Phys · neurological
Epilepsy
Also known as seizure disorder · epilepsy · convulsion · fits · focal seizure · generalised tonic-clonic seizure · GTCS · absence seizure · myoclonic seizure · temporal lobe epilepsy · juvenile myoclonic epilepsy · JME · Lennox-Gastaut syndrome · West syndrome · status epilepticus
Consultant-physician-depth guide to seizure classification (ILAE 2017), epilepsy syndromes, pathophysiology of neuronal hyperexcitability, EEG and MRI investigation, syndrome-based antiepileptic drug selection, status epilepticus management, and women-with-epilepsy care for FRACP DWE and DCE preparation.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Epilepsy
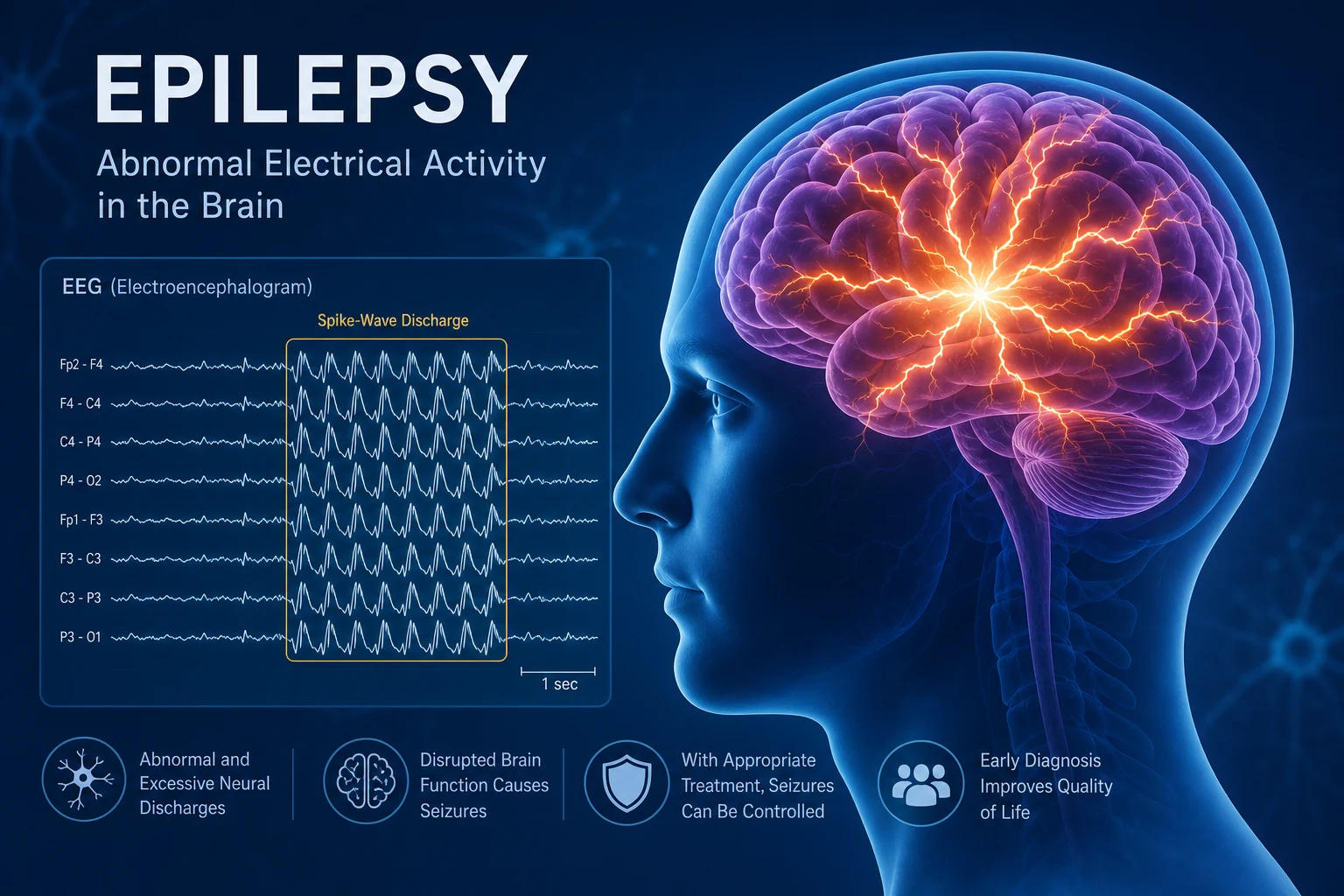
The answer first
Epilepsy is a disorder of recurrent, unprovoked seizures. A seizure is the transient clinical manifestation of abnormal, excessive, hypersynchronous cortical neuronal firing. The two questions that determine everything in epilepsy are: is this truly an epileptic seizure (and not syncope, dissociation, or another mimic)? and what is the seizure type, syndrome, and cause? — because that triad drives both prognosis and drug choice. [1]
The clinical decision rules a registrar must own at viva: [1]
- Diagnose before you treat. The single biggest source of "drug-resistant epilepsy" referred to specialty clinics is misdiagnosis — typically syncope or psychogenic non-epileptic seizures labelled as epilepsy. A detailed witness history is more powerful than any single investigation.
- Classify before you choose a drug. The 2017 ILAE classification splits seizures first by onset: focal, generalised, or unknown [1]. Focal seizures are treated with focal agents (lamotrigine, levetiracetam, lacosamide, carbamazepine); idiopathic generalised epilepsies require broad-spectrum agents (valproate, levetiracetam, topiramate). Prescribing carbamazepine or oxcarbazepine to a patient with juvenile myoclonic epilepsy can worsen the myoclonus and absences — a high-yield DWE trap.
- Status epilepticus is a time-critical emergency. Per the ILAE 2015 definition, a convulsive seizure becomes status epilepticus at 5 minutes (treatment threshold t1) and risks neuronal injury by 30 minutes (consequence threshold t2) [3]. Treat aggressively with benzodiazepines first, escalate to a second-line agent after two doses, then to anaesthetic infusion if refractory.
- Valproate is the most effective generalised AED — and the most dangerous in pregnancy. Avoid valproate in any woman of childbearing potential unless a formal Pregnancy Prevention Programme is met. The fetal malformation risk rises to 30 to 40 per cent and there is significant neurodevelopmental harm (IQ reduction, autism spectrum) [5].
