Phys · hepatic
Autoimmune Liver Disease
Also known as primary biliary cholangitis · primary biliary cirrhosis · PBC · primary sclerosing cholangitis · PSC · autoimmune hepatitis · AIH · lupoid hepatitis · antimitochondrial antibody · AMA · interface hepatitis · florid duct lesion · overlap syndrome · ursodeoxycholic acid · obeticholic acid · cholangiocarcinoma
Consultant-physician-depth guide to the three major autoimmune liver diseases — primary biliary cholangitis (AMA-positive, cholestatic LFTs, florid duct lesion, ursodeoxycholic acid ± obeticholic acid), primary sclerosing cholangitis (IBD association, beaded cholangiogram, cholangiocarcinoma and colorectal cancer risk, no effective medical therapy, ERCP for dominant strictures) and autoimmune hepatitis (interface hepatitis, high IgG, prednisolone plus azathioprine), with the AIH-PBC and AIH-PSC overlap syndromes, the diagnostic algorithm for abnormal LFTs, and the transplant indications. Structured for FRACP DWE and DCE preparation.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Autoimmune Liver Disease

The answer first
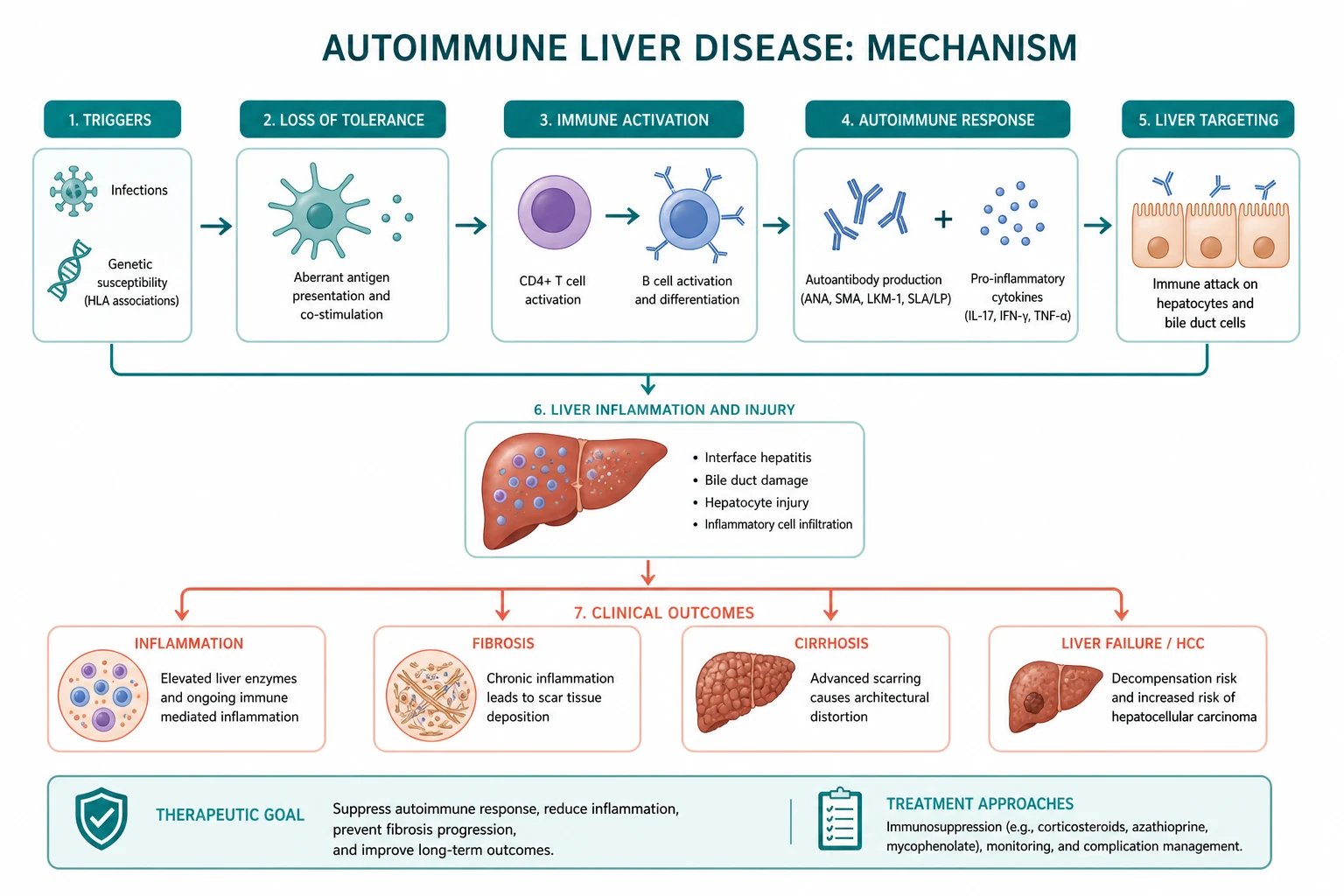
Autoimmune liver disease is a family of three conditions in which the immune system attacks the liver. They are not interchangeable. Each has a different target, a different laboratory fingerprint, and a different treatment. Know the discriminator for each, and the whole topic becomes one decision tree. [1]
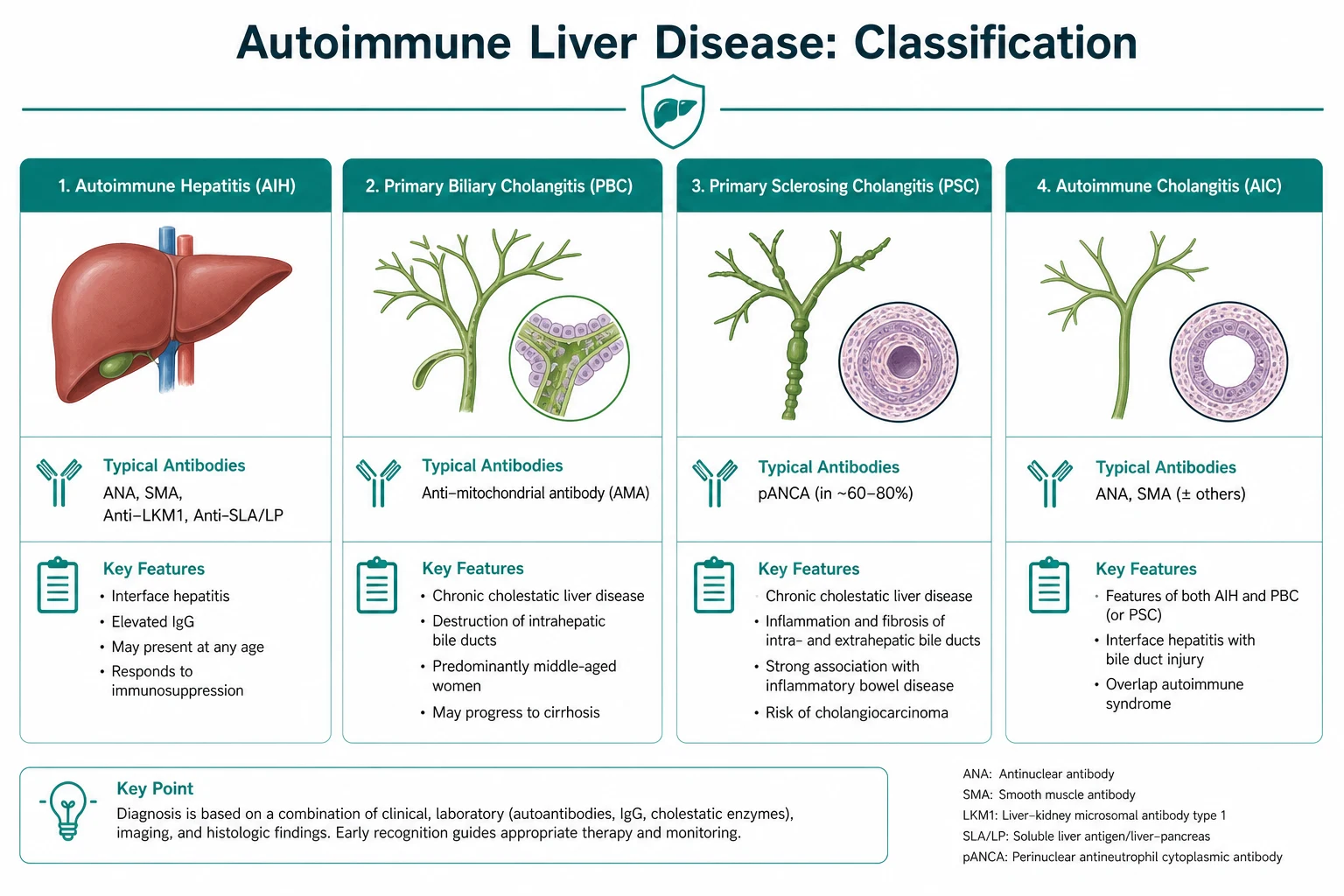
- Primary biliary cholangitis (PBC) — a T-cell attack on the small interlobular bile ducts, in a middle-aged woman, with a raised ALP, a raised IgM, and an antimitochondrial antibody (AMA) that is positive in around 95 percent. Treatment is ursodeoxycholic acid (first line for all stages) with obeticholic acid or a fibrate added for the inadequate responder.
- Primary sclerosing cholangitis (PSC) — a fibrosing inflammation of the medium and large bile ducts, in a young or middle-aged man with ulcerative colitis, with a raised ALP, a normal IgM, a negative AMA, and a beaded, multifocal-stricturing cholangiogram. There is no effective medical therapy; management is cancer surveillance and ERCP for dominant strictures.
