Phys · hepatic
Chronic Liver Disease and Cirrhosis
Also known as cirrhosis · chronic liver disease · portal hypertension · decompensated cirrhosis · liver cirrhosis · hepatic cirrhosis · end-stage liver disease · MASLD cirrhosis · alcohol-related liver disease · ALD
Consultant-physician-depth guide to chronic liver disease and cirrhosis — aetiology, fibrosis pathophysiology, the four decompensating events, and staging-driven management from primary prophylaxis to transplant assessment. Structured for FRACP DWE and DCE preparation.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Chronic Liver Disease and Cirrhosis
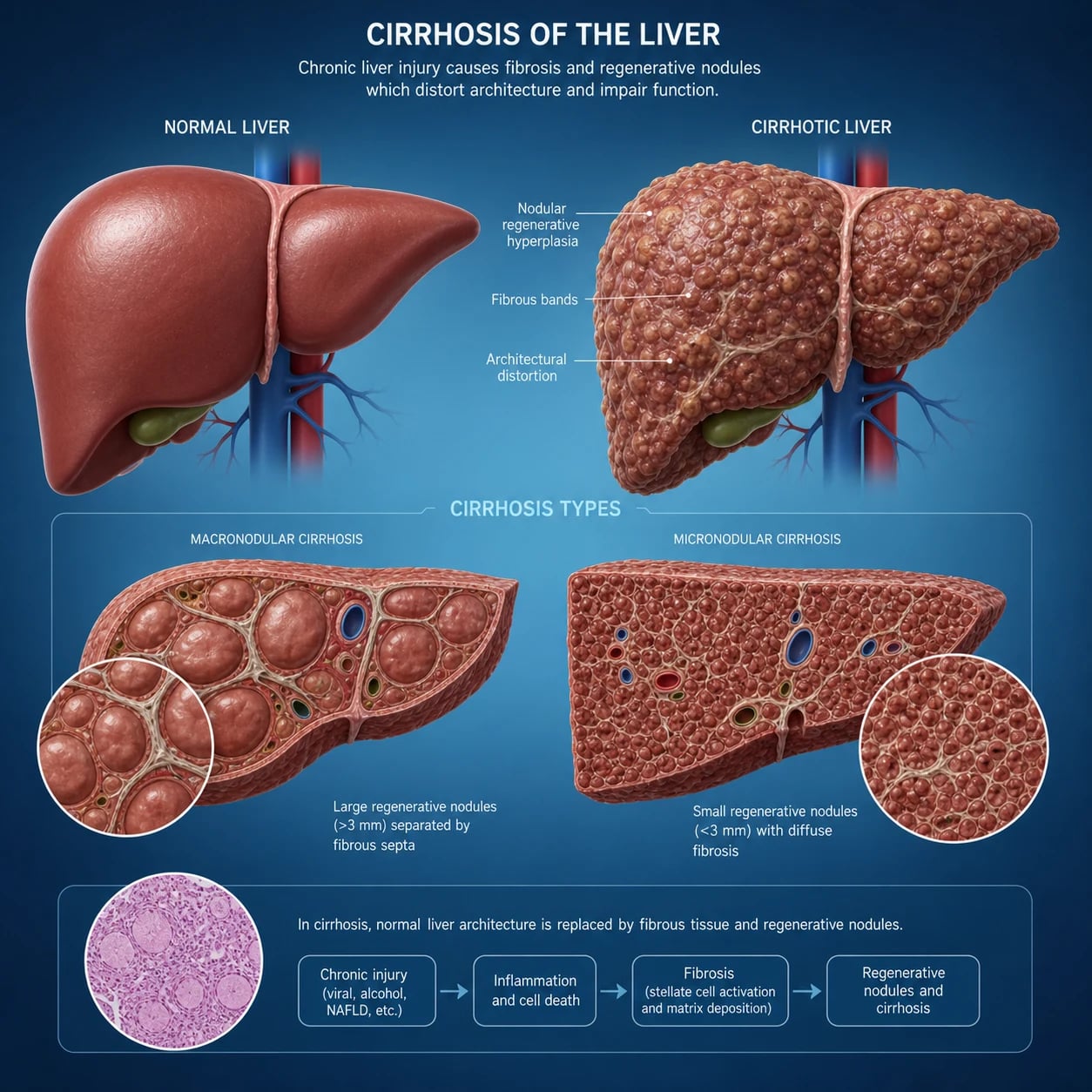
The answer first
Cirrhosis is the late stage of hepatic fibrosis in which regenerative nodules and fibrous septa distort the hepatic architecture and produce the structural and haemodynamic consequences of chronic liver disease. It is not a single disease — it is the common endpoint of many chronic liver injuries. The single most important concept for the exam is that cirrhosis is staged, and the stage drives everything. [1]
Cirrhosis has four clinical stages, and the patient moves in one direction without reversal of the underlying cause: [1]
- Compensated without varices — portal pressure below the threshold for clinically significant portal hypertension (CSPH).
- Compensated with varices — CSPH present (HVPG greater than or equal to 10 mmHg), no decompensation.
- Decompensated — first decompensating event: ascites, variceal bleed, encephalopathy or jaundice.
