Phys · infectious
Antimicrobial Stewardship and Resistance
Also known as antimicrobial stewardship · antibiotic stewardship · antimicrobial resistance · AMR · multidrug resistance · MDR organism · MRSA · VRE · ESBL · carbapenem-resistant Enterobacteriaceae · CRE · carbapenemase · KPC · NDM · OXA-48 · Clostridioides difficile · Clostridium difficile · C. diff · fidaxomicin · bezlotoxumab · faecal microbiota transplant · procalcitonin · penicillin allergy delabeling · intrinsic resistance · acquired resistance
Consultant-physician-depth guide to antimicrobial stewardship and resistance — the central principle that every antibiotic prescription has consequences, captured by the five rights (right drug, right dose, right route, right duration, right indication). Covers the mechanisms of resistance (intrinsic versus acquired; enzymatic inactivation, target modification, reduced permeability and efflux, bypass; plasmids, transposons, integrons; rpoB mutations in TB), the key resistant organisms and their first-line, alternative and last-resort agents (MRSA, VRE, ESBL Enterobacteriaceae, CRE, MDR Pseudomonas and Acinetobacter), Clostridioides difficile infection (NAP1/027 hypervirulent strain, two-step diagnostic algorithm, fidaxomicin first-line, bezlotoxumab, faecal microbiota transplant), infection control (five moments of hand hygiene, contact precautions, screening), antibiotic allergy delabeling (90 per cent of penicillin allergy labels are wrong), and procalcitonin-guided antibiotic cessation — structured for FRACP DWE and DCE preparation.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Antimicrobial Stewardship and Resistance

The one-minute consultant answer
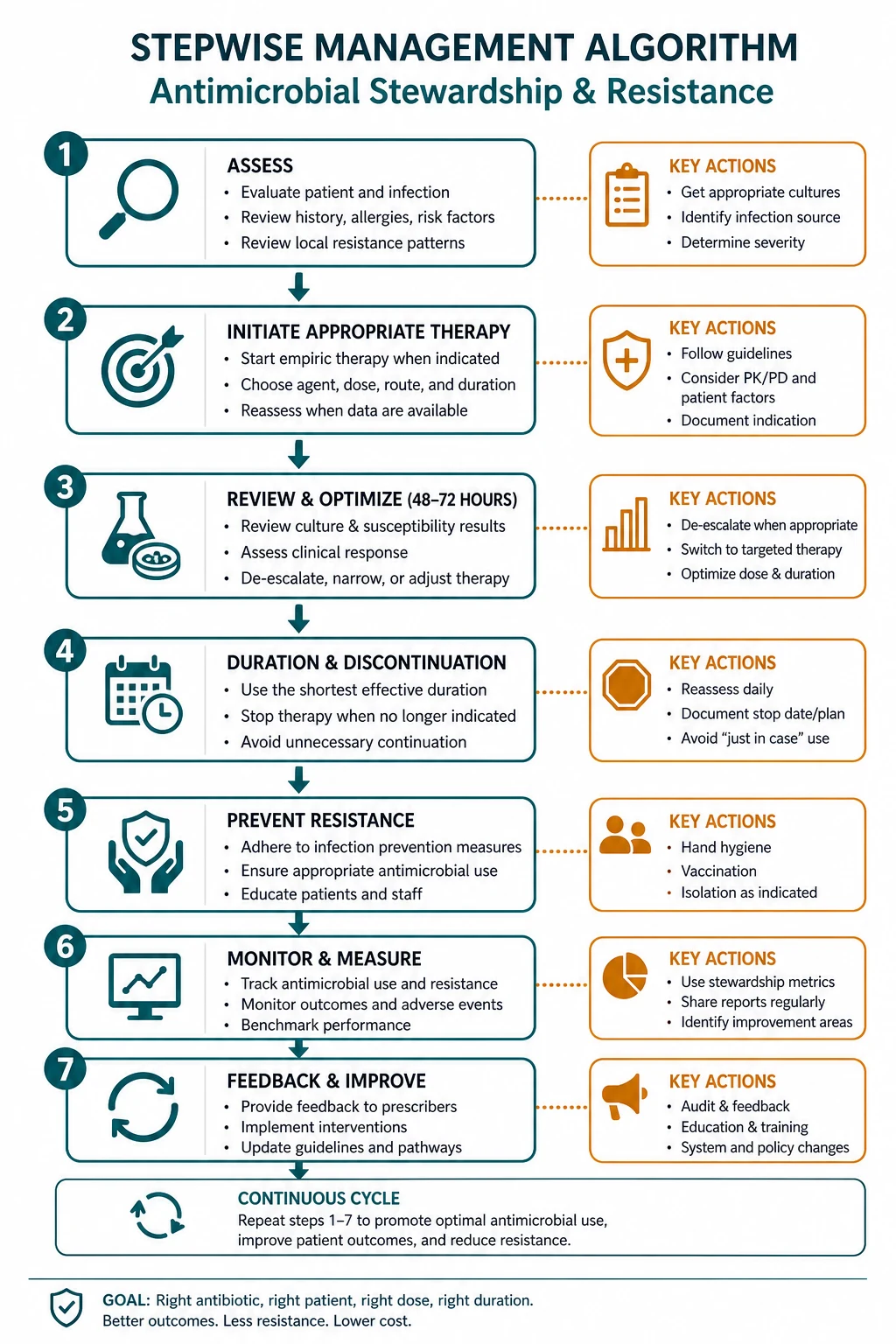
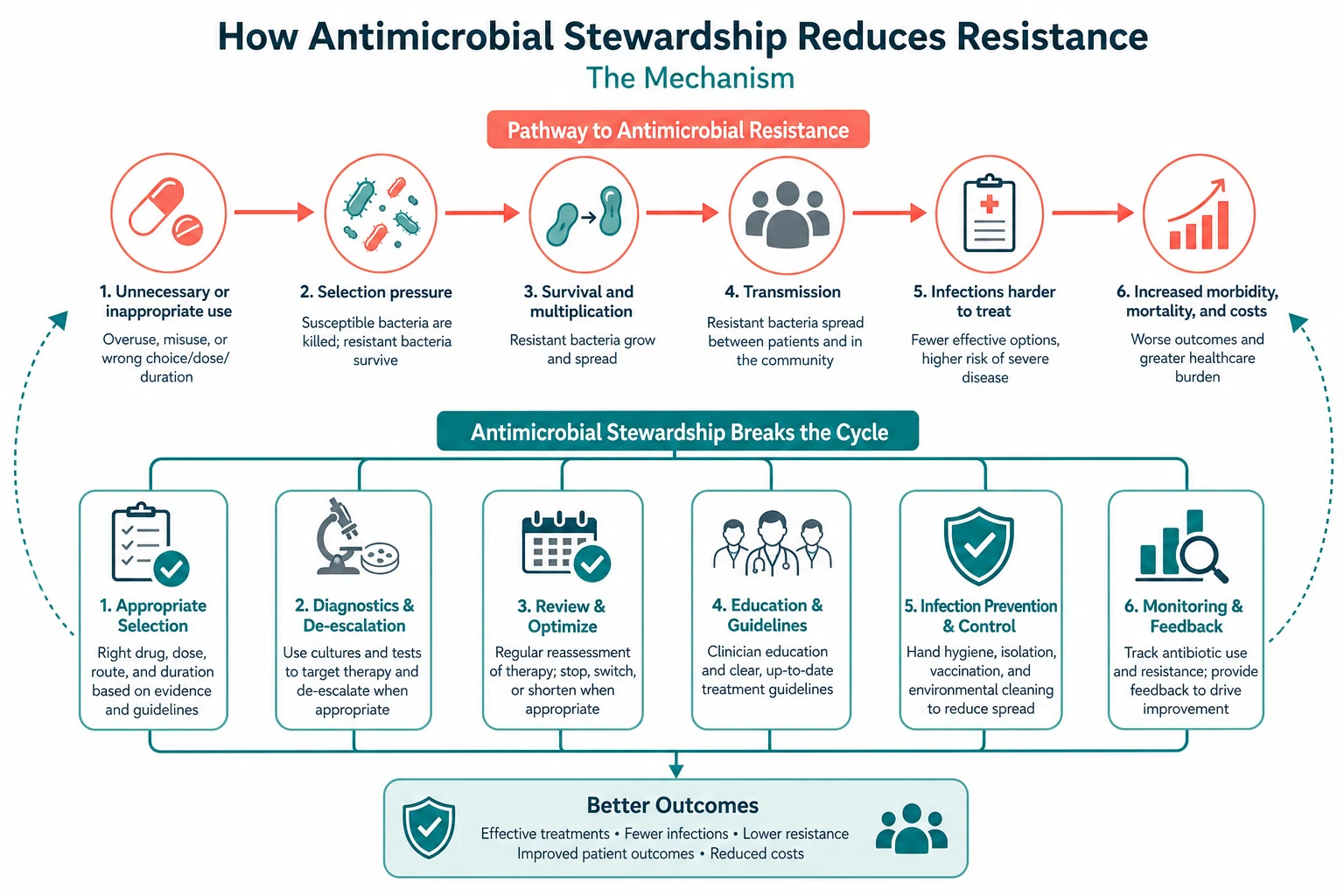
Every antibiotic prescription has consequences — for the patient in front of you, for the ward they are on, and for the population that will need antibiotics after them. Antimicrobial stewardship is the discipline of prescribing so that those consequences are beneficial and not harmful. The 2016 IDSA/SHEA guideline frames it through the five rights — right drug, right dose, right route, right duration, right indication — delivered through two core programme interventions: pre-authorisation (restricting certain drugs to infectious diseases approval) and