- Autoimmune hepatitis (AIH) — an attack on the hepatocytes, in a young woman (or anyone), with a hepatocellular LFT pattern (transaminases two to ten times the upper limit), a high IgG, and ANA/SMA (type 1) or anti-LKM1 (type 2). Histology shows interface hepatitis with plasma cells. Treatment is prednisolone plus azathioprine. [1]
The single organising principle: classify the disease by the LFT pattern and the autoantibody panel before reaching for a treatment. A cholestatic pattern with a positive AMA is PBC until proven otherwise. A cholestatic pattern with IBD and a normal AMA is PSC until proven otherwise. A hepatocellular pattern with a high IgG is AIH until proven otherwise. The overlap syndromes sit at the intersections. [1]
DWE high-yield: The three diseases are distinguished by the combination of (1) the LFT pattern, (2) the key antibody, (3) the immunoglobulin, and (4) the histology. Most aetiology MCQ stems can be answered from those four data points alone. PBC and PSC are cholestatic (raised ALP); AIH is hepatocellular (raised ALT/AST). PBC has AMA and raised IgM; PSC has neither; AIH has ANA/SMA or anti-LKM1 and raised IgG [1][6].

PBC — primary biliary cholangitis
PBC is the autoimmune destruction of the small interlobular bile ducts. It affects around 1 in 1000 women over the age of 40, with a female-to-male ratio of around 9 to 1. The name changed in 2014 from "primary biliary cirrhosis" to "primary biliary cholangitis" (both keep the PBC abbreviation) because most patients are now diagnosed before cirrhosis develops, and the old name carried an inaccurate and distressing implication of established cirrhosis [1].
How PBC presents
The classic patient is a woman aged 40 to 60 who presents with fatigue and pruritus. The fatigue is the most common symptom (reported by up to 80 percent) and is the most disabling; it is under-recognised and has no effective specific therapy. The pruritus is often worse at night, typically spares the face and extremities, and precedes jaundice by months to years. Many patients are now detected asymptomatically on a routine blood panel showing an isolated raised ALP, and the diagnosis is then confirmed by the AMA [1].
