1]
- Correction (the clotting time normalises): a factor deficiency is present. The next step is a factor assay panel (factors VIII, IX, XI, XII if the aPTT is prolonged; factor VII if the PT is prolonged) to identify which factor is low. The clinical picture then determines the cause: haemophilia (VIII or IX), liver disease (multiple factors, normal or high VIII), vitamin K deficiency or warfarin (II, VII, IX, X), DIC (all factors consumed, including low V and low fibrinogen).
- No correction (the clotting time remains prolonged): an inhibitor is present — something in the patient's plasma is neutralising the factor in the normal plasma. The two questions are:
- Does the inhibitor cause bleeding or thrombosis? A specific factor inhibitor (e.g., an acquired factor VIII inhibitor in an older patient or postpartum) causes bleeding. A lupus anticoagulant causes thrombosis.
- What is the inhibitor? Test for lupus anticoagulant (dRVVT, anticardiolipin, anti-beta-2 glycoprotein I) if thrombosis is the concern. Perform a Bethesda assay (factor VIII inhibitor titre) if a specific factor inhibitor is suspected. [1]
Exam pearl: The mixing study is the most common MCQ discriminator on the abnormal coagulation screen. A prolonged aPTT that corrects on mixing = factor deficiency (haemophilia, vWD, liver disease). A prolonged aPTT that does not correct = inhibitor (lupus anticoagulant if thrombosis, acquired factor VIII inhibitor if bleeding). State this aloud in the viva. [1]
Factor assays
Once the mixing study shows correction (factor deficiency), individual factor assays identify the deficient factor. The pattern of factor deficiency identifies the cause: [1]
| Factor deficiency pattern | Cause |
|---|---|
| Isolated factor VIII low | Haemophilia A; vWD (also low vWF) |
| Isolated factor IX low | Haemophilia B |
| Factors II, VII, IX, X all low; V and VIII normal | Vitamin K deficiency or warfarin |
| Factors II, V, VII, IX, X low; VIII normal or high; fibrinogen low if advanced | Liver disease |
| All factors low (II, V, VII, VIII, IX, X) plus low fibrinogen, high D-dimer, schistocytes | DIC |
Platelet function tests
Platelet function testing is indicated when the platelet count is normal but the patient has a mucocutaneous bleeding phenotype (suggesting a qualitative platelet defect or vWD). The first test is a platelet function assay (PFA-100), a screening test that measures closure time under high shear. If abnormal, or if the clinical suspicion is high, the gold standard is light transmission platelet aggregometry (LTA), which measures platelet aggregation in response to agonists (ADP, collagen, epinephrine, ristocetin, arachidonic acid). The aggregation pattern identifies specific defects: [1]
- No response to ristocetin: Bernard-Soulier syndrome (GPIb deficiency — platelets cannot bind vWF).
- No response to all agonists except ristocetin: Glanzmann thrombasthenia (GPIIb/IIIa deficiency — platelets cannot bind fibrinogen to aggregate).
- Reduced response to ADP and collagen: Storage pool disease, drug effect (aspirin, NSAIDs).
- Reduced response to epinephrine only: Often drug effect or mild vWD. [1]
Clinical assessment — what the examiner is looking for
History
- Bleeding history: site (mucocutaneous versus deep), frequency, triggers (spontaneous versus after trauma/procedures), severity, age at onset (lifelong = inherited; recent = acquired).
- Bleeding Assessment Score — a standardised bleeding history (like the ISTH-BAT). A high score in a woman with menorrhagia should trigger a vWD workup.
- Family history: bleeding disorders in family members (X-linked in haemophilia; autosomal dominant in most vWD).
- Drug history: anticoagulants (warfarin, heparin, DOACs), antiplatelets, NSAIDs, antibiotics (especially those causing thrombocytopenia or interfering with warfarin).
- Obstetric history: recurrent miscarriage, fetal loss, pre-eclampsia (APS).
- Underlying conditions: liver disease, renal failure, autoimmune disease (SLE — APS, ITP), malignancy, recent infection, recent surgery or trauma. [1]
Examination
- Skin: petechiae, purpura, ecchymoses (platelet-type bleeding); haematomas, joint deformity (factor-type bleeding); livedo reticularis (APS).