- Late decompensated — recurrent or multiple decompensating events, sepsis, organ failure. [1]
DCE trap: The biggest management pivot in hepatology is the move from compensated to decompensated cirrhosis. In the compensated phase you prevent the first decompensation (treat the cause, start a non-selective beta-blocker if CSPH). In the decompensated phase you treat complications and assess for transplant. Name the stage first in every long case. [1]
Aetiology — exhaust the cause
Always identify and treat the cause, because aetiology-specific therapy can halt or reverse fibrosis even in established cirrhosis. The mnemonic CIRCULATION-in-reverse is less useful than a structured screen: [1]
| Category | Cause | Key test |
|---|---|---|
| Viral | Hepatitis B (HBV), hepatitis C (HCV) | HBsAg, anti-HBs, anti-HBc; HCV Ab ± RNA |
| Metabolic | MASLD (formerly NAFLD); haemochromatosis; Wilson disease; alpha-1-antitrypsin deficiency | Metabolic criteria, ferritin/genetics, ceruloplasmin, A1AT phenotype |
| Alcohol | Alcohol-related liver disease (ALD) | AUDIT-C, history, AST/ALT ratio greater than 2, GGT |
| Autoimmune | Autoimmune hepatitis (AIH) | ANA, SMA, anti-LKM1, IgG |
| Cholestatic | Primary biliary cholangitis (PBC); primary sclerosing cholangitis (PSC) | AMA, IgM (PBC); MRCP, p-ANCA (PSC) |
| Vascular | Budd-Chiari, right heart failure, nodular regenerative hyperplasia | Doppler US, MR/CT venography |
| Drug/toxin | Methotrexate, amiodarone, anabolic steroids | Exposure history |
MASLD nomenclature (2023 update)
A 2023 multisociety Delphi consensus renamed non-alcoholic fatty liver disease as metabolic dysfunction-associated steatotic liver disease (MASLD), and NASH as MASH [7]. MASLD requires hepatic steatosis plus at least one of five cardiometabolic criteria (overweight/obesity, type 2 diabetes, hypertension, dyslipidaemia, prediabetes). A new hybrid category, MetALD, covers patients with MASLD who also drink above the MASLD threshold but below the alcohol-related liver disease threshold. The exam expects you to know both the old and new names — "NAFLD/NASH" still appears in older questions, but "MASLD/MASH" is the current term.
DWE high-yield: MASLD is now the leading cause of cirrhosis worldwide and the fastest-growing indication for liver transplant. Expect a stem describing a patient with metabolic syndrome, elevated AST/ALT and an incidental nodular liver. [1]
Pathophysiology of fibrosis
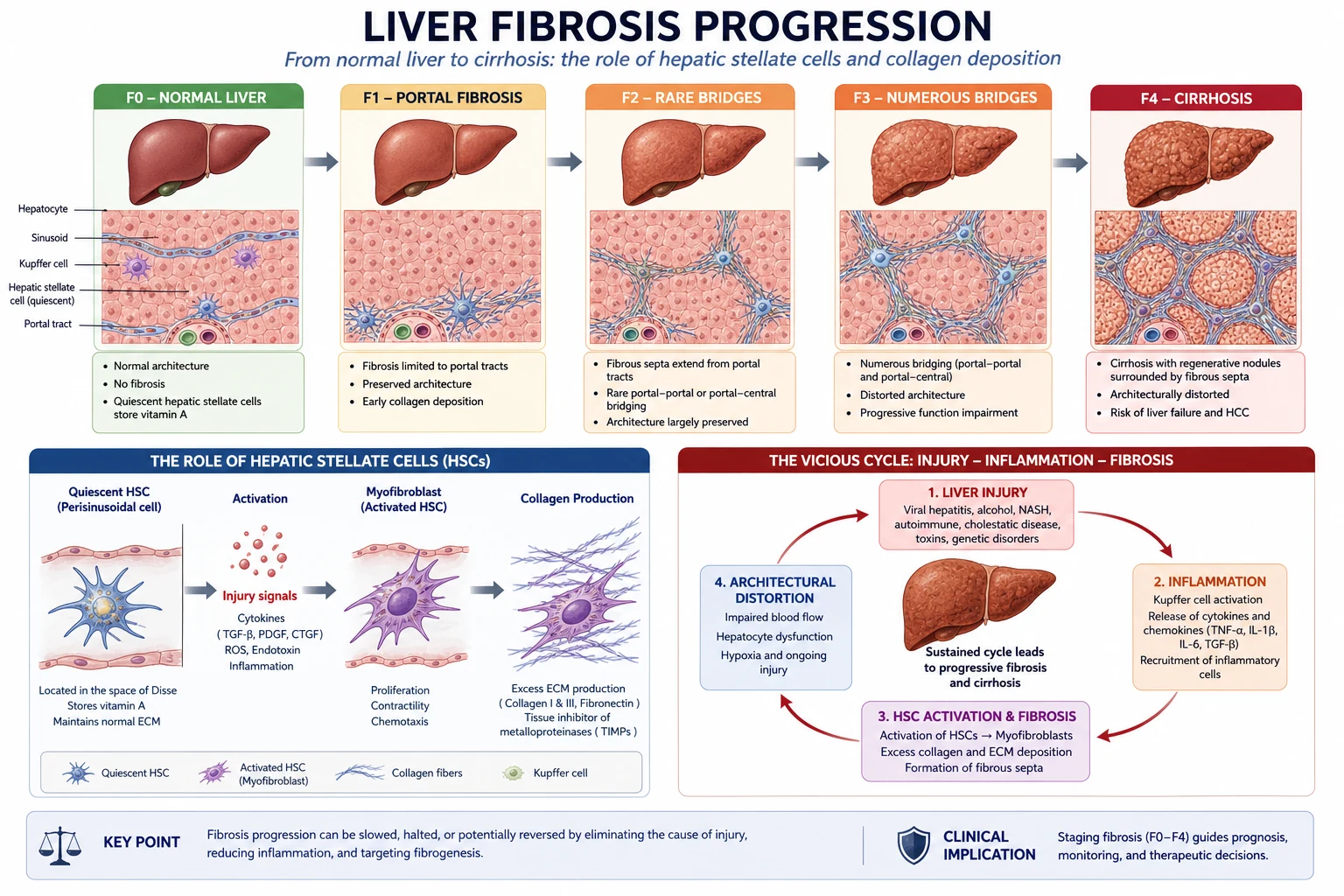
Cirrhosis is a fibrogenic disease, not simply cell death. The central cell is the hepatic stellate cell (Ito cell), which stores vitamin A in health. The cascade runs: [1]
- Chronic injury (viral, metabolic, toxic, immune) causes hepatocyte apoptosis and necrosis.