The organising principle is syndrome-based, patient-centred management: classify the seizure and syndrome, identify the cause, choose the drug by syndrome and patient factors (sex, age, comorbidity, reproductive plans), counsel on driving and lifestyle, and escalate to surgery early in drug-resistant focal epilepsy. [1]
Classification — the ILAE 2017 operational framework
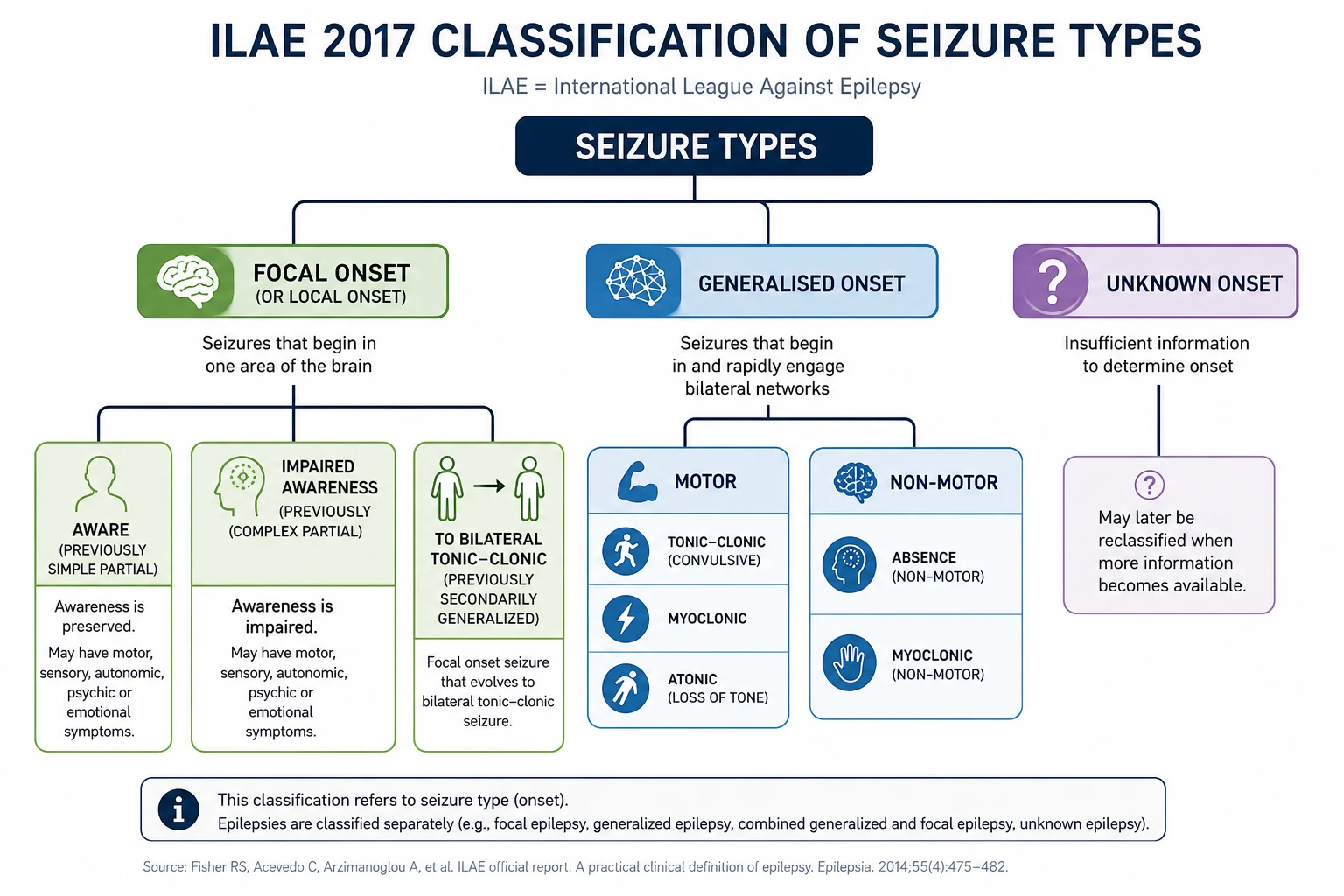
The 2017 ILAE position paper replaced the 1981 classification with a practical, three-level framework [1][2]. Master it — examiners will test you on it at every level.
Level 1 — Seizure type (by onset)
| Onset | Subtype | Distinguishing features |
|---|---|---|
| Focal | Focal aware (consciousness preserved) | Formerly "simple partial". Patient is fully responsive; can report the event |
| Focal impaired awareness (consciousness affected) | Formerly "complex partial". Staring, automatisms, postictal confusion | |
| Focal motor (focal aware or impaired) | Clonic, tonic, atonic, myoclonic, epileptic spasms, hyperkinetic, automatisms | |
| Focal non-motor (autonomic, cognitive, emotional, sensory) | Déjà vu, rising epigastric sensation, olfactory/gustatory hallucination, fear | |
| Focal to bilateral tonic-clonic | Formerly "secondarily generalised". The focal onset may be a brief aura before the convulsion | |
| Generalised | Motor: tonic-clonic, clonic, tonic, myoclonic, atonic, epileptic spasms | Onset is bilaterally synchronous from the start — no focal onset |
| Non-motor (absence): typical, atypical, myoclonic absence, eyelid myoclonia | Brief (seconds), staring, no postictal confusion; subtle eyelid flicker or automatisms | |
| Unknown | Motor (tonic-clonic, epileptic spasms), non-motor | Used when onset cannot be observed; reassess after investigation |
DWE high-yield: The most tested terminology change is "focal to bilateral tonic-clonic" replacing "secondarily generalised". The second most tested is "focal aware" and "focal impaired awareness" replacing "simple partial" and "complex partial". Examiners will accept the new terms in 2026. [1]
Level 2 — Epilepsy type
After classifying seizure type, classify the epilepsy as focal, generalised, combined focal and generalised, or unknown. This is a clinical classification informed by EEG, imaging, and history. [1]
Level 3 — Epilepsy syndrome
Where possible, make a syndromic diagnosis — this is what determines prognosis and drug choice. A syndrome is defined by a cluster of age of onset, seizure type(s), EEG signature, imaging, comorbidities, and often a known genetic aetiology. [1]
Level 4 — Aetiology (the six ILAE categories)
| Aetiology | Examples | Why it matters |
|---|---|---|
| Structural | Hippocampal sclerosis, stroke, tumour (low-grade glioma, DNET), focal cortical dysplasia, traumatic brain injury, vascular malformation, mesial temporal sclerosis | Surgical remediable syndrome — early MRI identification changes the trajectory |
| Genetic | Idiopathic generalised epilepsies (JME, childhood absence); channelopathies — SCN1A (Dravet), KCNQ2, GABA-A receptor (GABRA1); chromosomal (Down, ring chromosome 20) | Drug choice, prognosis, family counselling, genetic testing |
| Infectious | Neurocysticercosis (commonest worldwide), viral encephalitis (HSV), bacterial meningitis sequelae, cerebral malaria, tuberculosis | Treat the cause; commonest globally preventable cause |
| Metabolic | Hypoglycaemia, hyponatraemia, hepatic encephalopathy, uraemia, pyridoxine dependency, mitochondrial disorders (MELAS, MERRF) | Acute symptomatic seizures — correct the metabolic disturbance, often do not need long-term AEDs |
| Immune | Anti-NMDA receptor encephalitis, anti-LGI1, anti-GABA-B, anti-GAD, Hashimoto encephalopathy, Rasmussen encephalitis | Urgent immunotherapy (steroids, IVIG, plasma exchange, rituximab, cyclophosphamide); early antibody testing |
| Unknown | Cause not identified despite workup | Often the largest single group at first diagnosis; reassess over time |
DCE viva trap: When asked "what is the cause of this patient's epilepsy?", do not jump to "genetic" for idiopathic generalised epilepsy. The ILAE framework requires you to give the aetiology category explicitly. The commonest structural cause of focal epilepsy in adults is mesial temporal sclerosis; the commonest cause worldwide is neurocysticercosis in endemic regions. A first seizure in an older adult (over 60) demands a structural workup — stroke and tumour together account for the majority of new-onset epilepsy in this age group. [1]
Pathophysiology — why seizures happen and why they self-sustain
A seizure is the clinical expression of a sudden, excessive, hypersynchronous discharge of a population of cortical neurons. The fundamental disturbance is a loss of the normal balance between excitation (glutamate acting on NMDA, AMPA, and kainate receptors) and inhibition (GABA acting on GABA-A and GABA-B receptors). [1]
The four mechanisms of seizure generation
- Neuronal hyperexcitability — individual neurons fire too readily. This may be due to a gain-of-function mutation in a sodium channel (SCN1A, SCN2A, SCN8A), loss of inhibition (GABA-A subunit mutations), or acquired changes (gliosis, inflammation, denervation hypersensitivity after stroke).
- Hypersynchronisation — the firing of one neuron recruits its neighbours through recurrent excitatory collaterals. Normally this recruitment is damped by GABAergic interneurons and the blood–brain barrier's controlled ionic environment. When damping fails, recruitment cascades into a population burst.