prospective audit with feedback (reviewing ongoing therapy and advising the treating team) [1]. The bedside moves are the 48-hour review (reassess the empiric choice against culture and sensitivity and de-escalate), the IV-to-oral switch (when the patient is stable and can absorb), and the defined stop date (written on every prescription at the start).
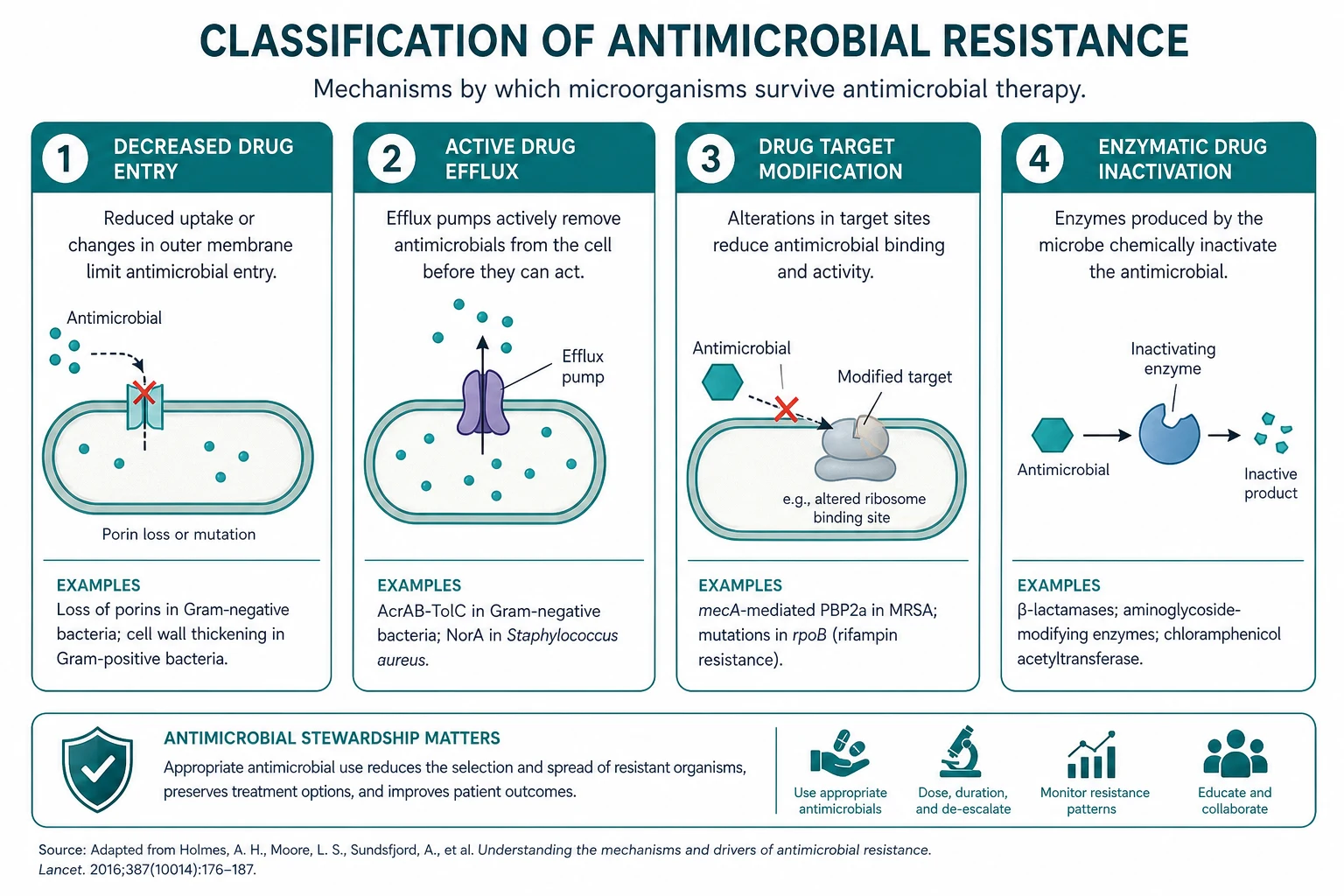
Resistance arises because bacteria acquire, exchange and accumulate resistance genes — by mutation (a spontaneous change in chromosomal DNA, the basis of rifampicin resistance in TB through rpoB mutation), and by horizontal gene transfer through plasmids, transposons and integrons. Four molecular mechanisms do the work: enzymatic inactivation (beta-lactamases, including ESBLs and carbapenemases such as KPC and NDM), target modification (PBP2a in MRSA; D-alanyl-D-lactate in VRE; altered DNA gyrase in fluoroquinolone resistance), reduced permeability and efflux (porin OprD loss in Pseudomonas conferring carbapenem resistance; AcrAB-TolC efflux pumps), and bypass pathways (alternative penicillin-binding proteins). Intrinsic resistance is species-inherent and chromosomal (Pseudomonas impermeability; Enterococcus intrinsically resistant to cephalosporins; Listeria intrinsically resistant to cephalosporins), while acquired resistance emerges under antibiotic pressure. [1]
The resistant organisms every physician must know — and the agents that treat them — are: MRSA (vancomycin, linezolid, daptomycin, ceftaroline), VRE (linezolid, daptomycin, tigecycline), ESBL Enterobacteriaceae (carbapenems — meropenem, ertapenem), CRE (colistin, tigecycline, ceftazidime-avibactam, meropenem-vaborbactam; aztreonam plus avibactam for NDM), multidrug-resistant Pseudomonas and Acinetobacter (ceftolozane-tazobactam, colistin), and Clostridioides difficile (fidaxomicin first-line, oral vancomycin for severe disease, bezlotoxumab for recurrence prevention, faecal microbiota transplant for multiple recurrences) [2]. And the stewardship wins that change outcome the most are the unglamorous ones — stopping the antibiotic when the indication has resolved, narrowing it when the culture returns, switching from IV to oral when the patient can absorb, and delabeling the penicillin allergy that was never real (90 per cent of labels are wrong) [9].