- Inflammation recruits Kupffer cells and releases cytokines — most importantly TGF-beta, the master fibrogenic cytokine.
- Stellate cells activate, transdifferentiate into myofibroblast-like cells, proliferate and secrete massive quantities of type I and III collagen.
- Collagen deposits in the space of Disse, creating basement membranes (capillarisation of sinusoids) that impair oxygen and substrate exchange.
- Architectural distortion — regenerative nodules surrounded by fibrous septa — disrupts hepatic architecture and increases intrahepatic vascular resistance.
- Portosystemic shunting develops as portal blood is forced through collaterals (oesophageal, gastric, rectal, retroperitoneal). [1]
Two consequences follow that govern every complication: [1]
- Portal hypertension — because outflow is obstructed. Clinically significant portal hypertension (CSPH) is defined as a hepatic venous pressure gradient (HVPG) greater than or equal to 10 mmHg. Varices form and bleed when HVPG exceeds 12 mmHg.
- Hepatic synthetic dysfunction — because functional hepatocyte mass is lost. This drives coagulopathy, hypoalbuminaemia, jaundice and impaired drug metabolism. [1]
The fibrosis is potentially reversible if the cause is removed early enough. Removing the cause (HCV cure, alcohol abstinence, venesection in haemochromatosis) can regress fibrosis by one or two METAVIR stages and even convert decompensated to compensated cirrhosis in some patients. [1]
DWE high-yield mechanism question: "Why does cirrhosis cause both portal hypertension and liver failure?" Answer: architectural distortion (regenerative nodules plus fibrous septa) raises intrahepatic resistance to produce portal hypertension, while loss of functional hepatocyte mass and sinusoidal capillarisation produce synthetic failure. [1]
Clinical presentation
Stigmata of chronic liver disease (the short-case core)
Elicit these systematically in any suspected chronic liver disease. Group them to retrieve them under exam pressure. [1]
Hands and limbs:
- Palmar erythema — thenar/hypothenar sparing of the central palm.
- Dupuytren contracture — especially alcohol-related disease.
- Clubbing — advanced disease, hypertrophic osteoarthropathy.
- Asterixis (liver flap) — metabolic flap of hepatic encephalopathy; a negative findings must be actively documented.
- Leuconychia — hypoalbuminaemia.
- Peripheral oedema — hypoalbuminaemia. [1]
Face and chest:
- Spider naevi (central arteriole with radiating vessels, blanches with pressure) — oestrogen excess. More than a few is significant.
- Gynaecomastia — true glandular breast tissue (distinguish from lipomastia); oestrogen/testosterone imbalance.
- Loss of axillary and pubic hair, testicular atrophy — hypogonadism.
- Parotid enlargement — alcohol-related disease.
- Fetor hepaticus — sweet musty breath from mercaptans. [1]
Abdomen:
- Hepatomegaly or a shrunken hard liver.
- Splenomegaly — portal hypertension (the single most specific sign of portal HTN).
- Ascites — shifting dullness, fluid thrill.
- Caput medusae — recanalised umbilical vein.
- Hepatic bruit or friction rub (hepatocellular carcinoma). [1]
DCE short-case trap: Examiners test whether you can discriminate signs that mean chronic liver disease from signs that mean portal hypertension from signs that mean hepatic failure. Spider naevi and palmar erythema = chronic liver disease. Splenomegaly and caput = portal hypertension. Asterixis, jaundice and fetor = hepatic failure. State which group each sign belongs to as you present. [1]
The decompensating events
Decompensation is the transition to a new, worse prognosis. The four decompensating events, in order of frequency: [1]
- Ascites — the most common first decompensation; 1-year mortality roughly 20% and 5-year mortality around 50% once it appears.
- Variceal bleeding — presents with haematemesis or melaena; 6-week mortality around 15 to 20%.
- Hepatic encephalopathy — spectrum from subtle cognitive change to coma.