- Failure of inhibitory control — the GABAergic interneurons that normally terminate a burst are overwhelmed, fatigued, or functionally downregulated. In status epilepticus this is mechanistically critical: GABA-A receptors are internalised from the neuronal membrane within minutes of sustained seizure activity, making the seizure progressively benzodiazepine-resistant.
- Recruitment of excitatory receptors — simultaneously, NMDA receptors are recruited to the synaptic membrane, sustaining the seizure through calcium influx and excitotoxicity. [1]
DWE high-yield: The receptor trafficking explanation is the molecular basis for time-dependent treatment of status epilepticus. Benzodiazepines work early (when GABA-A receptors are abundant) and lose efficacy late (after internalisation). This is why delayed treatment is harder to terminate — the longer status epilepticus persists, the more pharmacoresistant it becomes. The ESETT and RAMPART trials are anchored in this physiology [7][8].
The pathophysiology of specific syndromes
Mesial temporal lobe epilepsy with hippocampal sclerosis. This is the commonest surgically remediable epilepsy syndrome. The sequence is thought to be: an early cerebral insult (prolonged febrile convulsion in childhood, HSV encephalitis, head injury) damages the hippocampus → gliosis, neuronal loss, and mossy fibre sprouting → recurrent excitatory circuits form within the dentate gyrus → spontaneous focal seizures arise from the sclerotic hippocampus. The seizures characteristically begin with a rising epigastric aura, progress to impaired awareness with orofacial automatisms (lip-smacking, chewing), and may evolve to a focal to bilateral tonic-clonic seizure. MRI shows hippocampal atrophy with T2/FLAIR hyperintensity; interictal EEG shows anterior temporal spikes. [1]
Idiopathic generalised epilepsy (IGE) — JME and childhood absence. These are polygenic channelopathies affecting thalamocortical circuits. The hallmark EEG finding is the 3 Hz spike-and-wave discharge of absence epilepsy and the 4 to 6 Hz polyspike-and-wave of juvenile myoclonic epilepsy, generated by abnormal oscillatory loops between the thalamus and cortex. The clinical seizure types (absence, myoclonus, tonic-clonic) are the cortical read-out of this abnormal oscillation. The genetics are complex — multiple susceptibility loci including CACNA1H, GABRA1, EFHC1. [1]
Dravet syndrome (severe myoclonic epilepsy of infancy). A severe genetic epilepsy caused by de novo loss-of-function mutations in SCN1A (encoding the alpha-1 sodium channel subunit) in about 80 per cent of cases. Onset is in the first year with prolonged febrile seizures, evolving into afebrile myoclonic and focal seizures, ataxia, and intellectual disability. Sodium-channel-blocking AEDs (lamotrigine, carbamazepine, phenytoin) worsen Dravet syndrome — a high-yield MCQ point. [1]
Epilepsy syndromes — the must-know list
| Syndrome | Age of onset | Hallmark features | EEG signature | First-line drug |
|---|---|---|---|---|
| West syndrome (infantile spasms) | 4-8 months | Brief flexor spasms in clusters (jackknife), developmental regression, hypsarrhythmia | Hypsarrhythmia (chaotic high-voltage slow waves with multifocal spikes) | ACTH or prednisolone; vigabatrin (especially with tuberous sclerosis) [10] |
| Childhood absence epilepsy (CAE) | 4-10 years | Brief (5-10 s) staring spells, no postictal state, multiple per day, provoked by hyperventilation | 3 Hz generalised spike-and-wave | Ethosuximide (absences only); valproate or lamotrigine if tonic-clonic seizures coexist |
| Juvenile myoclonic epilepsy (JME) | 12-18 years | Morning myoclonic jerks, generalised tonic-clonic seizures on waking, sometimes absences; lifelong, seizure-provoking triggers include sleep deprivation and alcohol | 4-6 Hz irregular polyspike-and-wave | Valproate (most effective); levetiracetam or lamotrigine in women of childbearing age |
| Temporal lobe epilepsy (TLE) | Any (most common adult focal) | Aura (epigastric rising, déjà vu, olfactory), orofacial automatisms, postictal confusion; mesial temporal sclerosis on MRI | Anterior temporal spikes | Lamotrigine, levetiracetam, lacosamide; surgery if drug-resistant |
| Frontal lobe epilepsy | Any | Brief nocturnal motor seizures with bizarre hyperkinetic automatisms (bicycling, shouting), minimal postictal confusion, frequent clusters | Often normal interictal EEG | Lamotrigine, levetiracetam, carbamazepine; surgery if lesional |
| Lennox-Gastaut syndrome | 1-8 years | Triad: multiple seizure types (tonic, atonic, atypical absence), cognitive impairment, slow spike-and-wave on EEG | Slow (1.5-2.5 Hz) generalised spike-and-wave | Valproate, lamotrigine, topiramate, rufinamide; corpus callosotomy for drop attacks |
| Dravet syndrome | Under 1 year | Febrile hemiclonic seizures in infancy → myoclonic, atypical absence, focal; intellectual disability; worsened by sodium-channel blockers | Multifocal spikes and generalised polyspike-wave | Sodium valproate, clobazam, stiripentol, fenfluramine, cannabidiol; avoid lamotrigine, carbamazepine, phenytoin |
DCE viva trap: When presenting a young person with myoclonus and tonic-clonic seizures, always ask explicitly about morning myoclonus and whether seizures occur on waking — these are the clues to JME. The single most dangerous error is treating JME with carbamazepine (a "focal" agent), which worsens myoclonus and absences. JME is lifelong — most patients require lifelong AED therapy. The SANAD II trial confirmed valproate is the most effective agent but is contraindicated in pregnancy [5].