The central framework — the five rights and the collateral damage of antibiotics

The five rights are the answer to every stewardship question: [1]
| Right | What it means | The question to ask at the prescription |
|---|---|---|
| Right indication | Is this infection, prophylaxis, or neither? | "Is there an infection to treat, and have I sent cultures first?" |
| Right drug | The narrowest-spectrum agent active against the likely or proven organism | "Have I covered the likely organisms, and can I narrow when cultures return?" |
| Right dose | Adequate to achieve pharmacokinetic/pharmacodynamic targets at the site, adjusted for organ function | "Is the dose right for the patient's weight, renal and hepatic function, and the site of infection?" |
| Right route | IV only when the patient is unstable, cannot absorb orally, or the oral agent is unavailable | "Can this patient swallow and absorb, and does an oral equivalent exist?" |
| Right duration | The shortest duration supported by evidence | "What is my stop date, and what will I review to confirm it?" |
The collateral damage of antibiotics is the phrase the 2016 guideline uses for the predictable harms of every broad-spectrum course: Clostridioides difficile infection, selection of resistant organisms (MRSA, VRE, ESBL, CRE), antibiotic-associated adverse events (rash, nephrotoxicity, hepatotoxicity, QT prolongation, fluoroquinolone tendinopathy and aortic dissection), and the microbiome disruption whose full consequences are still being mapped. The stewardship mindset is to weigh these harms against the benefit at every prescription — not to deny antibiotics when they are needed, but to prescribe them so that the benefit exceeds the harm. [1]
The two core programme interventions
The IDSA/SHEA guideline identifies two interventions that consistently reduce inappropriate antibiotic use and improve outcomes [1]:
- Pre-authorisation — restricting the use of certain broad-spectrum agents (carbapenems, intravenous fluoroquinolones, daptomycin, linezolid, antifungals) to cases approved by the infectious diseases or microbiology team. This front-ends the stewardship conversation and limits the spread of resistance-driving drugs. Its limitation is that it does not address the duration or the continuing appropriateness of approved therapy.
- Prospective audit with feedback — an infectious diseases pharmacist or physician reviews patients on targeted antibiotics (usually after 48 to 72 hours of therapy, the natural "antibiotic timeout" point) and provides a recommendation to the treating team. This addresses the continuing need, the spectrum, the dose and the duration. It is the more powerful of the two interventions because it operates on the whole course of therapy, not just the start. [1]
Most effective programmes combine the two. A third set of complementary strategies — facility-specific treatment guidelines, IV-to-oral switch protocols, dose optimisation with therapeutic drug monitoring, and antibiotic timeouts built into the chart — operationalises the five rights at the bedside. [1]
Mechanisms of resistance — intrinsic versus acquired, mutation versus transfer

The examiner's first question on resistance is the distinction between intrinsic and acquired. [1]
Intrinsic resistance is the natural, species-inherent property of an organism — it is chromosomal, predictable, and shared by all members of the species. It is why you never test or expect certain drugs to work: [1]
| Organism | Intrinsically resistant to | Why |
|---|---|---|
| Pseudomonas aeruginosa | ampicillin, amoxicillin-clavulanate, first- and second-generation cephalosporins, ertapenem, tetracyclines, tigecycline | Impermeable outer membrane (low porin expression) plus efflux pumps exclude these agents |
| Enterococcus spp. | all cephalosporins (inherently lack the PBP target), aminoglycosides (low-level), trimethoprim-sulfamethoxazole (in vivo) | Lack the PBP that cephalosporins bind; can use alternative PBPs |
| Klebsiella pneumoniae | ampicillin, amoxicillin | Constitutive production of chromosomal SHV-1 beta-lactamase |
| Listeria monocytogenes | all cephalosporins | Does not express the PBP target for cephalosporins — the classic reason meningitis empiric regimens add ampicillin in the immunocompromised and the elderly |
| Stenotrophomonas maltophilia | all carbapenems (produces L1 metallo-beta-lactamase), most aminoglycosides | Intrinsic carbapenemase |
Acquired resistance emerges in a previously susceptible organism under antibiotic pressure, by one of two genetic routes — mutation or horizontal gene transfer. [1]
Mutation is a spontaneous change in chromosomal DNA. It occurs at a characteristic rate per organism per cell division. The clinically important example is Mycobacterium tuberculosis: spontaneous mutation to rifampicin resistance occurs at approximately 1 in 10 to the 8 cell divisions, and to isoniazid at approximately 1 in 10 to the 6. Because cavitary pulmonary TB carries bacillary loads of 10 to the 9 to 10 to the 12, any single-drug regimen WILL select a resistant mutant. This single fact is the molecular basis of multidrug anti-tuberculous therapy (treat with four drugs so that mutants resistant to one are killed by the others) and the reason adding a single drug to a failing regimen creates functional monotherapy and accelerates resistance — the cardinal sin in TB management. Other mutation-driven resistances include fluoroquinolone resistance (gyrA and parC mutations in DNA gyrase and topoisomerase IV) and rifampicin resistance (rpoB mutations in staphylococci and TB). [1]
Horizontal gene transfer moves resistance genes between organisms — even between unrelated species — by three vehicles. Plasmids are self-replicating extrachromosomal DNA circles that can carry multiple resistance genes (for example, a single plasmid carrying ESBL, aminoglycoside-modifying enzyme and fluoroquinolone-resistance determinants) and can transfer between E. coli, Klebsiella and other Enterobacteriaceae in the same gut. Transposons are mobile genetic elements ("jumping genes") that move within and between DNA molecules, carrying resistance cassettes; the best known is Tn4401 carrying the KPC carbapenemase gene. Integrons are site-specific recombination systems that capture and express gene cassettes, allowing rapid accumulation of resistance determinants in a single genetic locus. Together these three vehicles explain why resistance spreads faster than mutation alone could produce. [1]
The four molecular mechanisms of acquired resistance
Once the resistance gene exists (by mutation or transfer), it works by one of four mechanisms: [1]
- Enzymatic inactivation. The organism produces an enzyme that destroys or modifies the antibiotic before it reaches its target. The beta-lactamases are the dominant example — they hydrolyse the beta-lactam ring. ESBLs (extended-spectrum beta-lactamases) hydrolyse penicillins, all cephalosporins and aztreonam but are inhibited by beta-lactamase inhibitors (clavulanate, tazobactam) and remain susceptible to carbapenems. Carbapenemases (KPC, NDM, OXA-48, VIM, IMP) additionally hydrolyse the carbapenems. Aminoglycoside-modifying enzymes (acetyltransferases, adenyltransferases, phosphotransferases) explain high-level aminoglycoside resistance in enterococci and Gram-negatives.