- Jaundice — bilirubin greater than 50 micromol/L marks decompensation. [1]
After the first decompensation, 1-year mortality rises to roughly 20 to 30%. Acute-on-chronic liver failure (ACLF) — decompensation plus organ failure within 4 weeks — carries up to 50% 28-day mortality. [1]
Investigations
Confirm and stage fibrosis
| Investigation | Purpose |
|---|---|
| Transient elastography (FibroScan) | Non-invasive liver stiffness; stiffness greater than 12 to 15 kPa suggests cirrhosis; Baveno VII uses stiffness to define CSPH |
| FIB-4 and APRI scores | Serum-based fibrosis staging; FIB-4 less than 1.45 reliably excludes advanced fibrosis in those under 65 |
| Liver biopsy | Gold standard for fibrosis staging (METAVIR 0 to 4) when non-invasive tests are indeterminate or to confirm AIH, Wilson, PBC |
| CT or MRI liver | Defines nodularity, splenomegaly, collaterals; MRI/CT screens for HCC |
| Upper endoscopy | Detects and grades varices; all newly diagnosed cirrhosis patients should have a screening OGD |
Identify the aetiology
| Test | Aetiology screened |
|---|---|
| HBsAg, anti-HBs, anti-HBc, HCV Ab | Viral hepatitis |
| Ferritin, transferrin saturation, HFE genetics | Haemochromatosis |
| Caeruloplasmin, 24-hour urinary copper, slit-lamp (Kayser-Fleischer rings) | Wilson disease (under 40 years) |
| Alpha-1-antitrypsin level and phenotype | A1AT deficiency |
| ANA, SMA, anti-LKM1, IgG, IgM | Autoimmune hepatitis (IgG), PBC (IgM) |
| Anti-mitochondrial antibody (AMA) | PBC |
| MRCP | PSC (multifocal strictures, "pruned tree" appearance) |
| AUDIT-C, gamma-GT, AST/ALT ratio greater than 2 | Alcohol-related disease |
Assess severity and prognosis
Child-Pugh score (CPT) uses bilirubin, albumin, INR, ascites and encephalopathy: [1]
| Parameter | 1 point | 2 points | 3 points |
|---|---|---|---|
| Bilirubin (micromol/L) | less than 34 | 34 to 51 | greater than 51 |
| Albumin (g/L) | greater than 35 | 28 to 35 | less than 28 |
| INR | less than 1.7 | 1.7 to 2.3 | greater than 2.3 |
| Ascites | none | mild (diuretic-responsive) | moderate to severe (refractory) |
| Encephalopathy | none | grade 1 to 2 | grade 3 to 4 |
Child-Pugh A = 5 to 6 (1-year survival around 100%), B = 7 to 9 (around 80%), C = 10 to 15 (around 45%). [1]
MELD-Na is the score that governs transplant priority. It uses bilirubin, INR, creatinine and sodium, with the sodium term adding discriminating power. Typical thresholds: MELD less than 15 — transplant not usually indicated; 15 to 25 — referral and workup; greater than 25 — high priority; greater than 35 — very high 90-day mortality, urgent. [1]
DWE trap: Child-Pugh governs prognosis estimation and trial eligibility; MELD-Na governs transplant allocation. A classic MCQ asks which score determines transplant priority — the answer is MELD-Na, not Child-Pugh. [1]
Staging-driven management
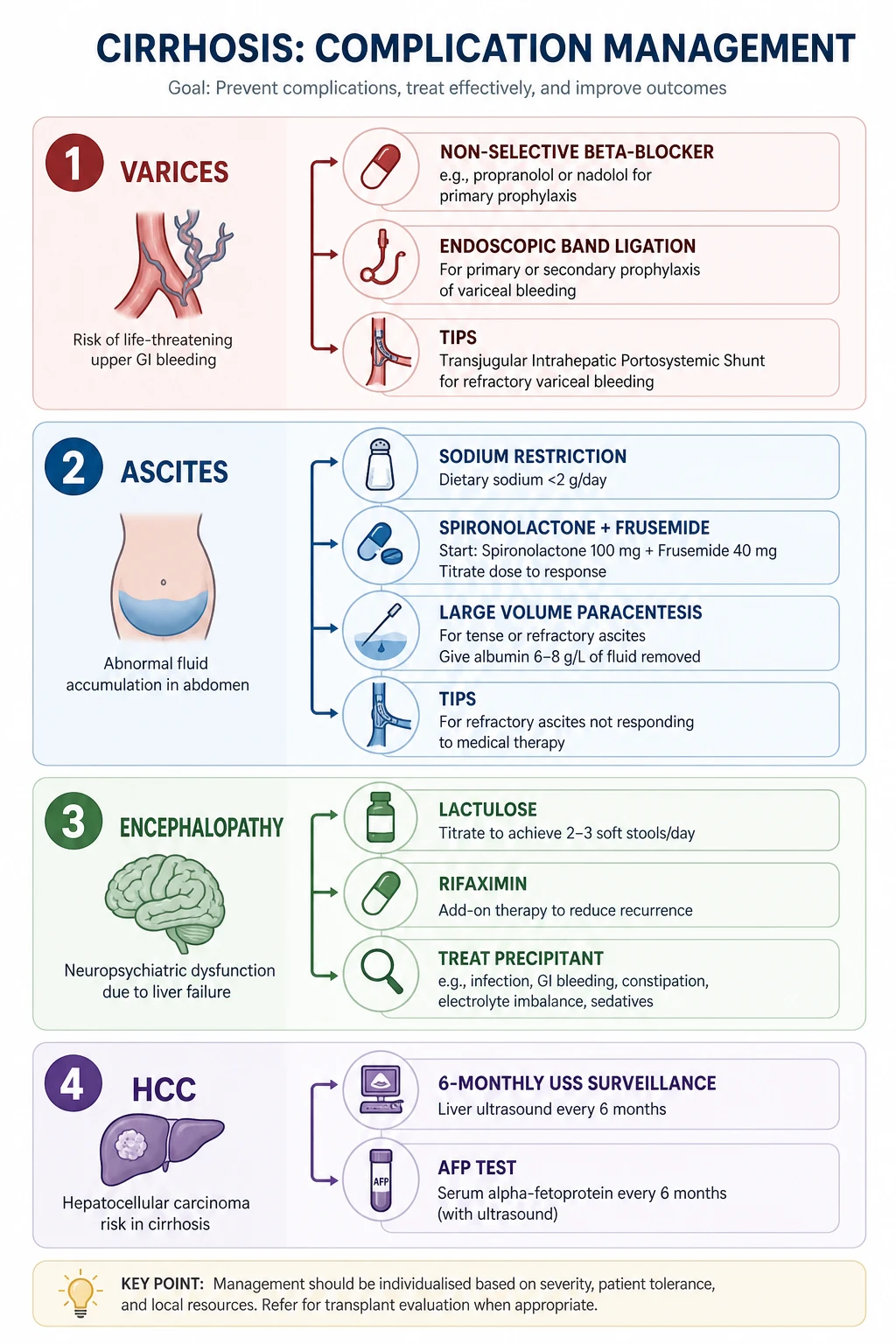
The management algorithm pivots on the stage. In compensated cirrhosis the goal is to prevent the first decompensation — treat the aetiology and start a non-selective beta-blocker when clinically significant portal hypertension is present. In decompensated cirrhosis the goal shifts to treating each complication (portal hypertension, ascites, encephalopathy, hepatorenal syndrome, HCC surveillance) and assessing transplant candidacy early. Every complication below is managed with this staging logic in mind. [1]
Complication 1 — portal hypertension and varices

The management of portal hypertension is layered across three phases: prevent first decompensation, prevent first bleed (primary prophylaxis), and prevent rebleeding (secondary prophylaxis). [1]
Preventing the first decompensation (PREDESCI)
The PREDESCI trial showed that in patients with compensated cirrhosis and CSPH (HVPG greater than or equal to 10 mmHg) but no high-risk varices, long-term non-selective beta-blocker (NSBB) therapy reduced the composite of decompensation or death from 27% to 16% (HR 0.51) over a median of 37 months [1]. The benefit was driven mainly by a reduction in ascites. Baveno VII incorporated this: NSBBs (or carvedilol) are recommended for compensated patients with CSPH defined non-invasively by liver stiffness and platelet count [3].