Differential diagnosis of transient loss of consciousness
Misdiagnosis of epilepsy is common — up to 20 to 30 per cent of patients referred to "drug-resistant epilepsy" clinics have an alternative diagnosis. The witness account is the discriminator. [1]
| Mimic | Discriminating clinical features | Investigations |
|---|---|---|
| Vasovagal syncope | Prodrome (nausea, sweating, tunnel vision, warmth), brief loss of tone (collapse), brief (less than 30 s), rapid recovery, pallor. Brief tonic stiffening or a few myoclonic jerks may occur (convulsive syncope) — do NOT confuse with epilepsy | ECG, tilt table, orthostatic BP |
| Cardiogenic syncope | No or brief prodrome, occurs during exertion (aortic stenosis, HCM, arrhythmia) or sitting, family history of sudden death, palpitations | ECG (long QT, Brugada, WPW), echocardiogram, Holter, exercise test |
| Psychogenic non-epileptic seizures (PNES / dissociative attacks) | Eyes closed, motor activity out of phase and variable, pelvic thrusting, side-to-side head movement, gradual onset, prolonged, no postictal confusion, recall of the event, normal prolactin and lactate | Video-EEG monitoring (gold standard); strongly consider in "drug-resistant" epilepsy |
| Convulsive syncope | Brief cerebral hypoperfusion → myoclonic jerks; recovery quick; pallor dominates | ECG, tilt table |
| Migraine with brainstem aura (basilar) | Gradual progression of brainstem symptoms over 5-60 min, positive visual phenomena, throbbing headache | Clinical; MRI to exclude posterior circulation TIA |
| Transient ischaemic attack | Negative symptoms (loss of function), no convulsion, no postictal confusion, sudden onset; "limb shaking TIA" is a carotid stenosis warning | Carotid imaging, MRI |
| Sleep disorders (narcolepsy, parasomnias) | Confusional arousals, REM behaviour disorder, narcolepsy with cataplexy; occur from sleep | Polysomnography, MSLT |
| Metabolic (hypoglycaemia, hyponatraemia, hypocalcaemia) | Confusional prodrome, autonomic features, precipitating cause (insulin, diuretics, parathyroid disorder) | Glucose, U&E, calcium, magnesium |
DWE trap: A "first seizure" with pallor, sweating, brief stiffening, and rapid recovery — especially on standing, after a painful stimulus, or in a hot environment — is syncope with convulsive features, not epilepsy. Treating such a patient with an AED is the classic error. The keys are the prodrome (syncopal: nausea, warmth, visual fade; epileptic: aura, deja vu, autonomic), the recovery (syncopal: rapid within a minute; epileptic: prolonged postictal confusion and sleepiness), and the posture at onset (syncope occurs standing or sitting; seizures occur in any posture). [1]
Investigations
Acute — first seizure presentation
| Investigation | Purpose | Comment |
|---|---|---|
| Blood glucose | Exclude hypoglycaemia | Bedside finger-prick immediately — hypoglycaemia causes seizures and brain injury |
| U&E, calcium, magnesium, LFTs | Identify metabolic precipitant | Hyponatraemia, hypocalcaemia, uraemia, hepatic encephalopathy |
| Toxicology screen (where indicated) | Drug withdrawal (alcohol, benzodiazepines), intoxication (amphetamines, cocaine) | Alcohol withdrawal is a common adult cause |
| ECG (12-lead) | Exclude cardiac syncope — long QT, Brugada, arrhythmogenic RV cardiomyopathy | Mandatory in every first-seizure workup; cardiac syncope misdiagnosed as epilepsy is a fatal error |
| CT brain (non-contrast, emergency) | Exclude acute intracranial haemorrhage, mass effect, large stroke | For emergency presentations, especially focal signs, head trauma, immunocompromise, fever, or age over 40 |
| MRI brain (epilepsy protocol, elective) | Identify structural cause — hippocampal sclerosis, focal cortical dysplasia, tumour, vascular malformation | Preferable to CT; performed after the acute episode |
| Lumbar puncture | If fever, meningism, immunocompromise, or autoimmune encephalitis suspected | Cell count, protein, glucose, microbial PCR; consider autoimmune antibody panel and voltage-gated potassium channel complex |
| Prolactin (paired ictal and baseline) | Historical — modestly elevated after generalised tonic-clonic seizures (60 per cent) | Now considered low-yield; video-EEG is more discriminating. Do not rely on prolactin alone |
Routine interictal EEG
A single routine 20- to 30-minute interictal EEG has a sensitivity of only 30 to 50 per cent for epilepsy. Interictal epileptiform discharges (IEDs) are found in about 50 per cent of patients with epilepsy on the first EEG, rising to 80 to 90 per cent with repeated recordings (including sleep-deprived EEG). Critically, about 0.5 to 4 per cent of healthy adults have IEDs on a single EEG — a positive EEG does not prove epilepsy; it must be interpreted in clinical context. [1]
Specific EEG signatures and what they mean: [1]
- Focal spikes/sharp waves — supports focal epilepsy; localises the irritative zone. Anterior temporal spikes point to temporal lobe epilepsy.