On examination, the signs of chronic cholestasis are the clue: excoriations (from scratching), hyperpigmentation (from melanin deposition), xanthelasma (yellow periorbital deposits) and less commonly tuberous and tendinous xanthomata (from the hypercholesterolaemia of cholestasis, which paradoxically does NOT increase cardiovascular mortality). In advanced disease, the stigmata of cirrhosis appear — spider naevi, palmar erythema, hepatosplenomegaly, ascites, and the signs of portal hypertension. [1]
The immunopathology
PBC is a T-cell-mediated destruction of the small bile ducts, triggered by a loss of tolerance to the E2 subunit of the pyruvate dehydrogenase complex (PDC-E2) on biliary epithelial cells. The AMA targets PDC-E2 and the related 2-oxo-acid dehydrogenase enzymes. Why the immune attack is confined to the bile ducts when PDC-E2 is present in every mitochondrion remains incompletely understood, but the biliary epithelial cell appears to handle the antigen differently, presenting it in a pro-inflammatory context [1].
The histological hallmark is the florid duct lesion — a granulomatous destruction of the small interlobular bile duct. As ducts are lost, cholestasis deepens, fibrosis progresses, and cirrhosis develops over years to decades. [1]
Making the diagnosis
The diagnosis of PBC requires two of three criteria: a cholestatic LFT pattern (raised ALP), a positive AMA (or PBC-specific ANA — anti-gp210 or anti-sp100), and a compatible liver biopsy. In practice, the AMA plus the cholestatic pattern makes the diagnosis and a biopsy is not required; biopsy is reserved for when the diagnosis is uncertain, when there is a suspected overlap with AIH, or to stage fibrosis [1].
| Investigation | Result in PBC |
|---|---|
| ALP | Raised (the dominant abnormality) |
| Gamma-GT | Raised (confirms a hepatic source for the ALP) |
| Bilirubin | Normal early; rises late (a poor prognostic marker) |
| ALT/AST | Normal or mildly raised |
| AMA | Positive in around 95 percent |
| PBC-specific ANA (anti-gp210, anti-sp100) | Positive in AMA-negative PBC |
| IgM | Raised (a discriminator from PSC and AIH) |
| IgG | Normal (a discriminator from AIH) |
| Cholesterol | Raised (but not atherogenic) |
| Ultrasound | Normal ducts (excludes obstruction) |
| Liver biopsy | Florid duct lesion; staged Ludwig 1 to 4 |
Treatment — UDCA first, for every stage
Ursodeoxycholic acid (UDCA) is first-line therapy for ALL stages of PBC, at 13 to 15 mg/kg/day in divided doses, lifelong. The 1994 Lindor NEJM trial in 145 patients showed that UDCA slows histological progression, reduces the need for transplant, and improves survival free of transplant [2]. UDCA is a hydrophilic bile acid that displaces the toxic hydrophobic bile acids from the pool, is immunomodulatory, and stimulates choleresis. It is well tolerated; diarrhoea is the main side effect. Every PBC patient should be on UDCA unless there is a contraindication (there rarely is).
DWE high-yield: The answer to "first-line treatment for PBC" is ursodeoxycholic acid, 13 to 15 mg/kg/day, for all stages. This is the one drug every PBC patient gets, regardless of histological stage or symptoms. [1]
Second-line — obeticholic acid and fibrates for the inadequate responder
Around 30 to 40 percent of patients have an inadequate biochemical response to UDCA, defined by the Barcelona, Paris-I, Paris-II or Toronto criteria (each uses the ALP, bilirubin and AST after 6 to 12 months of UDCA). These patients have a higher risk of progression and need second-line therapy [3].
- Obeticholic acid (OCA) is a farnesoid X receptor (FXR) agonist. The 2016 POISE trial (Nevens NEJM) showed it improved the composite of ALP and bilirubin in inadequate responders [3]. The dose is 5 mg daily for the first 6 months, then 10 mg daily if the target is not met. Two key cautions: OCA commonly worsens pruritus (the commonest reason to reduce the dose or stop), and in advanced (decompensated) cirrhosis it can precipitate liver failure and death — it is contraindicated in Child-Pugh B and C cirrhosis, and the frequency is reduced in stage 4.
- Fibrates (fenofibrate, bezafibrate) are an alternative or adjunct, particularly for pruritus as well as the biochemistry. The combination of UDCA plus a fibrate is increasingly used in the inadequate responder, though the evidence is weaker than for OCA.
Surveillance and complications
- Bone health — osteoporosis is the commonest bone disease in PBC (and PSC), driven by cholestasis, hypogonadism and (in overlap AIH) corticosteroids. Baseline and biennial DEXA, calcium, vitamin D, and a bisphosphonate as needed.
- Fat-soluble vitamins — deficiency of A, D, E and K develops with severe cholestasis. Check and replace annually in advanced disease.
- Portal hypertension and varices — screen by endoscopy at the diagnosis of cirrhosis and every 1 to 3 years.
- Hepatocellular carcinoma — six-monthly ultrasound plus alpha-fetoprotein once cirrhosis is established (PBC cirrhosis carries an HCC risk; non-cirrhotic PBC does not).
- Associated autoimmune disease — Sjogren syndrome, autoimmune thyroid disease, rheumatoid arthritis and CREST syndrome are common co-morbidities; screen for and manage them. [1]
Transplant
Liver transplant is indicated for decompensated cirrhosis (ascites, encephalopathy, variceal bleed, refractory pruritus or a rising bilirubin despite therapy) and for HCC within transplant criteria. Post-transplant survival is excellent (above 80 percent at 5 years). PBC can recur in the graft (around 11 to 42 percent), though recurrence rarely causes graft loss; UDCA is continued post-transplant [1].