This is a paradigm shift — NSBBs now prevent decompensation, not just bleeding. [1]
Primary prophylaxis (prevent first variceal bleed)
For patients with medium or large varices, or small varices with red wale signs or Child-Pugh B/C: [1]
- NSBB — propranolol 20 to 40 mg BID (titrate to heart rate 55 to 60), nadolol 40 mg OD, or carvedilol 6.25 mg OD (preferred by many units because it also blocks alpha-1 receptors and lowers portal pressure more effectively).
- Endoscopic variceal ligation (EVL) — alternative, especially if NSBB contraindicated or not tolerated. [1]
Either NSBB or EVL is acceptable; combination is not used for primary prophylaxis. Carvedilol is increasingly favoured. [1]
Secondary prophylaxis (prevent rebleed)
After any variceal bleed, both NSBB and serial EVL are used in combination. EVL alone has a high rebleed rate; adding an NSBB reduces it. Repeat banding every 2 to 4 weeks until variceal eradication, then surveillance OGD every 3 to 6 months. [1]
Acute variceal bleeding (emergency)
A resuscitation and treatment bundle, delivered simultaneously: [1]
- Airway, breathing, circulation — two large-bore cannulae, crossmatch, restricted transfusion (target haemoglobin around 70 to 80 g/L; over-transfusion raises portal pressure and worsens bleeding).
- Vasoactive drug at admission — terlipressin 2 mg IV every 4 hours (ANZ first-line; reduces splanchnic blood flow), octreotide or somatostatin as alternatives. Continue for 2 to 5 days. [1]3. Prophylactic antibiotic — ceftriaxone 1 g IV OD for up to 7 days. Antibiotics reduce mortality independently of infection risk — a frequent MCQ answer.
- Endoscopic band ligation within 12 hours.
- Early TIPS (transjugular intrahepatic portosystemic shunt) in high-risk patients — Child-Pugh C (score 10 to 13) or Child-Pugh B with active bleeding at endoscopy — within 72 hours. The García-Pagán trial showed early covered-TIPS reduced treatment failure and improved survival in this group [2].
- Balloon tamponade (Sengstaken-Blakemore) only as a bridge to definitive therapy if uncontrolled bleeding.
DWE high-yield: The two interventions proven to reduce mortality in acute variceal bleeding are antibiotics and early TIPS in high-risk patients. Terlipressin and band ligation reduce bleeding and rebleeding but have weaker mortality evidence. [1]
Complication 2 — ascites and SBP
Ascites evaluation: the SAAG
The serum-ascites albumin gradient (SAAG) = serum albumin minus ascitic albumin. It classifies the mechanism: [1]
| SAAG | Mechanism | Examples |
|---|---|---|
| 11 g/L or higher (high SAAG) | Portal hypertension | Cirrhosis, right heart failure, Budd-Chiari |
| less than 11 g/L (low SAAG) | Non-portal hypertensive | Peritoneal carcinomatosis, TB peritonitis, nephrotic syndrome |
Cirrhotic ascites is high-SAAG by definition. A diagnostic tap is mandatory on every new admission with ascites — send cell count, culture, albumin and cytology. [1]
Management of uncomplicated ascites
- Sodium restriction — less than 88 mmol (2 g) per day. [1]- Diuretics — spironolactone and frusemide in a 100:40 ratio (e.g. spironolactone 100 mg plus frusemide 40 mg, titrating upward maintaining the ratio). The ratio maintains normokalaemia. Single-agent spironolactone causes painful gynaecomastia; the frusemide helps avoid hyperkalaemia.
- Water restriction only if hyponatraemia (sodium less than 125 mmol/L).
- Stop NSAIDs, ACE inhibitors, ARBs — they worsen renal perfusion. [1]
Refractory ascites
Ascites that cannot be mobilised or recurs rapidly despite maximal diuretics. Two management pillars: [1]
- Large-volume paracentesis (LVP) — with albumin replacement (8 g of albumin per litre removed above 5 L) to prevent post-paracentesis circulatory dysfunction.