- Target modification. The antibiotic's binding site is altered so the drug no longer binds. PBP2a in MRSA (encoded by the mecA gene on the staphylococcal cassette chromosome mec, SCCmec) has low affinity for all beta-lactams — the basis of methicillin resistance and the reason cephalosporins (except ceftaroline, which has been engineered to bind PBP2a) do not work in MRSA. D-alanyl-D-lactate in the peptidoglycan of VRE (vanA and vanB operons) eliminates the hydrogen bond that vancomycin needs to bind — the basis of vancomycin resistance. rpoB mutations in M. tuberculosis change the beta-subunit of RNA polymerase so rifampicin cannot bind. GyrA mutations in DNA gyrase cause fluoroquinolone resistance.
- Reduced permeability and increased efflux. The drug cannot reach its target because of porin loss or active pumping out. OprD porin loss in Pseudomonas aeruginosa confers carbapenem (especially imipenem) resistance by preventing drug entry. The AcrAB-TolC efflux pump in Enterobacteriaceae pumps out multiple drug classes, producing multidrug resistance in a single step.
- Bypass pathways. The organism develops or acquires an alternative pathway or target that makes the drug's original target irrelevant. Enterococci use alternative PBPs that vancomycin cannot inhibit (the mechanism of VRE); some bacteria acquire dihydrofolate reductase variants resistant to trimethoprim. [1]
The exam trap: the question will name an organism and a drug, and the answer is the mechanism. MRSA and beta-lactams — target modification (PBP2a). VRE and vancomycin — target modification (D-Ala-D-Lac). ESBL E. coli and ceftriaxone — enzymatic inactivation (beta-lactamase). Pseudomonas and imipenem after long therapy — porin loss (OprD). TB and rifampicin — rpoB mutation. [1]
The key resistant organisms — what they are and how to treat them

Methicillin-resistant Staphylococcus aureus (MRSA)
MRSA is Staphylococcus aureus that has acquired the mecA (or mecC) gene encoding PBP2a, a penicillin-binding protein with low affinity for all beta-lactams. Resistance extends to all penicillins, all cephalosporins (except ceftaroline), and all carbapenems. The 2011 IDSA guideline sets the management framework [8].
First-line agents for serious MRSA infection:
- Vancomycin — the workhorse for MRSA bacteraemia, endocarditis, osteomyelitis and pneumonia. Loading dose 25 to 30 mg/kg, then 15 to 20 mg/kg every 8 to 12 hours targeting a trough of 15 to 20 mg/L (or AUC-guided dosing targeting an AUC of 400 to 600 mg.h/L). Nephrotoxicity (especially with concomitant piperacillin-tazobactam) and infusion reactions (red-man syndrome, histamine-mediated and rate-related, managed by slowing the infusion) are the main adverse effects. [1]- Linezolid — an oxazolidinone with excellent oral bioavailability (100 per cent), the preferred agent for MRSA pneumonia (better lung penetration than vancomycin) and complicated skin-soft-tissue infection. 600 mg IV or orally every 12 hours. Duration limited to 28 days because of thrombocytopenia (monitor full blood count weekly), peripheral and optic neuropathy, and serotonin syndrome with concomitant SSRIs, MAOIs or serotonergic foods.
- Daptomycin — a lipopeptide, bactericidal, the preferred agent for MRSA bacteraemia and endocarditis where vancomycin is failing or the MIC is high. 6 mg/kg IV daily (8 to 10 mg/kg for endocarditis). NEVER use for pneumonia — it is inactivated by pulmonary surfactant. Monitor creatine kinase weekly (myopathy is the main toxicity; hold statins during therapy).
- Ceftaroline — a fifth-generation cephalosporin engineered to bind PBP2a, the only beta-lactam active against MRSA. 600 mg IV every 12 hours. Used for skin-soft-tissue and community-acquired pneumonia. [1]
The MRSA bacteraemia non-negotiables: at least two sets of blood cultures before antibiotics; minimum 14 days of therapy for uncomplicated bacteraemia (4 to 6 weeks for endocarditis, osteomyelitis or metastatic foci); echocardiography (transthoracic first, transoesophageal if high suspicion or prosthetic valve) to exclude endocarditis; search for and remove any removable focus (the central line — remove it; the prosthetic joint — discuss with orthopaedics; the epidural abscess — neurosurgery); and repeat blood cultures every 48 to 72 hours until clearance, because persistent bacteraemia beyond 7 days predicts a deep focus (endocarditis, septic thrombophlebitis, osteomyelitis) and a worse outcome. [1]
Vancomycin-resistant enterococci (VRE)
VRE are Enterococcus (usually E. faecium, less often E. faecalis) that have acquired the vanA or vanB operon, which replaces the D-alanyl-D-alanine target of vancomycin with D-alanyl-D-lactate, eliminating the critical hydrogen bond. The result is resistance to vancomycin and, often, to multiple other agents (ampicillin, high-level aminoglycosides). [1]
The critical distinction is colonisation versus infection. Most VRE are isolated from urine, wounds or rectal surveillance swabs and reflect colonisation — they do not require antibiotic therapy. True VRE infection (bacteraemia, endocarditis, intra-abdominal, central nervous system) is far less common but carries 20 to 40 per cent mortality in bacteraemia. [1]
Treatment options for VRE infection:
- Linezolid 600 mg IV or orally every 12 hours — first-line for serious VRE infection; bacteriostatic, so avoided in endocarditis. Same toxicity profile and 28-day limit as for MRSA.
- Daptomycin 8 to 12 mg/kg IV daily — the preferred agent for VRE bacteraemia and endocarditis (bactericidal); dose is higher than for MRSA because enterococci have a higher MIC.
- Tigecycline 100 mg loading then 50 mg IV every 12 hours — a glycylcycline with broad anaerobic and Gram-negative cover; used for complicated intra-abdominal and skin-soft-tissue infection but NOT bacteraemia because serum levels are too low. [1]
ESBL-producing Enterobacteriaceae
Extended-spectrum beta-lactamases (ESBLs) are enzymes (the commonest are the TEM, SHV and CTX-M families, with CTX-M-15 now globally dominant) that hydrolyse penicillins, all cephalosporins and aztreonam. They are inhibited in vitro by beta-lactamase inhibitors (clavulanate, tazobactam), which is why the laboratory reports susceptibility to piperacillin-tazobactam — but for serious ESBL bacteraemia, a carbapenem outperforms piperacillin-tazobactam even when the isolate is susceptible in vitro, and is the agent of choice. ESBLs are commonly found in E. coli and Klebsiella pneumoniae, and the classical patient has a community-onset urinary tract infection or bacteraemia with a history of recent antibiotics, recent healthcare exposure, or travel to a high-prevalence region. [1]
Treatment of ESBL infection:
- Carbapenem — meropenem 1 g IV every 8 hours, imipenem 500 mg IV every 6 hours, or ertapenem 1 g IV daily (the convenient once-daily option, but lacking Pseudomonas cover) — for serious ESBL bacteraemia, pneumonia, intra-abdominal or complicated UTI.
- Nitrofurantoin, fosfomycin or an aminoglycoside — for lower-tract (uncomplicated) ESBL UTI, where these narrower agents may be effective and carbapenem-sparing.