PSC — primary sclerosing cholangitis
PSC is the fibrosing inflammation of the medium and large intra- and extra-hepatic bile ducts, producing the characteristic multifocal strictures and beading on cholangiography. It is the autoimmune liver disease with the highest cancer burden and the least effective medical therapy [4][5].
How PSC presents
The classic patient is a man in his 30s or 40s with ulcerative colitis. Around 70 percent of PSC patients have IBD (predominantly ulcerative colitis, less commonly Crohn colitis), and the PSC-IBD phenotype is a characteristic pan-colitis with rectal sparing and backwash ileitis, often clinically quiescent. Conversely, around 2.5 to 7.5 percent of patients with ulcerative colitis develop PSC, and these patients should have their LFTs monitored. [1]
PSC often presents asymptomatically, with a raised ALP found on IBD surveillance. Symptomatic presentation is with pruritus, fatigue, right upper quadrant pain, or recurrent bacterial cholangitis once strictures form. Acute bacterial cholangitis — fever, rigors and jaundice in a patient with known strictures — is the dangerous acute presentation and demands urgent antibiotics and biliary drainage. [1]
The immunopathology
PSC is immune-mediated, with a strong genetic component (an association with HLA-B8, DR3 and DR4). The mechanism is incompletely understood — hypotheses include a loss of tolerance to gut-derived antigens in a patient with a leaky, inflamed gut (explaining the IBD link), and a dysbiotic gut microbiome. The result is an "onion-skin" concentric periductal fibrosis around the medium and large ducts, with stricturing and upstream cholestatic injury. [1]
Making the diagnosis
The diagnosis of PSC requires a cholestatic LFT pattern (raised ALP), a characteristic cholangiogram, and exclusion of secondary causes of sclerosing cholangitis. [1]
| Investigation | Result in PSC |
|---|---|
| ALP | Raised (the dominant abnormality) |
| Bilirubin | Normal early; rises with strictures, cholangitis or cholangiocarcinoma |
| AMA | Negative (a key discriminator from PBC) |
| ANA, SMA, anti-LKM1 | May be positive (PSC can have an AIH-overlap component) |
| IgG4 | Normal (a key discriminator from IgG4-sclerosing cholangitis) |
| IgM | Normal |
| MRCP | Multifocal strictures and beading of intra- and extra-hepatic ducts (the gold standard) |
| ERCP | Reserved for therapy of a dominant stricture (brushings, dilatation, stenting) |
| Liver biopsy | Rarely diagnostic ("onion-skin" periductal fibrosis); used for small-duct PSC and overlap |
MRCP is the first-line imaging and confirms the diagnosis non-invasively. ERCP is reserved for therapy — balloon dilatation, stenting, or brush cytology of a dominant stricture — because of the pancreatitis and cholangitis risks of a purely diagnostic ERCP [4].
DWE high-yield trap: Small-duct PSC is the variant confined to the small intrahepatic ducts, with a normal cholangiogram but a characteristic biopsy. It carries a better prognosis and a lower cholangiocarcinoma risk than classic large-duct PSC. An examiner may test this distinction. [1]
The must-not-miss mimic — IgG4-related sclerosing cholangitis
Before labelling a patient PSC, check a serum IgG4. IgG4-related sclerosing cholangitis is a steroid-responsive mimic of PSC, occurring in older men, with a raised serum IgG4, dense lymphoplasmacytic infiltration with storiform fibrosis on biopsy, and a frequent association with autoimmune pancreatitis (a sausage-shaped pancreas on imaging). The treatment is corticosteroids, the opposite of PSC (which is not steroid-responsive). Missing IgG4-sclerosing cholangitis denies the patient an effective therapy [5].
Management — surveillance, not a drug
The single most important exam point about PSC: there is no effective medical therapy. High-dose UDCA (28 to 30 mg/kg/day) was tested in a 2009 trial and showed worse outcomes; it is not recommended. Low-dose UDCA is sometimes used for its biochemical effects but does not alter survival. No drug has been shown to change the natural history. Management is surveillance and complications management [4][5].

Surveillance is the core of PSC management: [1]
| Surveillance target | Modality | Frequency |
|---|---|---|
| Cholangiocarcinoma and gallbladder carcinoma | MRI/MRCP plus CA 19-9 | Annually |
| Colorectal cancer (in those with IBD) | Colonoscopy with biopsies | Annually from diagnosis |
| Hepatocellular carcinoma (in cirrhosis) | Ultrasound plus alpha-fetoprotein | Six-monthly |
| Bone health | DEXA | At diagnosis, then every 2 to 3 years |
| Fat-soluble vitamins | A, D, E, K levels | Annually in advanced disease |
Dominant stricture management — a dominant stricture (a stenosis of the common bile duct or a major hepatic duct) presents with worsening jaundice, cholangitis, worsening pruritus, or a rising CA 19-9. Arrange ERCP with balloon dilatation ± stenting, and send brush cytology (and consider cholangioscopy with targeted biopsy) to exclude cholangiocarcinoma. A dominant stricture is the commonest reason for ERCP in PSC and the commonest presentation of cholangiocarcinoma. [1]
The cancer burden
PSC carries the highest malignancy burden of any liver disease: [1]
- Cholangiocarcinoma — lifetime risk 5 to 20 percent, with 30 to 50 percent of cases diagnosed within the first year of the PSC diagnosis (because the PSC presentation is often the cancer presentation). A 2024 meta-analysis of 51 studies and 26 482 patients reported a pooled incidence of 9.31 per 1000 person-years [10].
- Gallbladder carcinoma — increased; annual ultrasound with the MRI/MRCP.
- Colorectal cancer — markedly increased in PSC-IBD, independent of the duration or activity of the colitis. Annual colonoscopy from diagnosis.
- Hepatocellular carcinoma — in established cirrhosis.