- TIPS — relieves ascites by decompressing the portal system, but trades ascites for hepatic encephalopathy risk; reserved for selected patients with preserved liver function. [1]
Spontaneous bacterial peritonitis (SBP)
SBP is infection of ascitic fluid without an obvious intra-abdominal source. Diagnose with ascitic polymorphonuclear leucocyte count of 250 cells/microlitre or higher. [1]
Treatment:
- Cefotaxime 2 g IV every 8 hours (or ceftriaxone 2 g IV OD) for 5 to 7 days.
- Intravenous albumin — 1.5 g/kg on day 1 and 1 g/kg on day 3. The Sort trial proved albumin reduces renal impairment (33% to 10%) and in-hospital mortality (29% to 10%) in SBP [4]. This dosing is a high-yield MCQ fact.
- Switch to oral secondary prophylaxis (norfloxacin 400 mg OD or ciprofloxacin) if high recurrence risk.
Secondary prophylaxis is given after a first SBP episode; primary prophylaxis (norfloxacin) is for patients with low ascitic protein (less than 15 g/L) plus impaired renal or hepatic function. [1]
DCE long-case trap: Any cirrhotic patient who deteriorates — new encephalopathy, rising creatinine, fever, abdominal pain — must have a diagnostic tap. Missing SBP is a catastrophic and common error. [1]
Complication 3 — hepatic encephalopathy
Hepatic encephalopathy (HE) is a reversible syndrome of impaired brain function due to liver failure and portosystemic shunting. Ammonia is central but not the whole story; inflammation, oxidative stress and altered neurotransmission contribute. [1]
West Haven grading
| Grade | Features |
|---|---|
| Minimal (covert) | No overt symptoms; psychometric or neurophysiological abnormalities only |
| Grade 1 | Mild lack of awareness, sleep disturbance, euphoria or anxiety, shortened attention |
| Grade 2 | Lethargy, apathy, disorientation, asterixis, obvious personality change |
| Grade 3 | Somnolent but arousable, gross disorientation, bizarre behaviour, marked asterixis |
| Grade 4 | Coma |
Asterixis is the key bedside sign for grade 2 to 3 — ask the patient to hold the hands dorsiflexed with arms outstretched for 30 seconds and watch for the irregular flapping. [1]
Management
- Identify and treat the precipitant. The four common precipitants: infection (SBP), gastrointestinal bleed, constipation, electrolyte disturbance (especially from diuretic over-diuresis). Other triggers: sedatives, TIPS, dehydration, alcohol withdrawal.
- Lactulose (a non-absorbable disaccharide) — 15 to 30 mL two to four times daily, titrated to two to three soft bowel motions per day. It lowers ammonia by acidifying the colon (trapping ammonia as ammonium) and via its cathartic effect.
- Rifaximin 550 mg BID — add for secondary prophylaxis of recurrent HE. The Bass trial showed rifaximin added to lactulose reduced breakthrough HE and HE hospitalisations over 6 months [5]. Most patients in the trial were already on lactulose, so rifaximin is an add-on, not a replacement.
- Treat grade 3 to 4 in a high-dependency setting — airway protection, head elevation, avoid sedatives.
DWE high-yield contrast: Lactulose is first-line for both treatment and primary prevention. Rifaximin is for secondary prevention (after a second episode) added to lactulose, not monotherapy. [1]
Complication 4 — hepatorenal syndrome (HRS)
HRS is a functional, potentially reversible renal failure caused by intense splanchnic and renal vasoconstriction in advanced cirrhosis. The kidneys are structurally normal — they fail because of the haemodynamic milieu. [1]
Diagnostic criteria (ICA-AKI definition)
Cirrhosis with ascites plus all of:
- Acute kidney injury: rise in creatinine by 26.5 micromol/L within 48 hours, or a 50% rise from baseline within 7 days.
- No response after 48 hours of diuretic withdrawal and albumin expansion (1 g/kg per day, up to 100 g). [1]- Absence of shock.
- No current or recent nephrotoxic drugs.
- No structural kidney disease (normal urine sediment, proteinuria less than 500 mg/day, normal renal ultrasound). [1]
Always exclude volume-responsive prerenal AKI and ATN first — withdraw diuretics and give albumin for 48 hours before calling it HRS. [1]
Phenotypes (formerly type 1 / type 2)
- HRS-AKI (formerly type 1) — rapidly progressive; doubling of creatinine to more than 221 micromol/L in less than 2 weeks; median survival untreated around 2 to 3 weeks. This is a transplant urgency.
- HRS-AKD/CKD (formerly type 2) — slower, stable moderate renal impairment associated with refractory ascites; median survival around 4 to 6 months. [1]
Treatment
- Terlipressin plus albumin — terlipressin 1 to 2 mg IV every 4 to 6 hours (or continuous infusion) plus albumin 20 to 40 g/day. Titrate to a rising blood pressure and falling creatinine. The CONFIRM trial showed terlipressin achieved verified HRS reversal in 32% versus 17% with placebo [6]. Watch for respiratory failure (11% vs 2% in CONFIRM) and ischaemia — monitor oxygen saturation and perfusion.
- Noradrenaline is the ICU alternative where terlipressin is unavailable or in fluid overload.
- Liver transplant is the definitive treatment. HRS-AKI is a strong indication — refer urgently, because renal recovery depends on the liver recovering.