- NOT piperacillin-tazobactam for serious ESBL bacteraemia, even if reported susceptible — outcomes are worse. [1]
Carbapenem-resistant Enterobacteriaceae (CRE)
CRE are Enterobacteriaceae (Klebsiella, E. coli, Enterobacter) that have acquired a carbapenemase — an enzyme that hydrolyses all carbapenems (and usually all other beta-lactams). The three main families are: [1]
| Carbapenemase | Epidemiology | Treatment |
|---|---|---|
| KPC (Klebsiella pneumoniae carbapenemase, class A) | Commonest in the Americas, parts of Europe (Italy, Greece), Israel | Ceftazidime-avibactam or meropenem-vaborbactam (both inhibit KPC); colistin or tigecycline as alternatives |
| NDM (New Delhi metallo-beta-lactamase, class B) | Indian subcontinent, Balkans, and increasingly global | Aztreonam plus avibactam (NDM does not hydrolyse aztreonam, and avibactam protects aztreonam from co-produced ESBLs and AmpC); colistin; ceftazidime-avibactam does NOT work alone |
| OXA-48-like (class D) | Middle East, North Africa, Turkey, now Europe | Ceftazidime-avibactam; colistin or tigecycline as alternatives |
The classic patient is one with recent hospitalisation abroad (especially in southeast Asia, the Indian subcontinent, the Middle East, the Balkans), long-term care facility residence, prior broad-spectrum antibiotic exposure, or known prior CRE colonisation. CRE bacteraemia carries 40 to 50 per cent mortality, rising above 60 per cent in ventilator-associated pneumonia. The two immediate responses are infectious diseases consultation for combination-therapy decision and infection-control escalation (single-room contact isolation, screening of contacts, ward outbreak investigation). [1]
Treatment of CRE infection:
- Ceftazidime-avibactam 2.5 g IV every 8 hours (for KPC and OXA-48) — has transformed KPC-CRE management and avoids colistin nephrotoxicity. [1]- Meropenem-vaborbactam 4 g IV every 8 hours (for KPC) — an alternative carbapenem-beta-lactamase inhibitor combination.
- Aztreonam plus avibactam — for NDM-producing CRE, the regimen that works (NDM does not hydrolyse aztreonam).
- Colistin (polymyxin E) — the last-resort agent; loading 4 to 5 MU, then approximately 9 MU daily in two divided doses, renal-adjusted. Nephrotoxicity in 40 to 60 per cent and neurotoxicity are dose-limiting; combination therapy (colistin plus a carbapenem at high dose, or colistin plus tigecycline) is often used for severe CRE infection on the principle that two active agents outperform one.
- Tigecycline — useful for soft-tissue and intra-abdominal CRE infection; NOT for bacteraemia (low serum levels) or UTI (minimal urinary excretion). [1]
Multidrug-resistant Pseudomonas and Acinetobacter
Pseudomonas aeruginosa is intrinsically difficult (impermeable outer membrane, efflux pumps, AmpC beta-lactamase) and acquires resistance by multiple mechanisms simultaneously — porin OprD loss (carbapenem resistance), overexpressed efflux pumps (fluoroquinolone and multiple drug resistance), acquired beta-lactamases, and target modification. MDR Pseudomonas is a hospital pathogen of the critically ill (ventilator-associated pneumonia, catheter-related bacteraemia, burn wound infection). Treatment options include ceftolozane-tazobactam and ceftazidime-avibactam (newer beta-lactam/beta-lactamase inhibitor combinations developed for MDR Pseudomonas), high-dose antipseudomonal beta-lactams (piperacillin-tazobactam, cefepime, meropenem) with an aminoglycoside, and colistin as last resort. The choice is driven by the local antibiogram because resistance mechanisms vary by institution. [1]
Acinetobacter baumannii is a hospital-acquired pathogen of the critically ill and of military trauma patients, often susceptible only to colistin, tigecycline (for soft-tissue, not bacteraemia) and ampicillin-sulbactam (which has intrinsic anti-Acinetobacter activity through PBP3 binding, even though sulbactam has no activity against other Gram-negatives). When MDR Acinetobacter appears on a ward it is an infection-control emergency. Combination therapy (colistin plus a carbapenem or sulbactam) is often used for severe infection; mortality is high. [1]
Clostridioides difficile infection — the paradigm of collateral damage
C. difficile infection (CDI) is the most measurable harm of antibiotic use, and the stewardship intervention that most reliably reduces its incidence is reducing the duration and spectrum of antibiotic exposure. The pathophysiology is worth knowing cold because it explains every management decision: antibiotic disruption of the normal colonic microbiota allows ingested or resident C. difficile spores to germinate, vegetate, and elaborate toxin A (enterotoxic) and toxin B (cytotoxic), which glycosylate and inactivate Rho-family GTPases in colonic epithelial cells, causing actin cytoskeleton disruption, cell death, neutrophilic colitis and pseudomembrane formation. [1]
The NAP1/BI/027 hypervirulent strain
The epidemiology of CDI changed in the early 2000s with the emergence of the NAP1/BI/027 strain (the names reflect the three typing methods: North American Pulsed-field type 1 by PFGE; BI by restriction endonuclease analysis; 027 by PCR ribotyping). The seminal description by Loo and colleagues analysed a predominantly clonal multi-institutional outbreak in Quebec with high morbidity and mortality — attributable mortality of 6.9 per cent at 30 days and 16.7 per cent at one year in patients aged 60 and above [7]. The strain carries three features that drive its hypervirulence: a deletion in the tcdC gene (a negative regulator of toxin production, leading to higher toxin A and B levels), production of binary toxin (whose exact role remains debated), and fluoroquinolone resistance (which gave it a selective advantage during the fluoroquinolone era). The clinical lesson is that any patient with severe or fulminant CDI — particularly with WBC above 30 or shock — may carry the hypervirulent strain and requires aggressive therapy.
Diagnosis — the two-step algorithm
The recommended approach is a two-step algorithm that maximises sensitivity without overdiagnosing colonisation: [1]
- Screening test — either glutamate dehydrogenase (GDH), a highly sensitive but non-specific marker of C. difficile presence (it detects the organism, not the toxin), or nucleic acid amplification testing (NAAT) for the toxin gene (equally sensitive).
- Confirmation — a toxin enzyme immunoassay (EIA) that detects free toxin A and B in the stool (the marker of active toxin production and therefore of true disease). [1]
Interpretation:
- GDH-positive and toxin-positive — confirmed CDI. Treat.
- GDH-positive and toxin-negative — possible colonisation OR low-level toxin production below the assay threshold. Use clinical judgement and/or NAAT; do not treat asymptomatic patients.