Transplant
Liver transplant is indicated for decompensated cirrhosis, for HCC within transplant criteria, for recurrent bacterial cholangitis, and for intractable pruritus (a transplant indication specific to cholestatic disease). Five-year survival exceeds 80 percent. PSC can recur in the graft (around 10 to 25 percent), and recurrent PSC is a more common cause of graft loss than recurrent PBC [4].
AIH — autoimmune hepatitis
AIH is the immune-mediated attack on the hepatocytes, producing a hepatocellular pattern of liver injury. It responds dramatically to immunosuppression, which makes it one of the most satisfying diagnoses in hepatology — but untreated, it progresses rapidly to cirrhosis (5- and 10-year survival of around 50 and 10 percent without therapy) [6][7].
How AIH presents
AIH has a bimodal age distribution (peaks in the teenage/young-adult years and around the menopause) and a female predominance (around 4 to 1). It can present in three ways: [1]
- Chronic insidious onset (the majority) — fatigue, malaise, arthralgia, a hepatocellular LFT pattern found incidentally, and occasionally amenorrhoea.
- Acute presentation — a viral-hepatitis-like illness with high transaminases, or even acute liver failure with coagulopathy and encephalopathy. AIH can present de novo as ALF, and this is a key exam point.
- Cirrhosis at presentation — an incidental finding or a decompensation, in a patient who has had silent disease for years. [1]
Extrahepatic autoimmune features are common and support the diagnosis — autoimmune thyroid disease, rheumatoid arthritis, Sjogren syndrome, ulcerative colitis, type 1 diabetes, and (a high-yield exam association) vitiligo, alopecia and nail dystrophy. [1]
The two types — defined by the autoantibody
| Feature | Type 1 AIH | Type 2 AIH |
|---|---|---|
| Defining antibody | ANA and/or anti-smooth muscle antibody (anti-actin) | Anti-liver-kidney microsomal type 1 (anti-LKM1, anti-CYP2D6) |
| Age | Any age; the common adult form | Children and young adults |
| Sex | Female predominant | Female predominant |
| Course | Variable; often responsive | More aggressive; higher risk of acute liver failure and rapid progression to cirrhosis |
| IgG | Raised | Raised (though can be normal in type 2) |
| Other antibodies | Anti-actin, anti-SLA/LP (anti-soluble liver antigen — highly specific for AIH and relapse-prone) | Anti-LC1 (anti-liver cytosol) |
Type 1 is the common adult form; type 2 is predominantly paediatric and more aggressive. A third antibody, anti-SLA/LP (anti-soluble liver antigen / liver-pancreas), is highly specific for AIH and identifies a relapse-prone subset — it is worth checking in seronegative cases [6].
The immunopathology
The histological hallmark of AIH is interface hepatitis — a lymphoplasmacytic infiltrate that spills from the portal tracts into the limiting plate of the lobule, destroying hepatocytes at the interface. The plasma cell-rich infiltrate is the classic feature, though its absence does not exclude AIH. The mechanism is a loss of self-tolerance, with a CD4 T-cell-driven attack on hepatocyte antigens, supported by autoantibody production and a genetic predisposition (HLA-DR3 and DR4) [7].
Making the diagnosis — the simplified score
The diagnosis of AIH requires the right clinical context (a hepatocellular pattern, exclusion of viral and drug causes), supportive autoantibodies, a high IgG, and a compatible biopsy. The simplified diagnostic criteria (Hennes 2008) are the bedside tool an examiner expects a candidate to use [8].
| Variable | Points |
|---|---|
| ANA or SMA 1:40 | 1 |
| ANA or SMA 1:80, OR anti-LKM1 1:40 or greater | 2 |
| IgG above the upper limit of normal | 1 |
| IgG more than 1.1 times the upper limit | 2 |
| Histology compatible with AIH | 1 |
| Histology typical of AIH (interface hepatitis, plasma cells) | 2 |
| Absence of viral hepatitis | 2 |
| Total 6 or more | Probable AIH |
| Total 7 or more | Definite AIH |
The simplified score has a sensitivity of 88 percent and specificity of 97 percent at the cut-off of 6. The key point: exclusion of viral hepatitis is mandatory (HBV, HCV, HEV) — drug-induced liver injury (nitrofurantoin, minocycline, methyldopa, herbal supplements) can cause an AIH-like picture and must be excluded [8].
DWE high-yield trap: Around 10 to 20 percent of AIH patients are seronegative (negative ANA, SMA and anti-LKM1) at presentation. A high IgG with interface hepatitis on biopsy, in the right clinical context and with viral and drug causes excluded, still makes the diagnosis. Do not dismiss a hepatocellular pattern with a high IgG just because the antibodies are negative. Check anti-SLA/LP in the seronegative case [6].