DCE long-case trap: In a cirrhotic patient with rising creatinine, the first move is stop diuretics, give 48 hours of albumin, exclude SBP — only then call it HRS and start terlipressin. Jumping straight to terlipressin without excluding volume-responsive AKI is a common error. [1]
Hepatocellular carcinoma surveillance
Cirrhosis is the major risk factor for hepatocellular carcinoma (HCC). All patients with cirrhosis (and selected high-risk groups such as chronic HBV in Asian or African men) should undergo surveillance: [1]
- 6-monthly liver ultrasound, with or without alpha-fetoprotein (AFP).
- Any nodule found triggers CT or MRI with multiphase contrast for the characteristic arterial enhancement and washout pattern.
- Treatment options by Milan criteria (one lesion up to 5 cm, or up to three lesions each up to 3 cm, without vascular invasion or extrahepatic spread): resection, ablation (RFA/MWA), transarterial chemoembolisation (TACE), or liver transplant. [1]
Surveillance detects HCC at an earlier, treatable stage and improves survival after adjusting for lead-time bias. [1]
Coagulopathy in cirrhosis
The old teaching was that cirrhosis causes a bleeding diaphysis — "check the INR." The modern "rebalance theory" recognises that cirrhosis reduces both procoagulant and anticoagulant factors in parallel, so the net haemostatic balance is roughly normal. INR measures only the procoagulant side and overestimates bleeding risk. The practical consequences: [1]
- Do not routinely correct INR with fresh frozen plasma before procedures in stable cirrhosis — it does not prevent bleeding.
- Cirrhotic patients are actually at increased thrombotic risk (portal vein thrombosis, DVT/PE). Consider thromboprophylaxis and treat portal vein thrombosis with low-molecular-weight heparin or DOACs (dose-adjusted).
- Use viscoelastic testing (ROTEM/TEG) where available for peri-procedural assessment. [1]
DWE high-yield paradigm shift: A cirrhotic patient with an INR of 2.0 is not automatically a coagulopathic bleeder. The rebalance theory means INR is a poor predictor of bleeding in cirrhosis. [1]
Liver transplant assessment
Transplant is the definitive treatment for decompensated cirrhosis, HCC within Milan criteria, and selected acute liver failure. Assessment is multidisciplinary and considers: [1]
- Severity — MELD-Na governs priority; transplant generally indicated at MELD greater than or equal to 15.
- Aetiology and reversibility — treat the cause before listing (alcohol abstinence, usually 6 months; HCV cure).
- Contraindications — uncontrolled sepsis, extrahepatic malignancy, severe cardiopulmonary disease, ongoing alcohol or substance use, poor social support, uncontrolled psychiatric illness.
- HCC downstaging — lesions within Milan criteria (or downstaged to Milan) qualify for transplant.
- Quality of life and frailty — sarcopenia and frailty predict post-transplant outcomes. [1]
Timing matters: refer early, because workup takes weeks to months and the patient may deteriorate while waiting. [1]
Prognosis
| Stage | 1-year mortality |
|---|---|
| Compensated cirrhosis | roughly 1 to 2% |
| First decompensation | roughly 20 to 30% |
| Refractory ascites | roughly 50% |
| HRS-AKI untreated | roughly 50% at 1 month |
| ACLF grade 3 | up to 80% at 28 days |
The trajectory is dominated by the number and type of decompensating events. Once a patient has had ascites, a bleed and encephalopathy, 1-year mortality exceeds 50% without transplant. [1]
DCE long-case approach
Opening statement (SASPOP)
"Mr Chen is a 58-year-old retired carpenter who presents with increasing abdominal distension, leg swelling and two days of drowsiness. He has a 20-year history of heavy alcohol use and known alcohol-related cirrhosis. [1]
His main problems are:
- Decompensated alcohol-related cirrhosis — Child-Pugh C, MELD 24
- Tense ascites — likely with spontaneous bacterial peritonitis
- Grade 3 hepatic encephalopathy — precipitated by infection
- Acute kidney injury — probable hepatorenal syndrome
- Portal hypertension with medium oesophageal varices on secondary prophylaxis
- Alcohol dependence — needs addiction medicine input
- HCC surveillance overdue" [1]
Integrated management plan
- Resuscitate and protect the airway — grade 3 encephalopathy, nursing in semi-recumbent position, consider HDU.
- Diagnostic paracentesis — send cell count, culture, albumin; treat SBP with cefotaxime plus albumin if PMN count 250 or higher.
- Renal — stop diuretics and nephrotoxins; 48 hours of albumin 1 g/kg/day; if no response, diagnose HRS-AKI and start terlipressin plus albumin. [1]4. Encephalopathy — lactulose titrated to two to three soft stools daily; add rifaximin 550 mg BID; correct precipitant (the SBP).
- Portal hypertension — continue NSBB if blood pressure allows; plan repeat OGD for variceal surveillance.
- Nutrition and abstinence — high-protein, high-energy diet (sarcopenia worsens outcomes); thiamine replacement; addiction medicine referral.
- Transplant referral — MELD 24, decompensated with HRS — urgent transplant assessment once the acute episode stabilises and alcohol abstinence is established. [1]
DCE examiner probing questions you must anticipate:
- "What is his Child-Pugh and MELD, and which governs transplant?" → MELD governs priority; Child-Pugh informs prognosis.
- "His creatinine is rising despite fluids — what now?" → Stop diuretics, give albumin 48 hours, exclude SBP, then terlipressin for HRS.
- "Should you give fresh frozen plasma for his INR of 2.1 before paracentesis?" → No — the rebalance theory; INR does not predict bleeding in cirrhosis.
- "What is the role of rifaximin?" → Secondary prophylaxis of HE, added to lactulose, not monotherapy. [1]
DCE short-case approach: abdominal examination
Instruction: "Examine this patient's abdominal system." [1]
Systematic routine
- End of bed — cachexia, muscle wasting (sarcopenia), spider naevi on chest, gynaecomastia, loss of body hair, parotid enlargement.