- GDH-negative — CDI effectively excluded. No further testing. [1]
Non-negotiable rules: do NOT perform a test of cure. Toxin can persist in the stool for weeks after clinical resolution, and treating an asymptomatic patient with a positive result is futile, selects resistance, and increases recurrence risk. The endpoint of therapy is clinical resolution (resolution of diarrhoea), not microbiological clearance. [1]
Severity and the drug choice
The 2018 IDSA/SHEA guideline (updated 2021) defines severity and the drug choice [2]:
| Severity | Definition | First-line treatment |
|---|---|---|
| Initial, non-severe | WBC below 15 and creatinine below 1.5 times baseline | Fidaxomicin 200 mg orally twice daily for 10 days (preferred) OR oral vancomycin 125 mg four times daily for 10 days |
| Initial, severe | WBC 15 or above OR creatinine 1.5 times baseline or above | Fidaxomicin 200 mg twice daily for 10 days OR oral vancomycin 125 mg four times daily for 10 days |
| Fulminant | Hypotension, shock, ileus, toxic megacolon | Oral vancomycin 500 mg four times daily PLUS intravenous metronidazole 500 mg every 8 hours; add a vancomycin rectal retention enema (500 mg in 100 mL normal saline every 6 hours) if ileus; urgent surgical review for toxic megacolon, perforation or refractory shock |
Metronidazole is no longer first-line for any CDI in adults. It remains a third-line or limited-access option (500 mg orally three times daily for 10 days) where fidaxomicin and oral vancomycin are unavailable, and it retains a role as IV adjunctive therapy in fulminant disease (where oral vancomycin may not reach the colon through an ileus). [1]
The reason fidaxomicin is preferred over vancomycin for initial and recurrent CDI is the recurrence benefit. The pivotal Louie trial randomised 629 adults to fidaxomicin 200 mg twice daily or vancomycin 125 mg four times daily for 10 days: fidaxomicin was non-inferior for clinical cure (88 versus 86 per cent) but superior for recurrence (15.4 versus 25.3 per cent, P=0.005) [6]. Fidaxomicin is a narrow-spectrum macrocycle with minimal systemic absorption and minimal microbiome disruption — which is precisely why it reduces recurrence. Its limitation is cost, which has driven variable uptake.
Recurrent CDI and faecal microbiota transplantation
Recurrence is the central clinical challenge of CDI: 15 to 25 per cent of patients recur after an initial episode, and the risk climbs to 40 to 65 per cent after multiple episodes because each episode further disrupts the colonic microbiome and each antibiotic course paradoxically predisposes to the next recurrence. [1]
Management of recurrent CDI:
- First recurrence: fidaxomicin twice daily for 10 days (preferred), OR a tapered-and-pulsed oral vancomycin course (125 mg four times daily for 10 to 14 days, then twice daily for a week, then daily, then every 2 to 3 days, over 6 to 8 weeks) — the taper-and-pulse principle works by allowing spores to germinate and then killing the vegetative forms.
- Second or subsequent recurrence: fidaxomicin, OR oral vancomycin taper followed by faecal microbiota transplantation (FMT).
- Bezlotoxumab — a single 10 mg/kg IV infusion of a human monoclonal antibody against toxin B, given WITH standard antibiotic therapy to patients at high recurrence risk (age 65 and above, immunocompromised, severe CDI, prior CDI within 6 months). The MODIFY I and MODIFY II trials showed bezlotoxumab reduced recurrence at 12 weeks (16.5 versus 26.6 per cent versus placebo) [4].
Faecal microbiota transplantation is the most effective treatment for multiply recurrent CDI. The mechanism is restoration of the colonic microbiome and its resistance to C. difficile colonisation. The pivotal van Nood randomised trial compared duodenal infusion of donor faeces with vancomycin (with or without bowel lavage) in patients with recurrent CDI; the trial was stopped early for efficacy because 81 per cent of the FMT group achieved cure after a single infusion (94 per cent after a second infusion), versus 31 per cent on vancomycin alone [5]. FMT can be delivered by nasoduodenal tube, colonoscope, or capsule; donor screening (for enteric pathogens, multi-resistant organisms, and now SARS-CoV-2) is mandatory, and regulatory oversight has tightened after rare pathogen-transmission events.
Infection control in CDI
Patients with CDI are placed in a single room with contact precautions (gown and gloves for all contact) until at least 48 hours after diarrhoea has resolved. Hand hygiene uses soap and water (not alcohol-based hand rub, which is less effective against C. difficile spores) before and after patient contact and after removing gloves. Environmental cleaning uses a sporicidal agent (hypochlorite-based bleach at 1000 to 5000 ppm available chlorine). Stethoscopes, thermometers and blood-pressure cuffs are dedicated to the patient. The patient is not moved to a shared room until resolution. Antibiotic stewardship (reducing the inciting antibiotic) is itself an infection-control measure in CDI. [1]
Infection control — hand hygiene, isolation, screening
Resistance spreads in hospitals along three highways: the hands of healthcare workers, the shared environment and equipment, and the movement of patients between facilities. The infection-control response breaks each of these. [1]
The five moments of hand hygiene
The WHO framework defines five moments when hand hygiene is mandatory: [1]
- Before touching a patient — protects the patient from the healthcare worker's flora.
- Before a clean or aseptic procedure — protects the patient from colonising organisms introduced into a sterile site.
- After body fluid exposure risk — protects the healthcare worker and the environment.
- After touching a patient — protects the healthcare worker and other patients.
- After touching the patient's surroundings — protects against the contaminated environment. [1]
Alcohol-based hand rub is the default for the five moments (it is rapid, effective against most vegetative bacteria and many viruses, and skin-friendly). The exceptions are C. difficile (spores are resistant to alcohol — use soap and water), norovirus (soap and water is preferred), and visibly soiled hands (wash before rubbing). Hand hygiene adherence above 80 per cent is associated with measurable reductions in MRSA, VRE and MDR Gram-negative transmission. [1]
Contact precautions and isolation
A patient colonised or infected with a resistant organism is placed in a single room with contact precautions — gown and gloves for all contact, dedicated equipment, and hand hygiene before and after. The duration of precautions depends on the organism: MRSA and VRE precautions are often continued for the duration of the admission (and sometimes beyond, on re-admission), because decolonisation is unreliable; CRE precautions are typically continued indefinitely, because gut carriage is prolonged. The psychological and practical costs of isolation (reduced nurse contact, less mobilisation, more depression) are real and must be weighed — isolation is a serious intervention, not a reflexive one. [1]
Screening for resistant organisms
Active surveillance cultures on admission identify colonised patients who would otherwise spread the organism unknowingly. The high-yield groups are: patients transferred from another healthcare facility (especially overseas or interstate), patients with known prior colonisation, patients admitted to intensive care or long-term care, patients with recent broad-spectrum antibiotic exposure, and patients admitted during a known ward outbreak. Nasal swabs screen for MRSA; rectal swabs or stool screen for VRE, ESBL Enterobacteriaceae and CRE on selective chromogenic media. [1]
Antibiotic allergy — the label that denies first-line therapy
A penicillin allergy label is the commonest reason patients are denied first-line beta-lactam therapy and pushed to vancomycin, fluoroquinolones, clindamycin or aztreonam — with measurable consequences including higher mortality in sepsis and endocarditis, longer hospital stays, more C. difficile infection, more resistance selection, and more surgical site infection. The problem is that approximately 90 per cent of patients who carry a penicillin allergy label are not, in fact, allergic when formally evaluated [9]. The reasons are several: many labels originate from a childhood viral exanthem misattributed to the antibiotic; true IgE-mediated allergy wanes over years (approximately 50 per cent lose sensitivity within 5 years, up to 80 per cent within 10); and many reported reactions were never allergic in the first place (gastrointestinal intolerance, headache, a non-urticarial rash).