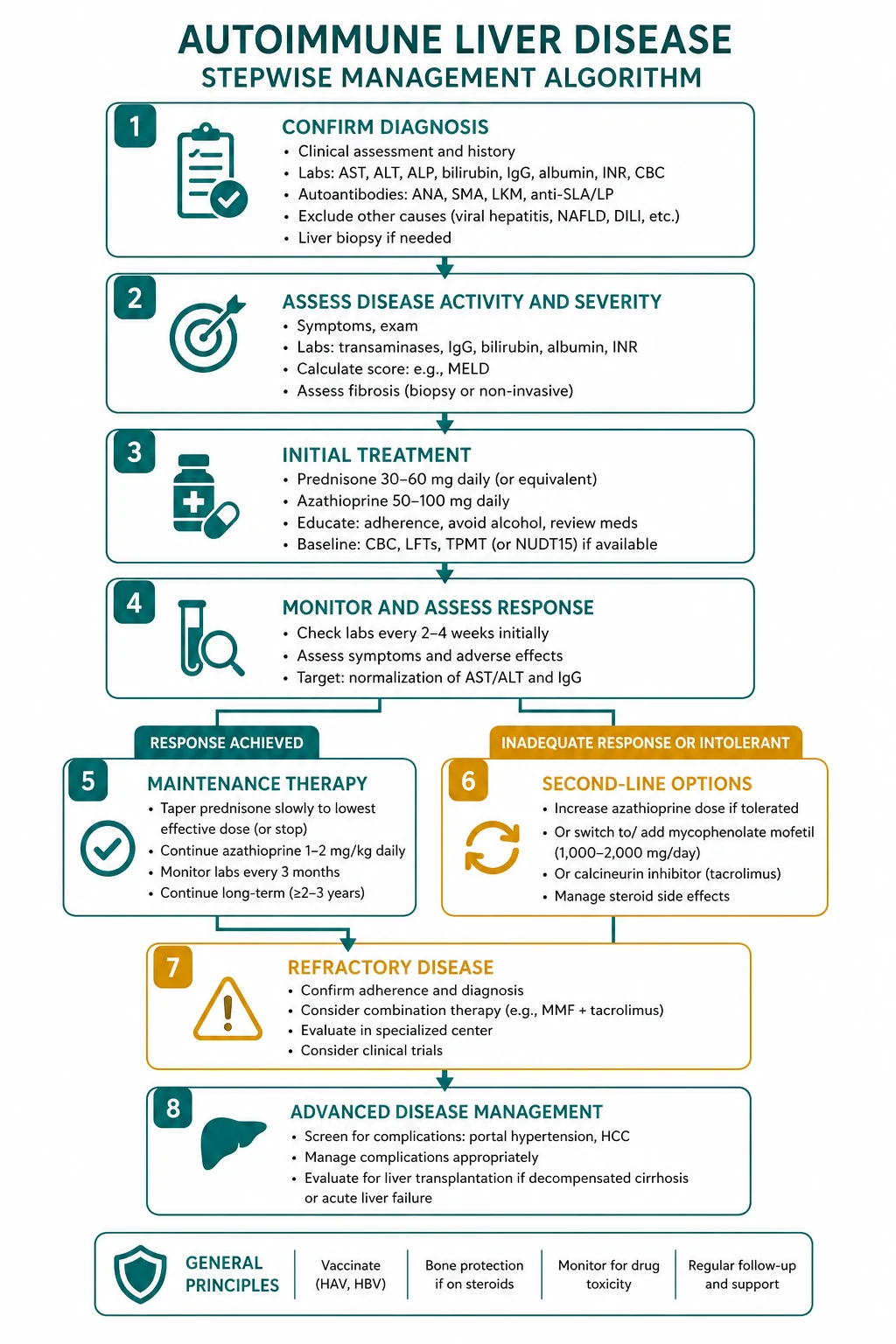
Treatment — prednisolone plus azathioprine
The standard regimen is induction with prednisolone plus azathioprine, then maintenance, then an attempt at withdrawal. [1]
- Induction — prednisolone 0.5 to 1 mg/kg/day (typically 40 to 60 mg daily), tapered over weeks to a maintenance dose, PLUS azathioprine 1 to 2 mg/kg/day added once the diagnosis is confirmed and the TPMT (thiopurine methyltransferase) activity has been checked (to avoid fatal myelosuppression in TPMT-deficient patients).
- Maintenance — aim for biochemical remission (normal transaminases and IgG) and histological remission (no interface hepatitis on a follow-up biopsy). Maintenance is typically for at least 24 months before attempting withdrawal.
- Withdrawal — taper over months; about 50 percent relapse and need lifelong maintenance. [1]
The alternatives: budesonide (a first-pass corticosteroid, 3 mg three times daily) is a steroid-sparing induction option in non-cirrhotic AIH (it is ineffective in cirrhosis because of portosystemic shunting). Mycophenolate mofetil (1 to 2 g daily) is the preferred second-line for azathioprine intolerance. Tacrolimus is used for refractory disease. An examiner wants prednisolone plus azathioprine as the standard regimen [6].
DWE high-yield: The answer to "first-line treatment for AIH" is prednisolone plus azathioprine, with TPMT checked before azathioprine. Budesonide is the steroid-sparing alternative in non-cirrhotic AIH. Mycophenolate is second-line for azathioprine intolerance. [1]
Monitoring and the long term
- Full blood count — weekly for the first month on azathioprine, then monthly, then three-monthly (myelosuppression).
- Liver function — at every visit (azathioprine hepatotoxicity, disease activity).
- Bone protection on prednisolone — calcium, vitamin D, a DEXA, and a bisphosphonate as needed.
- Blood glucose and blood pressure on prednisolone.
- Sun protection — azathioprine increases skin cancer risk.
- Vaccinations — influenza, pneumococcus, COVID-19, hepatitis A and B before immunosuppression; avoid live vaccines. [1]
Transplant
Liver transplant is indicated for acute liver failure (apply the King's College Criteria for non-paracetamol ALF), for decompensated cirrhosis, and for HCC within transplant criteria. AIH can recur in the graft (20 to 30 percent at 5 to 10 years), and recurrence is associated with an increased risk of graft loss — closer immunosuppression monitoring is required [6].
Overlap syndromes
Overlap syndromes are patients with features of two autoimmune liver diseases — most commonly AIH with PBC, or AIH with PSC. They are a recurring exam topic because they test whether a candidate can apply the diagnostic criteria and reason about a combined treatment strategy [9].
AIH-PBC overlap — the Paris criteria
The Paris criteria require two of three AIH features AND two of three PBC features: [1]
- AIH features (need 2 of 3): interface hepatitis; ALT 2 to 5 times the upper limit; IgG 2 times the upper limit OR positive ANA/SMA.
- PBC features (need 2 of 3): AMA positive; a compatible biopsy (florid duct lesion); raised IgM. [1]
The management is UDCA plus immunosuppression (prednisolone plus azathioprine). The IAIHG position statement (Boberg 2011) cautions that overlap is partly a research concept and that patients should be treated for their predominant feature, but in practice the combined strategy is used for the clear overlap case [9].