- Hands — palmar erythema, Dupuytren contracture, clubbing, leuconychia; assess for asterixis.
- Face — jaundice (sclera), fetor hepaticus, Kayser-Fleischer rings (Wilson), xanthelasma (PBC).
- Neck — left supraclavicular node (Virchow, metastatic HCC), JVP (cor pulmonale in portopulmonary hypertension).
- Chest — spider naevi, gynaecomastia; inspect for signs of chronic lung disease (A1AT, alpha-1-antitrypsin).
- Abdomen — inspect — distension (ascites), caput medusae, visible veins, surgical scars, cachexia.
- Palpate — hepatomegaly or shrunken liver; splenomegaly (the key sign of portal hypertension); ballotable organs.
- Percuss — liver span; shifting dullness for ascites.
- Auscultate — hepatic bruit (HCC), friction rub.
- Examine for oedema, testicular atrophy, and complete the peripheral stigmata. [1]
Presentation template
"I examined Mr Chen's abdominal system. He is cachectic with several spider naevi on the anterior chest wall, palmar erythema and a Dupuytren contracture of the right ring finger. There is gynaecomastia and loss of axillary hair. The abdomen is distended with shifting dullness, consistent with ascites. There is no palpable liver edge but the spleen is enlarged 4 cm below the costal margin, indicating portal hypertension. A caput medusae is present around the umbilicus. There is asterixis on wrist dorsiflexion. There is peripheral oedema to the mid-shin bilaterally. [1]
These findings are consistent with decompensated chronic liver disease with portal hypertension, ascites and hepatic encephalopathy. I would like to take a full alcohol and viral hepatitis history, perform a diagnostic ascitic tap and organise bloods including coagulation and liver function." [1]
Key DWE MCQ patterns
- Which score governs transplant priority? MELD-Na (not Child-Pugh).
- Albumin dose in SBP? 1.5 g/kg day 1, 1 g/kg day 3 (Sort trial). [1]3. First drug for secondary HE prophylaxis? Rifaximin 550 mg BID added to lactulose.
- SAAG in cirrhotic ascites? 11 g/L or higher (high SAAG = portal hypertension).
- Acute variceal bleed high-risk patient? Early TIPS within 72 hours (Child-Pugh C, or B with active bleeding).
- What reduces mortality in acute variceal bleed? Prophylactic antibiotics and early TIPS.
- Best test to stage fibrosis non-invasively? Transient elastography (FibroScan).
- HRS-AKI first step? Stop diuretics, 48 hours albumin, exclude SBP, then terlipressin. [1]
References
[1] PREDESCI (Villanueva, 2019) — NSBBs prevent first decompensation in compensated cirrhosis with CSPH (decompensation/death 16% vs 27%, HR 0.51). [2] García-Pagán (2010) — Early covered-TIPS within 72 hours in high-risk variceal bleed reduces failure and improves survival. [3] Baveno VII (de Franchis, 2022) — Consensus on non-invasive CSPH definition and NSBB use to prevent decompensation. [4] Sort (1999) — IV albumin in SBP reduces renal impairment (33% to 10%) and mortality (29% to 10%). [5] Bass (2010) — Rifaximin 550 mg BID reduces breakthrough HE when added to lactulose. [6] CONFIRM (Wong, 2021) — Terlipressin plus albumin achieves HRS reversal in 32% vs 17%; watch respiratory failure. [7] Rinella (2023) — Multisociety Delphi renaming NAFLD to MASLD and NASH to MASH. [8] Sarin (2005) — EVL plus propranolol versus EVL alone for primary prophylaxis of variceal bleeding.
AASLD Practice Guidance on cirrhosis complications (2021) and HCC (2023); EASL Clinical Practice Guidelines on decompensated cirrhosis; BSG/BASL guidelines on variceal bleeding; GESA clinical guidelines. [1]
References
- [1]Villanueva C, Albillos A, Genescà J, et al. β blockers to prevent decompensation of cirrhosis in patients with clinically significant portal hypertension (PREDESCI): a randomised, double-blind, placebo-controlled, multicentre trial Lancet, 2019.PMID 30910320
- [2]García-Pagán JC, Caca K, Bureau C, et al. Early use of TIPS in patients with cirrhosis and variceal bleeding N Engl J Med, 2010.PMID 20573925
- [3]de Franchis R, Bosch J, Garcia-Tsao G, et al. Baveno VII - Renewing consensus in portal hypertension J Hepatol, 2022.PMID 35120736
- [4]Sort P, Navasa M, Arroyo V, et al. Effect of intravenous albumin on renal impairment and mortality in patients with cirrhosis and spontaneous bacterial peritonitis N Engl J Med, 1999.PMID 10432325
- [5]Bass NM, Mullen KD, Sanyal A, et al. Rifaximin treatment in hepatic encephalopathy N Engl J Med, 2010.PMID 20335583
- [6]Wong F, Pappas SC, Curry MP, et al. Terlipressin plus Albumin for the Treatment of Type 1 Hepatorenal Syndrome N Engl J Med, 2021.PMID 33657294
- [7]Rinella ME, Lazarus JV, Ratziu V, et al. A multisociety Delphi consensus statement on new fatty liver disease nomenclature J Hepatol, 2023.PMID 37364790
- [8]Sarin SK, Wadhawan M, Agarwal S, et al. Endoscopic variceal ligation plus propranolol versus endoscopic variceal ligation alone in primary prophylaxis of variceal bleeding Am J Gastroenterol, 2005.PMID 15784021