Delabeling is therefore a core stewardship intervention. The approach: [1]
- Take a structured allergy history. Characterise the reaction — timing (immediate, within 1 hour, suggests IgE; delayed beyond 72 hours suggests T-cell-mediated), the manifestation (anaphylaxis, angioedema, urticaria, maculopapular rash, Stevens-Johnson syndrome, toxic epidermal necrolysis, DRESS, interstitial nephritis, haematological), the year, the drug, and any re-exposure.
- Stratify risk. Low-risk reactions (vague childhood rash, remote gastrointestinal intolerance, family history only, non-allergic symptoms) can proceed to a direct oral amoxicillin challenge (typically 250 to 500 mg under observation for 1 hour) without skin testing. High-risk reactions (anaphylaxis, angioedema, severe cutaneous adverse reactions such as SJS/TEN/DRESS, positive re-exposure) require formal skin-prick and intradermal testing followed by supervised drug provocation by an allergist.
- Remove the label and document it. Once a patient has tolerated a beta-lactam challenge, the label is removed from the chart, the medication record, and the patient's allergy card, and the patient (and their general practitioner) is informed. The benefit is lifelong — access to first-line beta-lactams for every future infection. [1]
The exam point: never accept a penicillin allergy label without characterising the reaction, and never deny a patient first-line beta-lactam therapy on the basis of an uncharacterised childhood label. In the acutely septic patient, if the reported reaction is non-severe and remote, the benefit of first-line beta-lactam cover (a cephalosporin or a carbapenem, which have low cross-reactivity of 1 to 2 per cent with penicillins) usually outweighs the small allergy risk. [1]
Procalcitonin-guided antibiotic cessation
Procalcitonin (PCT) is the 116-amino-acid precursor of calcitonin, produced by the parafollicular C cells of the thyroid in health but, during systemic bacterial infection, produced in large quantities by extra-thyroidal tissues (liver, kidney, lung, intestine) under the stimulation of interleukin-1, tumour necrosis factor-alpha and interleukin-6. It is suppressed by interferon-gamma, the dominant cytokine of viral infection — which gives it a degree of specificity for bacterial over viral infection. It rises within 3 to 6 hours of a systemic bacterial infection and falls with a half-life of approximately 24 hours once the infection is controlled. [1]
The clinical use is guidance to start and stop antibiotics, not diagnosis of infection. A low baseline PCT (below 0.1 micrograms per litre) in a patient with suspected acute respiratory infection argues against bacterial infection and supports withholding or early cessation of antibiotics. A serial fall of 80 per cent or more from baseline, or to below 0.5 micrograms per litre, safely supports stopping antibiotics. [1]
The evidence base is the 2017 Cochrane individual-participant-data meta-analysis by Schuetz and colleagues, which pooled 26 trials and 6708 participants across primary care, emergency, ward and ICU settings [3]. It found that PCT-guided therapy reduced all-cause mortality (adjusted odds ratio 0.83, 95 per cent confidence interval 0.70 to 0.99, P=0.037), reduced antibiotic exposure by 2.4 days (from 8.1 to 5.7 days), and reduced antibiotic-related side effects (16.3 versus 22.1 per cent), with no increase in treatment failure. The findings held across clinical settings and infection types.
The caveats. PCT is a guide, not a rule. It is falsely low in early infection (it has not yet risen), in localised infection without bacteraemia, and in patients on immunosuppression. It is falsely elevated in severe trauma, major surgery, cardiogenic shock, pancreatitis, and some malignancies. A single value in isolation is never a decision — serial trends are. The Cochrane evidence is strongest for acute respiratory infections and sepsis in stable patients; the strategy should not override clinical judgement in a deteriorating patient, and a rising PCT should prompt a search for an uncontrolled source, not reflexive antibiotic escalation without re-evaluation. [1]
The IV-to-oral switch — the most underused stewardship move
The IV-to-oral switch is one of the highest-yield, lowest-risk stewardship interventions, and it remains systematically underused. The principle is simple: once a patient is clinically stable and able to absorb orally, an oral agent with good bioavailability achieves the same serum concentration as the IV formulation, at lower cost, with fewer line complications and shorter length of stay. [1]
The criteria for switch:
- Clinically improving — afebrile for 24 to 48 hours, haemodynamically stable, improving white cell count and C-reactive protein.
- Able to absorb — tolerating oral intake, no ileus, no vomiting, no malabsorption, no nasogastric tube feeding failure.
- An oral equivalent with good bioavailability exists — the agents with near-100 per cent oral bioavailability are fluoroquinolones (ciprofloxacin, levofloxacin, moxifloxacin), linezolid, metronidazole, fluconazole, co-trimoxazole, doxycycline, clindamycin, and rifampicin. Beta-lactams generally have acceptable but variable bioavailability (amoxicillin 80 per cent; amoxicillin-clavulanate is reliable; most cephalosporins have poor oral bioavailability, with the exception of cephalexin and cefuroxime axetil). [1]
The switch is a medical decision, not a nursing protocol in most units, though nurse- and pharmacist-led switch protocols are effective and increasingly used. The exam point is to recognise the patient who meets the criteria and switch — failure to switch is a leading cause of prolonged unnecessary IV therapy. [1]
Surgical prophylaxis — the prescription that should never be prolonged
Surgical antibiotic prophylaxis is one of the commonest reasons for inappropriate antibiotic prescribing, and the principles are clear. A single preoperative dose, given within 60 minutes before incision (within 120 minutes for fluoroquinolones and vancomycin), at the correct dose for the patient's weight, covering the likely surgical-site organisms (Gram-positive cover for clean surgery, Gram-negative and anaerobic cover for colorectal and gynaecological surgery). Repeat the dose only for prolonged surgery (over 4 hours for most beta-lactams; over 3 hours for fluoroquinolones) or major blood loss (over 1.5 litres). Discontinue within 24 hours of surgery (within 48 hours for cardiac surgery). [1]
Continuing prophylaxis beyond 24 hours does not reduce surgical site infection — the evidence is robust — and it does increase C. difficile infection, resistance selection, line complications and cost. The stewardship audit of surgical prophylaxis duration is a high-yield programme intervention. [1]
Regional guideline deltas
| Aspect | ANZ (eTG, ASID) | UK (NICE, PHE) | US (IDSA/SHEA, CDC) |
|---|---|---|---|
| Stewardship structure | Hospital antimicrobial management teams; restricted broad-spectrum availability; mandatory ID consultation for certain agents; antibiograms | "Start Smart Then Focus" — review at 48 hours; Five Year AMR Strategy; national stewardship targets | Pre-authorisation and audit-and-feedback as the two core interventions; CDC Core Elements of Hospital Stewardship Programs |
| C. difficile first-line | Fidaxomicin preferred, oral vancomycin; metronidazole third-line | Fidaxomicin first-line (NICE); oral vancomycin; FMT for recurrence | Fidaxomicin or oral vancomycin (IDSA/SHEA 2021 update); bezlotoxumab for high recurrence risk |
| MRSA first-line | Vancomycin; linezolid for pneumonia; switch to cefazolin for MSSA | Vancomycin; linezolid for pneumonia; daptomycin NOT for pneumonia | Vancomycin; linezolid for pneumonia; daptomycin for bacteraemia |
| CRE management | Combination therapy; colistin with meropenem or tigecycline; ceftazidime-avibactam for KPC | Combination therapy; ceftazidime-avibactam for KPC and OXA-48; aztreonam-avibactam for NDM | Combination therapy; ceftazidime-avibactam (KPC, OXA-48); meropenem-vaborbactam (KPC); colistin as last resort |
| Penicillin allergy delabeling | Growing uptake; oral challenge protocols in many hospitals | National delabeling toolkit; pharmacist-led pathways | AAAAI and CDC endorse delabeling; allergy specialist input for high-risk |
High-yield exam discriminators
- Fidaxomicin or oral vancomycin (not metronidazole) for initial C. difficile — fidaxomicin preferred for its recurrence benefit; metronidazole is no longer first-line for any adult CDI.