AIH-PSC overlap
More common in children and young adults (autoimmune sclerosing cholangitis in paediatrics), the AIH-PSC overlap has the autoantibodies and histology of AIH with the cholangiographic findings of PSC. Management combines immunosuppression (for the AIH component, which does respond) with the PSC surveillance strategy. The immunosuppression does not alter the PSC natural history [9].
DWE trap: Do not diagnose overlap on the basis of a single positive antibody. The Paris criteria require two of three features from each disease. A patient with PBC (AMA positive, raised IgM, cholestatic) who happens to have a low-titre ANA does NOT have overlap — the ANA alone is non-specific and is found in many cholestatic and healthy patients [9].
The investigation strategy — abnormal LFTs
The first decision in any patient with unexplained abnormal LFTs is the pattern: is it cholestatic (ALP and gamma-GT raised disproportionately) or hepatocellular (ALT and AST raised disproportionately)? [1]

| Step | Cholestatic pattern | Hepatocellular pattern |
|---|---|---|
| 1 | Ultrasound — duct dilatation? | Ultrasound — parenchymal change, obstruction? |
| 2 | If ducts dilated → MRCP → obstruction (stone, stricture, tumour) | Viral serology — HBV, HCV, HEV |
| 3 | If ducts normal → AMA, IgM, IgG, IgG4 | Drug and toxin history — DILI |
| 4 | AMA positive, IgM raised → PBC | ANA, SMA, anti-LKM1, anti-SLA, IgG |
| 5 | AMA negative, IBD → MRCP → PSC | IgG raised, interface hepatitis → AIH (simplified score) |
| 6 | IgG4 raised → IgG4-sclerosing cholangitis | Wilson markers, alpha-1-antitrypsin, haemochromatosis, NAFLD |
| 7 | Biopsy if diagnosis uncertain or for staging | Biopsy for the simplified score and to stage |
The autoantibody panel (AMA, ANA, SMA, anti-LKM1) and the immunoglobulins (IgM for PBC, IgG for AIH) are the high-yield first-line tests in any patient with suspected autoimmune liver disease. Add an IgG4 in any cholestatic patient to exclude IgG4-sclerosing cholangitis. [1]
Management and surveillance — the integrated plan
Cancer surveillance
The cancer surveillance burden differs by disease: [1]
| Disease | Cholangiocarcinoma | Colorectal cancer | HCC |
|---|---|---|---|
| PBC | Not increased | Not increased | In cirrhosis — six-monthly US plus AFP |
| PSC | Annual MRI/MRCP plus CA 19-9 | Annual colonoscopy (in IBD) | In cirrhosis — six-monthly US plus AFP |
| AIH | Not increased | Not increased | In cirrhosis — six-monthly US plus AFP |
Bone health
Osteoporosis is the commonest bone disease in PBC and PSC, and is compounded by corticosteroids in overlap AIH. Baseline and biennial DEXA, with calcium, vitamin D, and a bisphosphonate as needed. Avoid over-treating with corticosteroids in the overlap patient — budesonide is the steroid-sparing option in non-cirrhotic AIH. [1]
Fat-soluble vitamin deficiency
Severe cholestasis (advanced PBC and PSC) causes deficiency of vitamins A, D, E and K. Check and replace annually in advanced disease — night blindness (A), osteomalacia (D), neuropathy and ataxia (E), and coagulopathy (K). [1]
Liver transplant — the indications
Liver transplant is the definitive treatment for the end-stage of all three diseases. The indications: [1]
| Indication | Detail |
|---|---|
| Decompensated cirrhosis | Ascites, spontaneous bacterial peritonitis, hepatic encephalopathy, variceal haemorrhage, hepatorenal syndrome, hepatopulmonary syndrome, or a rising bilirubin refractory to therapy |
| Hepatocellular carcinoma | Within transplant criteria (Milan criteria: single lesion under 5 cm, or up to 3 lesions each under 3 cm) |
| PSC-specific | Recurrent bacterial cholangitis, or intractable pruritus |
| AIH-specific | Acute liver failure (apply the King's College Criteria) |
Recurrence in the graft: PBC recurs in around 11 to 42 percent, PSC in 10 to 25 percent, and AIH in 20 to 30 percent. Recurrent PSC is the most likely to cause graft loss. UDCA is continued post-transplant in PBC to reduce recurrence risk [4][6].
The common exam traps
- Confusing PBC with PSC — PBC is a woman with AMA and raised IgM and a normal cholangiogram; PSC is a (usually male) patient with IBD, normal IgM and AMA, and a strictured, beaded cholangiogram. The cholangiogram is the discriminator.
- Treating PSC like PBC with UDCA — high-dose UDCA is harmful in PSC (the 2009 trial), there is no effective drug, and the management is surveillance and ERCP for dominant strictures.
- Missing AIH because the antibodies are negative — 10 to 20 percent are seronegative; a high IgG with interface hepatitis still makes the diagnosis. Check anti-SLA/LP.
- Over-diagnosing overlap syndrome — the Paris criteria require two of three features from each disease; do not diagnose overlap on a single non-specific antibody.
- Forgetting the colorectal cancer risk in PSC-IBD — annual colonoscopy from diagnosis, regardless of the duration or activity of the colitis.
- Prescribing OCA in decompensated cirrhosis — OCA can precipitate liver failure and death in Child-Pugh B and C cirrhosis; check the stage before starting.
- Missing IgG4-sclerosing cholangitis — a steroid-responsive mimic of PSC; check a serum IgG4 in any cholestatic patient.