- Generalised 3 Hz spike-and-wave — typical absence epilepsy (childhood absence).
- Generalised 4 to 6 Hz irregular polyspike-and-wave — juvenile myoclonic epilepsy.
- Slow (1.5 to 2.5 Hz) generalised spike-and-wave — Lennox-Gastaut syndrome.
- Hypsarrhythmia — chaotic high-voltage slow waves with multifocal spikes — West syndrome.
- Periodic lateralised epileptiform discharges (PLEDs) — focal structural lesion (stroke, herpes encephalitis, tumour); often reflects acute or subacute severe cortical injury.
- Electroclinical response to hyperventilation — provokes absence seizures in childhood absence epilepsy; a bedside test. [1]
Video-EEG monitoring
Indicated when the diagnosis remains uncertain, when seizure classification drives surgical decisions, or to distinguish psychogenic non-epileptic seizures. Inpatient monitoring (typically 5 to 7 days) captures the habitual events with concomitant EEG, allowing correlation of behaviour and electrical activity. It is the gold standard for diagnosis. [1]
MRI brain — epilepsy protocol
A standard brain MRI can miss hippocampal sclerosis and focal cortical dysplasia. The dedicated epilepsy protocol at 3 Tesla includes:
- Coronal T2 and FLAIR thin slices perpendicular to the long axis of the hippocampus
- Three-dimensional T1 volumetric acquisition for hippocampal volumetry
- Susceptibility-weighted imaging (SWI) for vascular malformations and haemosiderin
- Optional functional MRI and diffusion tensor imaging for surgical planning [1]
Key findings to recognise:
- Hippocampal sclerosis — hippocampal atrophy with T2/FLAIR hyperintensity, loss of internal architecture
- Focal cortical dysplasia — cortical thickening with blurring of the grey-white junction, often with transmantle sign
- Long-term epilepsy-associated tumours — ganglioglioma, dysembryoplastic neuroepithelial tumour (DNET), low-grade glioma — typically slow-growing, cystic, often temporal
- Cavernous malformation (cavernoma) — "popcorn" lesion with haemosiderin ring on T2* [1]
Genetic testing
Genetic testing is now indicated in:
- Early-onset epileptic encephalopathies (Dravet — SCN1A; West syndrome if atypical)
- Patients with intellectual disability and dysmorphism (chromosomal microarray)
- Family history of epilepsy with a clear Mendelian pattern
- Drug-resistant focal epilepsy without imaging lesion — occasionally a genetic cause is identified [1]
The genetic workup is increasingly a gene panel (next-generation sequencing of 100 to 500 epilepsy-related genes), with chromosomal microarray and whole-exome sequencing reserved for complex presentations. A genetic diagnosis directly changes management — for example, Dravet syndrome (SCN1A) demands avoidance of sodium-channel blockers and use of stiripentol, fenfluramine, and cannabidiol. [1]
Management — first seizure and the decision to treat
When does a single seizure become epilepsy?
A single unprovoked seizure is not epilepsy. The 2014 ILAE practical definition permits a diagnosis of epilepsy after one unprovoked seizure if the recurrence risk over the next 10 years is at least 60 per cent (e.g. abnormal EEG with IEDs, structural lesion on imaging, or a remote neurological insult such as stroke or traumatic brain injury). For most first-seizure patients without these risk factors, the 2-year recurrence risk is 30 to 40 per cent. [1]
The MESS trial — immediate versus deferred treatment
The MESS trial randomised 1443 patients with one or more unprovoked seizures to immediate or deferred AED treatment [6]. Findings:
- Immediate treatment reduced early seizure recurrence (2-year risk reduced from 39 to 32 per cent).
- No difference in long-term remission at 5 years — early treatment did not alter the natural history of epilepsy.
- Conclusion: the decision to treat is a shared decision weighing seizure recurrence risk, lifestyle factors (driving, occupation), and drug side effects against the modest short-term benefit. [1]
In practice:
- Treat after the first seizure if there is a structural lesion, EEG with IEDs, a remote neurological insult, a nocturnal seizure, or a clear family history.
- Defer treatment for a single, clearly provoked seizure (correctable metabolic cause, drug withdrawal) — treat the cause, not with an AED.
- Treat after a second unprovoked seizure — recurrence risk after two seizures is over 70 per cent. [1]
Antiepileptic drug selection — syndrome-based

The guiding principle: choose the drug by syndrome, not by seizure alone. The wrong drug can worsen the underlying epilepsy (carbamazepine in JME; sodium-channel blockers in Dravet). The SANAD and SANAD II trials underpin modern first-line choices [4][5].