- Oral vancomycin 500 mg four times daily PLUS IV metronidazole for fulminant CDI — add a rectal vancomycin enema for ileus; urgent surgical review for toxic megacolon. [1]3. Faecal microbiota transplantation for multiply recurrent CDI — 94 per cent cure after one or two infusions in the van Nood trial.
- Bezlotoxumab reduces recurrence when given with standard therapy in high-risk patients — single IV infusion, anti-toxin B monoclonal antibody.
- C. difficile test of cure is not done — toxin persists for weeks; clinical resolution is the endpoint.
- MRSA bacteraemia — at least 14 days, echocardiography, remove the focus, repeat blood cultures — a vancomycin MIC above 1.5 to 2 mg/L prompts a switch. [1]7. Linezolid for MRSA pneumonia; daptomycin for MRSA bacteraemia — daptomycin is NEVER used for pneumonia (inactivated by surfactant).
- Carbapenem for ESBL bacteraemia — NOT piperacillin-tazobactam, even if reported susceptible.
- CRE therapy driven by carbapenemase type — KPC: ceftazidime-avibactam; NDM: aztreonam-avibactam or colistin; OXA-48: ceftazidime-avibactam.
- Penicillin allergy delabeling — 90 per cent of labels are wrong — characterise the reaction, challenge low-risk patients, remove the label.
- Procalcitonin guides antibiotic cessation — 80 per cent fall or below 0.5 supports stopping; do not use a single value in isolation or as a diagnostic test.
- Adding a single drug to a failing TB regimen creates monotherapy and resistance — always add at least two (preferably three) new active agents.
- Surgical prophylaxis — single dose, repeat only for long surgery or major blood loss, stop within 24 hours.
- IV-to-oral switch when stable and absorbing — fluoroquinolones, linezolid, metronidazole, fluconazole, co-trimoxazole, doxycycline all have near-complete oral bioavailability. [1]
Communication and shared decision-making
- On stopping antibiotics: "Your tests show the infection has been treated and your body is recovering, so the antibiotics have done their job. Continuing them would only increase your risk of side effects and a serious bowel infection called C. difficile. We are stopping them today."
- On the C. difficile diagnosis: "The antibiotics you received disrupted the normal bacteria in your bowel and allowed a germ called C. difficile to overgrow and release a toxin that causes the diarrhoea. We treat it with a different antibiotic by mouth — fidaxomicin or vancomycin — and most people recover within a week. If it comes back, we have further options including a treatment that restores the healthy bowel bacteria."
- On penicillin allergy delabeling: "Only a small fraction of people who carry a penicillin allergy label are truly allergic, and the label denies you the best and safest first-line antibiotics. We can test you safely today with a small observed dose, and if you tolerate it we remove the label — which means better and safer treatment for you for the rest of your life."
- On isolation for a resistant organism: "We have found a germ in your tests that is resistant to some antibiotics, and we are caring for you in a single room with gowns and gloves to protect other patients from catching it. This is a precaution, not a punishment — we will review it regularly, and it does not change the quality of care you receive."
- On last-resort therapy for CRE: "The germ causing your infection is resistant to most antibiotics, and the drugs we are using can affect the kidneys. We will watch your kidney function closely, we have a clear plan with the infectious diseases team, and we will be honest with you about how things are going. The most important thing we can do is treat the source of the infection and support your body's recovery while the antibiotics work." [1]
Summary: the antimicrobial stewardship consultation in one paragraph
Apply the five rights — right drug, right dose, right route, right duration, right indication — to every prescription, and deliver them through the two core programme interventions of pre-authorisation and prospective audit with feedback. Send cultures before starting empiric therapy, start broad when the patient is unstable, and NARROW to the narrowest effective agent at the 48-hour review when culture and sensitivity return. Switch from IV to oral when the patient is stable and absorbing. Use procalcitonin as a guide to start and stop, not as a diagnostic test. For C. difficile, use fidaxomicin or oral vancomycin (not metronidazole) for initial disease, add IV metronidazole and consider surgery for fulminant disease, use bezlotoxumab and faecal microbiota transplantation for recurrence, and never perform a test of cure. For MRSA, use vancomycin for bacteraemia, linezolid for pneumonia, daptomycin (never for pneumonia) for bacteraemia failing vancomycin, treat for at least 14 days, echocardiograph, and remove the source. For ESBL use a carbapenem; for CRE choose therapy by carbapenemase type (KPC: ceftazidime-avibactam; NDM: aztreonam-avibactam; OXA-48: ceftazidime-avibactam), with colistin as last resort. Delabel the penicillin allergy — 90 per cent of labels are wrong — and never let an uncharacterised childhood rash deny a patient first-line beta-lactam therapy. Practise the five moments of hand hygiene, isolate and screen for resistant organisms, and limit surgical prophylaxis to a single preoperative dose. Every antibiotic you do not prescribe, every day you shorten, and every spectrum you narrow is a patient protected from collateral damage and a population whose antibiotics will keep working when they are needed next. [1]
References
- [1]Barlam TF, Cosgrove SE, Abbo LM, et al. Implementing an Antibiotic Stewardship Program: Guidelines by the Infectious Diseases Society of America and the Society for Healthcare Epidemiology of America Clin Infect Dis, 2016.PMID 27080992
- [2]McDonald LC, Gerding DN, Johnson S, et al. Clinical Practice Guidelines for Clostridium difficile Infection in Adults and Children: 2017 Update by the Infectious Diseases Society of America (IDSA) and Society for Healthcare Epidemiology of America (SHEA) Clin Infect Dis, 2018.PMID 29462280
- [3]Schuetz P, Wirz Y, Sager R, et al. Procalcitonin to initiate or discontinue antibiotics in acute respiratory tract infections Cochrane Database Syst Rev, 2017.PMID 29025194
- [4]Wilcox MH, Gerding DN, Poxton IR, et al. Bezlotoxumab for Prevention of Recurrent Clostridium difficile Infection N Engl J Med, 2017.PMID 28121498
- [5]van Nood E, Vrieze A, Nieuwdorp M, et al. Duodenal infusion of donor feces for recurrent Clostridium difficile N Engl J Med, 2013.PMID 23323867
- [6]Louie TJ, Miller MA, Mullane KM, et al. Fidaxomicin versus vancomycin for Clostridium difficile infection N Engl J Med, 2011.PMID 21288078
- [7]Loo VG, Poirier L, Miller MA, et al. A predominantly clonal multi-institutional outbreak of Clostridium difficile-associated diarrhea with high morbidity and mortality N Engl J Med, 2005.PMID 16322602
- [8]Liu C, Bayer A, Cosgrove SE, et al. Clinical practice guidelines by the infectious diseases society of america for the treatment of methicillin-resistant Staphylococcus aureus infections in adults and children Clin Infect Dis, 2011.PMID 21208910
- [9]Castells M, Khan DA, Phillips EJ Penicillin Allergy N Engl J Med, 2019.PMID 31826341