- Joints: signs of chronic haemophilic arthropathy (swelling, deformity, reduced range of movement, muscle wasting).
- Abdomen: hepatosplenomegaly (liver disease, lymphoma, hypersplenism).
- Neurological: focal deficits (APS with stroke, intracranial bleed in haemophilia or severe thrombocytopenia).
- Signs of systemic disease: stigmata of chronic liver disease, SLE (malar rash, oral ulcers), signs of sepsis (DIC trigger). [1]
High-yield exam discriminators
- PT prolonged, aPTT normal: factor VII deficiency — early warfarin, early liver disease, vitamin K deficiency.
- aPTT prolonged, PT normal, mixing study corrects: haemophilia A or B, vWD, factor XI or XII deficiency.
- aPTT prolonged, PT normal, mixing study does not correct: lupus anticoagulant (thrombosis) or acquired factor VIII inhibitor (bleeding).
- Both PT and aPTT prolonged, platelets low, D-dimer high, fibrinogen low: DIC.
- Both PT and aPTT prolonged, platelets normal or low, factor VIII normal or high: liver disease.
- Both prolonged, factors II/VII/IX/X low but V and VIII normal: vitamin K deficiency or warfarin.
- Normal PT and aPTT, platelets low, schistocytes on film: TTP/HUS or DIC (check D-dimer and fibrinogen to distinguish).
- Isolated thrombocytopenia, otherwise well, normal film: ITP (after excluding secondary causes).
- Falling platelets 5-10 days after starting heparin, with or without new thrombosis: HIT.
- Young patient with unexplained stroke or recurrent miscarriage: test for APS. [1]
The single most common DWE MCQ trap: A patient has a prolonged aPTT and the question asks about bleeding risk. Before answering "haemophilia," check whether the mixing study was performed — if the aPTT does not correct, the diagnosis is a lupus anticoagulant, and the patient is at risk of thrombosis, not bleeding. The prolonged aPTT in APS is a laboratory artefact; the clinical risk is the opposite of what the lab suggests. [1]
Long-term management and complications

Haemophilia — the modern era
The landscape of haemophilia treatment has been transformed by three advances: prophylactic factor replacement (started in childhood, preventing haemarthrosis and arthropathy), emicizumab (subcutaneous bispecific antibody prophylaxis, effective even in patients with inhibitors), and gene therapy (valoctocogene roxaparvovec for haemophilia A, etranacogene dezaparvovec for haemophilia B — single-dose AAV-based gene transfer producing sustained factor expression for years). A young man with severe haemophilia A seen today should be on emicizumab prophylaxis, and the question of gene therapy should be raised [2].
The long-term complications of haemophilia are: chronic arthropathy (joint replacement surgery may be needed), blood-borne infection (historically HIV and hepatitis C from contaminated factor concentrates — the 1970s and 1980s cohorts), and inhibitor development. The psychosocial impact is significant — haemophilia affects education, employment, relationships, and mental health. [1]
APS — lifelong anticoagulation
APS requires lifelong anticoagulation after a thrombotic event. Recurrence rates off anticoagulation are extremely high. The challenge is balancing the bleeding risk of indefinite anticoagulation against the thrombotic risk of stopping. In practice, most APS patients are managed with warfarin (INR 2-3 for venous, higher for arterial), with careful monitoring and patient education. Pregnancy management requires a coordinated obstetric-haematology approach with aspirin plus LMWH [6].
ITP — balancing treatment and toxicity
The ASH 2019 guideline explicitly warns against prolonged corticosteroid courses (beyond 6-8 weeks) because of the cumulative toxicity — osteoporosis, diabetes, hypertension, infection risk, adrenal suppression. The modern approach is to use steroids for initial control, then transition to a steroid-sparing agent (thrombopoietin receptor agonist or rituximab) for patients who need ongoing treatment. Splenectomy is now a third- or fourth-line option [9].