Focal epilepsy — first-line options
| Drug | Typical adult dose | Mechanism | Key adverse effects / cautions |
|---|---|---|---|
| Lamotrigine | Start 25 mg daily, titrate to 200-400 mg/day over 6 weeks | Sodium-channel blockade (slow inactivation) | Maculopapular rash, Stevens-Johnson syndrome — slow titration is mandatory; risk higher with valproate co-prescription (valproate inhibits lamotrigine metabolism). Well tolerated, broad spectrum |
| Lacosamide | 100 mg BD, titrate to 200-400 mg/day | Sodium-channel blockade (slow inactivation, distinct site) | PR interval prolongation — avoid in second-degree AV block or with other PR-prolonging drugs; dizziness, nausea. Useful add-on in refractory focal |
| Carbamazepine | 200 mg BD, titrate to 600-1800 mg/day | Sodium-channel blockade (fast inactivation) | Hyponatraemia (SIADH — check sodium at 1, 3, 6 months), rash (HLA-B*1502 in Han Chinese/South-East Asians — screen before use), enzyme induction (reduces OCP efficacy), leukopenia, agranulocytosis, aplastic anaemia, ataxia, diplopia. Historically first-line focal but SANAD preferred lamotrigine for tolerability |
| Oxcarbazepine | 300 mg BD, titrate to 1200-2400 mg/day | Sodium-channel blockade (eslicarbazepine metabolite) | Hyponatraemia (more frequent than carbamazepine), rash, fewer interactions than carbamazepine. Less enzyme-inducing |
| Eslicarbazepine acetate | 800 mg daily, titrate to 800-1600 mg/day | Sodium-channel blockade | Lower hyponatraemia risk than oxcarbazepine; once-daily dosing |
Generalised epilepsy — first-line options
| Drug | Adult dose | Indication | Key cautions |
|---|---|---|---|
| Sodium valproate | 500 mg BD, titrate to 20-30 mg/kg/day | Most effective for idiopathic generalised epilepsy (JME, generalised tonic-clonic). Confirmed superior to levetiracetam by SANAD II [5] | TERATOGEN — neural tube defects, cardiac and facial clefts; neurodevelopmental harm (IQ reduction 6-9 points, autism risk). Prohibited in women of childbearing potential without a Pregnancy Prevention Programme. Weight gain, hair thinning, tremor, polycystic ovarian syndrome, thrombocytopenia, hepatotoxicity (rare fatal), pancreatitis |
| Levetiracetam | 500 mg BD | Reasonable first-line alternative in women of childbearing age with JME | Reasonable safety in pregnancy (large registry data). Less effective than valproate for generalised seizures per SANAD II |
| Topiramate | 25 mg daily, titrate to 200-400 mg/day | Broad spectrum; useful for generalised and focal | Cognitive slowing ("dopamax"), word-finding difficulty, weight loss, paraesthesia, nephrolithiasis, acute angle-closure glaucoma (rare), teratogen (oral clefts). Avoid in pregnancy |
| Lamotrigine | 25 mg daily, titrate slowly | Reasonable in women of childbearing age; broad spectrum | Best safety profile in pregnancy of the older agents; less effective for myoclonus (may worsen). Slow titration to avoid rash |
Absence epilepsy — specific agents
- Ethosuximide — first-line for childhood absence epilepsy without generalised tonic-clonic seizures. Acts on T-type calcium channels of thalamic neurons. GI upset, nausea, hiccups. Does not protect against tonic-clonic seizures — add valproate or lamotrigine if these coexist.
- Valproate — first-line if absence coexists with generalised tonic-clonic seizures.
- Lamotrigine — alternative, particularly in women of childbearing age, but less effective.
- Avoid carbamazepine and phenytoin — these can worsen absences. [1]
DWE high-yield — women of childbearing age with IGE: The SANAD II trial confirmed valproate is superior but the regulatory prohibition in pregnancy is now absolute in Australia (TGA) and Europe (EMA). The pragmatic alternative is levetiracetam (most evidence for pregnancy safety) or lamotrigine (long safety track record), accepting that seizure control may be less complete. Counsel explicitly: incomplete seizure control in pregnancy carries its own risks (trauma, hypoxia, SUDEP), and the trade-off must be made jointly. [1]
Drug interactions and enzyme induction
A small number of AEDs are hepatic enzyme inducers and they complicate management substantially: [1]
- Strong inducers: carbamazepine, phenytoin, phenobarbitone, primidone
- Moderate inducer: oxcarbazepine, eslicarbazepine, rufinamide, perampanel (less predictable)
- Inhibitor: valproate (inhibits lamotrigine, phenobarbitone metabolism) [1]
Consequences:
- Combined oral contraceptive pill failure — enzyme inducers accelerate oestrogen and progestogen metabolism; use a non-hormonal or higher-dose (50 microgram ethinyloestradiol) method, or switch to a non-enzyme-inducing AED. The interaction persists for 4 weeks after stopping the inducer.
- Warfarin dose changes — enzyme inducers lower the INR; stopping them raises it.
- Reduced efficacy of doxycycline, atorvastatin, many antiretrovirals, corticosteroids, and some chemotherapeutics.
- Phenytoin toxicity — zero-order kinetics; a small dose increase can produce toxicity. Check free phenytoin levels in hypoalbuminaemia. [1]
AED adverse effects — the high-yield list
| AED | Adverse effect | When to check |
|---|---|---|
| Carbamazepine/oxcarbazepine | Hyponatraemia (SIADH) | Sodium at 1, 3, 6 months, then 6-monthly |
| Carbamazepine, phenytoin | Osteoporosis, osteomalacia (vitamin D acceleration) | Vitamin D, bone densitometry at baseline and 2-5 years |
| Phenytoin | Gum hypertrophy, hirsutism, coarse facies, cerebellar atrophy, megaloblastic anaemia (folate), peripheral neuropathy | FBC, folate annually |
| Valproate | Thrombocytopenia, hepatotoxicity, pancreatitis, weight gain, PCOS, hyperammonaemia | LFTs, FBC, ammonia if encephalopathic |
| Lamotrigine | Stevens-Johnson syndrome (especially first 8 weeks, higher with valproate co-prescription or rapid titration) | Counsel patient to stop and present if rash |
| Topiramate | Nephrolithiasis, acute angle-closure glaucoma, cognitive slowing, weight loss, metabolic acidosis | Renal function, bicarbonate; ophthalmology if visual symptoms |
| Levetiracetam | Behavioural disturbance, depression, suicidal ideation | Mental health screen at initiation |
| Perampanel | Serious psychiatric (aggression, psychosis, suicidality) | Avoid in psychiatric history |
Epilepsy surgery and neuromodulation
Drug-resistant epilepsy is defined as failure of two appropriately chosen and tolerated AEDs (monotherapy or combination) to achieve seizure freedom. This is the point at which surgical evaluation should be initiated — not after years of further drug trials. The average delay to surgical referral is 20 years in many countries, a profound failure of care. [1]
Temporal lobectomy — the evidence
The Wiebe randomised controlled trial (NEJM 2001) randomised 80 patients with temporal lobe epilepsy to surgery versus continued medical therapy [9]. At 1 year, 58 per cent of surgical patients were seizure-free compared with 8 per cent of medical patients. Surgery is the most effective intervention in all of neurology with a number-needed-to-treat for seizure freedom of approximately 2.