- Not checking TPMT before azathioprine — TPMT deficiency causes fatal myelosuppression; check before starting. [1]
Regional guideline anchoring
- ANZ (GESA) — UDCA 13 to 15 mg/kg/day for PBC, OCA for the inadequate responder, no effective therapy for PSC with surveillance emphasis, prednisolone plus azathioprine for AIH. The Paris and simplified criteria for overlap diagnosis. [1]- UK (BSG, NICE) — broadly the same; OCA (NICE TA) for the inadequate responder to UDCA in PBC within its licensed population; the Cambridge and Oxford PSC centres lead on surveillance.
- US (AASLD) — the 2018 PBC guidance and 2024 PSC guidance, the 2010 AIH guidance; OCA (FDA-approved 2016) for the inadequate responder.
- Europe (EASL) — the 2017 PBC guideline, the 2009 cholestatic liver diseases guideline. [1]
Drug doses are verified; the ANZ dose/brand is primary where it differs (it does not materially here). [1]
Summary
Autoimmune liver disease is a family of three. Classify by the LFT pattern (cholestatic vs hepatocellular), the key antibody (AMA for PBC, anti-LKM1/ANA/SMA for AIH), the immunoglobulin (IgM for PBC, IgG for AIH), and the histology (florid duct lesion for PBC, interface hepatitis with plasma cells for AIH, periductal onion-skin fibrosis for PSC). Treat PBC with UDCA (and OCA or a fibrate for the inadequate responder), manage PSC with surveillance and ERCP for dominant strictures (no effective drug), and treat AIH with prednisolone plus azathioprine. Transplant for decompensation, HCC, recurrent cholangitis or intractable pruritus. The overlap syndromes sit at the intersections and need a combined strategy. [1]
One-line answer for the viva: "I classify autoimmune liver disease by the LFT pattern and the autoantibody panel — a cholestatic pattern with AMA and raised IgM is PBC (treat with UDCA), a cholestatic pattern with IBD and a normal AMA is PSC (surveillance and ERCP, no drug), and a hepatocellular pattern with a high IgG is AIH (prednisolone plus azathioprine). The overlap syndromes need a combined strategy; the cholangiogram, the IgG4 and the simplified score are the discriminators I use when the diagnosis is not clear." [1]
Sources
Kaplan MM, Gershwin ME. Primary Biliary Cirrhosis. N Engl J Med 2005;353(12):1261-1273 [1]; Lindor KD, Dickson ER, Baldus WP, et al. Ursodiol for the Long-Term Treatment of Primary Biliary Cirrhosis. N Engl J Med 1994;330:1342-1347 [2]; Nevens F, Andreone P, Mazzella G, et al. A Placebo-Controlled Trial of Obeticholic Acid in Primary Biliary Cholangitis. N Engl J Med 2016;375(7):631-643 [3]; Hirschfield GM, Karlsen TH, Lindor KD, Adams DH. Primary Sclerosing Cholangitis. Lancet 2013;382(9904):1587-1599 [4]; Karlsen TH, Folseraas T, Hirschfield GM, et al. Primary Sclerosing Cholangitis. Nat Rev Dis Primers 2025 [5]; Manns MP, Czaja AJ, Gorham JD, et al. Diagnosis and Management of Autoimmune Hepatitis. Hepatology 2010;51(6):2193-2213 [6]; Krawitt EL. Autoimmune Hepatitis. N Engl J Med 2006;354(1):54-66 [7]; Hennes EM, Zeniya M, Czaja AJ, et al. Simplified Criteria for the Diagnosis of Autoimmune Hepatitis. Hepatology 2008;48(1):169-176 [8]; Boberg KM, Chapman RW, Hirschfield GM, et al. Overlap Syndromes: the IAIHG Position Statement. J Hepatol 2011;54(2):374-385 [9]; Hilsden M, et al. Incidence of Hepatobiliary Malignancies in PSC: Systematic Review and Meta-analysis. Clin Gastroenterol Hepatol 2025 [10].
AASLD Practice Guidance on Primary Biliary Cholangitis (2018); AASLD Practice Guidance on Primary Sclerosing Cholangitis (2024); AASLD Diagnosis and Management of Autoimmune Hepatitis (2010); EASL Clinical Practice Guidelines on PBC and PSC; GESA hepatology guidelines. [1]
References
- [1]Kaplan MM, Gershwin ME Primary biliary cirrhosis N Engl J Med, 2005.PMID 16177252
- [2]Lindor KD, Dickson ER, Baldus WP, et al. Ursodiol for the long-term treatment of primary biliary cirrhosis. The UDCA-PBC Study Group N Engl J Med, 1994.PMID 8152446
- [3]Nevens F, Andreone P, Mazzella G, et al. A Placebo-Controlled Trial of Obeticholic Acid in Primary Biliary Cholangitis N Engl J Med, 2016.PMID 27532829
- [4]Hirschfield GM, Karlsen TH, Lindor KD, Adams DH Primary sclerosing cholangitis Lancet, 2013.PMID 23810223
- [5]Karlsen TH, Folseraas T, Hirschfield GM, et al. Primary sclerosing cholangitis Nat Rev Dis Primers, 2025.PMID 40082445
- [6]Manns MP, Czaja AJ, Gorham JD, et al. Diagnosis and management of autoimmune hepatitis Hepatology, 2010.PMID 20513004
- [7]Krawitt EL Autoimmune hepatitis N Engl J Med, 2006.PMID 16394302
- [8]Hennes EM, Zeniya M, Czaja AJ, et al. Simplified criteria for the diagnosis of autoimmune hepatitis Hepatology, 2008.PMID 18537184
- [9]Boberg KM, Chapman RW, Hirschfield GM, et al. Overlap syndromes: the International Autoimmune Hepatitis Group (IAIHG) position statement on a controversial issue J Hepatol, 2011.PMID 21067838
- [10]Hilsden M, van der Meer AJ, Sclair SN, et al. Incidence of Hepatobiliary Malignancies in Primary Sclerosing Cholangitis: Systematic Review and Meta-analysis Clin Gastroenterol Hepatol, 2025.PMID 39709139