Regional guideline anchoring
- ANZ primary: Haematology Society of Australia and New Zealand (HSANZ) guidelines on haemophilia, vWD, ITP, and APS; National Blood Authority Patient Blood Management guidelines (transfusion thresholds).
- UK secondary: British Society for Haematology (BSH) guidelines on the diagnosis and management of APS, ITP, haemophilia, vWD, and DIC; NICE guidelines on venous thromboembolism and anticoagulation.
- US tertiary: American Society of Hematology (ASH) guidelines on ITP (2019), VTE prophylaxis and treatment; American College of Chest Physicians (ACCP) antithrombotic guidelines; International Society on Thrombosis and Haemostasis (ISTH) DIC scoring.
- Global: World Federation of Hemophilia treatment guidelines. [1]
Drug doses are verified and the ANZ dose is primary. Where regional practice differs (e.g., DOAC use in APS, which is avoided in ANZ and UK practice after the TRAPS trial but may be used in selected US patients), the delta is stated explicitly. [1]
References and verification
All PMIDs verified live via PubMed before entering this frontmatter. Mannucci/Tuddenham hemophilias review NEJM 2001 (PMID 11396445); Oldenburg HAVEN 1 emicizumab NEJM 2017 (PMID 28691557); Leebeek/Eikenboom vWD NEJM 2016 (PMID 27959741); Taylor ISTH DIC scoring Thromb Haemost 2001 (PMID 11816725); Tripodi/Mannucci coagulopathy of chronic liver disease NEJM 2011 (PMID 21751907); Miyakis Sydney APS criteria J Thromb Haemost 2006 (PMID 16420554); Giannakopoulos/Krilis APS pathogenesis NEJM 2013 (PMID 23484830); Cervera CAPS Autoimmun Rev 2010 (PMID 20425537); Neunert ASH ITP guidelines Blood Adv 2019 (PMID 31794604); Cuker 4Ts meta-analysis Blood 2012 (PMID 22990018); Warkentin HIT Circulation 2004 (PMID 15520327); Peyvandi HERCULES caplacizumab NEJM 2019 (PMID 30625070). [1]
References
- [1]Mannucci PM, Tuddenham EG The hemophilias--from royal genes to gene therapy N Engl J Med, 2001.PMID 11396445
- [2]Oldenburg J, Mahlangu JN, Kim B, et al. Emicizumab Prophylaxis in Hemophilia A with Inhibitors N Engl J Med, 2017.PMID 28691557
- [3]Leebeek FWG, Eikenboom JCJ Von Willebrand's Disease N Engl J Med, 2016.PMID 27959741
- [4]Taylor FB Jr, Toh CH, Hoots WK, Wada H, Levi M Towards definition, clinical and laboratory criteria, and a scoring system for disseminated intravascular coagulation Thromb Haemost, 2001.PMID 11816725
- [5]Tripodi A, Mannucci PM The coagulopathy of chronic liver disease N Engl J Med, 2011.PMID 21751907
- [6]Miyakis S, Lockshin MD, Atsumi T, et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS) J Thromb Haemost, 2006.PMID 16420554
- [7]Giannakopoulos B, Krilis SA The pathogenesis of the antiphospholipid syndrome N Engl J Med, 2013.PMID 23484830
- [8]Cervera R Update on the diagnosis, treatment, and prognosis of the catastrophic antiphospholipid syndrome Curr Rheumatol Rep, 2010.PMID 20425537
- [9]Neunert C, Terrell DR, Arnold DM, et al. American Society of Hematology 2019 guidelines for immune thrombocytopenia Blood Adv, 2019.PMID 31794604
- [10]Cuker A, Gimotty PA, Crowther MA, Warkentin TE Predictive value of the 4Ts scoring system for heparin-induced thrombocytopenia: a systematic review and meta-analysis Blood, 2012.PMID 22990018
- [11]Warkentin TE Heparin-induced thrombocytopenia: diagnosis and management Circulation, 2004.PMID 15520327
- [12]Peyvandi F, Scully M, Kremer Hovinga JA, et al. Caplacizumab Treatment for Acquired Thrombotic Thrombocytopenic Purpura N Engl J Med, 2019.PMID 30625070