Surgical procedures
| Procedure | Indication | Expected outcome |
|---|---|---|
| Anterior temporal lobectomy with amygdalohippocampectomy | Drug-resistant mesial temporal lobe epilepsy with hippocampal sclerosis | 60-70 per cent seizure freedom at 2 years |
| Lesionectomy | Focal lesion (DNET, ganglioglioma, cavernoma, focal cortical dysplasia) — seizure focus concordant | 50-80 per cent seizure freedom depending on completeness of resection |
| Hemispherotomy | Catastrophic unilateral epilepsy (Rasmussen encephalitis, Sturge-Weber, large perinatal infarct) in children | 60-80 per cent seizure freedom |
| Corpus callosotomy (anterior or complete) | Drop attacks (atonic, tonic) in Lennox-Gastaut syndrome — palliative | Reduces drop attacks; not for seizure cure |
| Multiple subpial transection | Focal epilepsy in eloquent cortex (Landau-Kleffner syndrome) — rare | Palliative |
Neuromodulation — the three options
| Device | Indication | Mechanism | Outcome |
|---|---|---|---|
| Vagus nerve stimulation (VNS) | Drug-resistant focal or generalised epilepsy not suitable for resective surgery | Intermittent electrical stimulation of the left cervical vagus nerve; modulates brainstem and thalamocortical networks | 50 per cent seizure reduction in 50 per cent of patients; few seizure-free |
| Responsive neurostimulation (RNS) | Focal epilepsy with one or two identifiable seizure foci in eloquent cortex — implanted at the focus | Closed-loop detection of abnormal electrical activity with responsive stimulation at the focus | Median 75 per cent seizure reduction at 9 years; 15 per cent seizure-free |
| Deep brain stimulation (DBS — anterior nucleus of thalamus) | Drug-resistant focal epilepsy with bitemporal or multifocal onset | Open-loop stimulation of anterior thalamic nucleus | Median 69 per cent seizure reduction at 5 years (SANTE trial) |
DCE long-case approach: When presenting a patient with long-standing focal epilepsy, the integrated surgical pathway is: (1) confirm drug-resistance (two failed AEDs); (2) phase 1 workup — video-EEG with ictal recording, epilepsy-protocol MRI, neuropsychology, FDG-PET, SPECT; (3) concordance — does the electrical, structural, and metabolic data all point to the same epileptogenic zone? (4) Phase 2 — invasive EEG with subdural grids or stereo-EEG if non-invasive data are discordant or the focus is near eloquent cortex; (5) multidisciplinary epilepsy surgery conference for decision. [1]
Status epilepticus

Definition and time thresholds
The ILAE 2015 Task Force defined status epilepticus (SE) as a condition resulting from failure of seizure termination mechanisms, with two operational time points [3]:
- t1 (treatment threshold): 5 minutes of continuous convulsive seizure — this is when treatment should begin
- t2 (long-term consequence threshold): 30 minutes — beyond this, neuronal injury, neuronal death, and alteration of neuronal networks are expected
A patient in convulsive SE at 5 minutes must be treated. Do not wait. [1]
Stepwise treatment protocol
| Time | Stage | Treatment | Dose and route |
|---|---|---|---|
| 0-5 min | Stabilisation | Airway, oxygen, IV access, glucose check, bloods (U&E, LFTs, FBC, glucose, calcium, magnesium, toxicology, AED levels), temperature | Finger-prick glucose; if hypoglycaemic, 50 mL 50 per cent dextrose IV (or 25 mL 10 per cent in children) with IV thiamine 100 mg if alcoholism suspected |
| 5-20 min | First-line: benzodiazepine | IV lorazepam (preferred — longer anticonvulsant duration than diazepam) | Lorazepam 0.1 mg/kg (4 mg typical adult) IV, repeated once at 5-10 min if still seizing |
| OR IM midazolam if no IV access | Midazolam 10 mg IM (5 mg if under 13 years or under 40 kg); shown non-inferior and superior to IV lorazepam in prehospital RAMPART trial [8] | ||
| OR (if IV available) IV diazepam or IV clonazepam | Diazepam 0.15 mg/kg (10 mg adult) IV — risk of repeat due to rapid redistribution | ||
| 20-40 min | Second-line: non-benzodiazepine AED | Choose ONE — levetiracetam, fosphenytoin, or valproate (all equivalent efficacy per ESETT) | Levetiracetam 60 mg/kg IV (max 4500 mg) over 5 min; OR fosphenytoin 20 mg PE/kg IV at 150 mg PE/min; OR valproate 40 mg/kg IV (max 3000 mg) over 10 min [7] |
| 40+ min | Refractory: anaesthetic infusion | Intubate and ventilate; continuous EEG monitoring; ICU admission | Propofol infusion 1-2 mg/kg bolus then 30-200 microgram/kg/min; OR midazolam infusion 0.2 mg/kg bolus then 0.05-2 mg/kg/hr; OR thiopentone — bolus 2-3 mg/kg then infusion 0.3-5 mg/kg/hr |
| Adjuncts in super-refractory SE | Ketamine infusion (NMDA antagonism); ketogenic diet; immunotherapy if autoimmune aetiology; consider surgical resection or corpus callosotomy if focal |
Critical practical points
- Do not under-dose the benzodiazepine. The commonest error is inadequate first-line dosing. Give a full weight-based dose, repeat once if still seizing, then move to second-line.
- The RAMPART trial changed prehospital practice. IM midazolam 10 mg was superior to IV lorazepam in the prehospital setting because IV access was slow to establish [8]. In a convulsing patient without IV access, give IM midazolam.
- The ESETT trial established equivalence of second-line agents. Levetiracetam, fosphenytoin, and valproate all terminated benzodiazepine-refractory SE in about half of patients [7]. Choose by patient factors: fosphenytoin in pregnancy or hepatic impairment (avoid valproate), levetiracetam in cardiac conduction disease or with arrhythmia risk (avoid fosphenytoin), valproate in primary generalised epilepsy.
- Fosphenytoin is preferred over phenytoin — water-soluble, can be given IM, lower infusion-site reaction and cardiac arrhythmia risk. Dose expressed as phenytoin equivalents (PE).
- Identify and treat the cause concurrently. Hypoglycaemia (give dextrose), hyponatraemia (3 per cent saline if severe with seizures), alcohol withdrawal (thiamine before dextrose), hypoxia, sepsis, CNS infection (empirical antibiotics and aciclovir), drug non-adherence, intracranial haemorrhage.
- Non-convulsive status epilepticus — consider in any patient with unexplained coma or fluctuating altered consciousness, especially after a convulsive SE has been "treated". Requires continuous EEG to diagnose. Treated with IV benzodiazepine or anaesthetic infusion as above, but more permissively (less aggressive airway intervention).
DWE high-yield MCQ pattern: A patient presents convulsing; no IV access. The correct answer is IM midazolam 10 mg (RAMPART). A patient has received two doses of IV lorazepam and continues to seize at 25 minutes; the correct answer is a second-line agent — IV levetiracetam 60 mg/kg (or fosphenytoin 20 mg PE/kg, or valproate 40 mg/kg), not a third benzodiazepine. [1]
Women with epilepsy — the complete preconception and pregnancy pathway
This is one of the highest-yield DWE/DCE topics and the area where the registrar's knowledge most directly affects patient outcomes. [1]
Preconception counselling
Begin the conversation before the woman wants to conceive — ideally at diagnosis. [1]
- Aim for seizure freedom before conception — uncontrolled generalised tonic-clonic seizures in pregnancy carry risks of maternal trauma, hypoxia, and SUDEP, and fetal risks.
- Review the AED regimen:
- Valproate is prohibited in women of childbearing potential unless a formal Pregnancy Prevention Programme is met (highly effective contraception, annual specialist review, signed risk acknowledgement form, and a documented clinical reason no alternative will work). The TGA in Australia and the EMA in Europe have issued these restrictions because of the 30 to 40 per cent major congenital malformation rate and the consistent neurodevelopmental harm (IQ reduction, autism spectrum) [5]. Switch to levetiracetam or lamotrigine well before conception.
- Lamotrigine is the AED with the most reassuring pregnancy safety data. Lamotrigine clearance increases substantially in pregnancy (oestrogen-induced glucuronidation) — lamotrigine levels fall by 50-70 per cent in the second and third trimesters and require dose escalation. Check lamotrigine trough levels trimesterly.
- Levetiracetam has substantial pregnancy safety registry data and is a reasonable alternative.
- Carbamazepine has the lowest teratogenicity of the older agents (about 2-3 per cent major malformation rate) but is restricted to focal epilepsy.
- Topiramate and phenytoin — avoid in pregnancy if possible.
- Folic acid 5 mg daily — start before conception and continue through the first trimester to reduce neural tube defect risk. This is recommended for all women with epilepsy on AEDs (especially valproate, carbamazepine, phenytoin). [1]4. Enzyme-inducing AEDs and contraception: carbamazepine, phenytoin, phenobarbitone, primidone, topiramate (high dose) reduce combined oral contraceptive pill efficacy. Use a copper IUD, levonorgestrel IUD, or depo-medroxyprogesterone. The progesterone-only pill is also unreliable with enzyme inducers.
- Genetic counselling if a hereditary epilepsy syndrome is present.
During pregnancy
- Specialist joint care — neurology and obstetric medicine, with the obstetric anaesthetist informed.
- Monitor AED levels trimesterly for lamotrigine, levetiracetam, and oxcarbazepine — doses often need to rise in pregnancy and fall postpartum.
- Avoid breakthrough seizures — the primary risk to mother and fetus is uncontrolled convulsive seizure.
- Vitamin K — enzyme-inducing AEDs (carbamazepine, phenytoin, phenobarbitone, primidone) induce vitamin K metabolism and may cause neonatal haemorrhagic disease of the newborn; the mother should receive oral vitamin K 10 mg daily in the last month of pregnancy, and the newborn receives standard IM vitamin K.
- Mode of delivery — not usually altered by epilepsy alone; obstetric indications only.
- Pain relief in labour — epidural is safe; pethidine metabolite norpethidine is proconvulsant — avoid. [1]
Breastfeeding
Breastfeeding is encouraged for women with epilepsy on most AEDs. The developmental and immunological benefits of breastfeeding outweigh the small additional drug exposure (which is less than the third-trimester exposure). Specific considerations:
- Valproate, levetiracetam, lamotrigine, carbamazepine — compatible with breastfeeding; infant sedation is rare.
- Phenobarbitone, primidone — significant infant sedation; consider alternatives.
- Counsel the mother to monitor the infant for excessive sedation, poor feeding, or poor weight gain. [1]
DCE long-case trap: When asked about a young woman with JME on valproate planning pregnancy, the structured viva answer must hit all of: (1) valproate prohibition — switch to levetiracetam or lamotrigine well before conception; (2) folic acid 5 mg daily; (3) monitor drug levels and anticipate dose rise in pregnancy for lamotrigine; (4) counsel that incomplete seizure control in pregnancy is also dangerous — balance; (5) lifelong JME — recurrence risk is high if drugs are withdrawn; (6) breastfeeding is safe with most AEDs. [1]
Driving regulations — ANZ, UK, US
Driving restrictions are a major patient concern and a frequent DCE discussion point. The principle across jurisdictions: a seizure-free period must elapse before driving is permitted, and the clinician has a role in counselling and (in some jurisdictions) reporting. [1]
| Jurisdiction | Private vehicle licence | Commercial vehicle licence | Clinician obligation |
|---|---|---|---|
| Australia (Austroads 2022) | 6 months seizure-free for a first unprovoked seizure or new epilepsy. 3 months if a clear provoking factor was treated (e.g. metabolic). 12 months after withdrawal of AEDs (for recurrence risk) | 10 years seizure-free (and off AEDs for the preceding 2 years) for a heavy vehicle licence | Counsel patient on the legal obligation to notify the licensing authority; in most states, the patient self-notifies. Mandatory clinician reporting applies in some jurisdictions (e.g. South Australia, Northern Territory) — check local law |
| New Zealand (NZTA) | 12 months seizure-free for new epilepsy; 3 months for a single provoked seizure | 10 years seizure-free | Counsel patient; mandatory notification in some clinical contexts |
| UK (DVLA) | 6 months seizure-free for new epilepsy (or 12 months if there is a continuing risk); 6 months after AED withdrawal | 10 years seizure-free off AEDs | Patient self-notifies DVLA; clinician documents advice given |
| USA (state-specific) | Typically 3-12 months seizure-free, varies by state | Federal (FMCSA): seizure-free and off AEDs | State-dependent mandatory reporting — six states (CA, DE, NV, NJ, OR, PA) require clinician reporting |
Always check the current regulation for your jurisdiction and document the conversation. The registrar's responsibility is to (a) inform the patient of the restriction and the legal obligation, (b) document this advice, (c) advise on notification to the licensing authority. [1]
SUDEP — sudden unexpected death in epilepsy
SUDEP is defined as sudden, unexpected, witnessed or unwitnessed, non-traumatic and non-drowning death in a patient with epilepsy, with or without evidence of a seizure, and excluding documented status epilepticus. The mechanism is thought to be postictal central apnoea and cardiac arrhythmia after a generalised tonic-clonic seizure. [1]
Risk factors (highest-yield):
- Generalised tonic-clonic seizures (the dominant risk factor)
- Nocturnal seizures (unwitnessed, longer delay to resuscitation)
- Frequent seizures (recurrence risk)
- Long-standing epilepsy, structural cause, intellectual disability
- Non-adherence to AEDs [1]
Counselling: discuss SUDEP with every patient with epilepsy and especially those at higher risk (generalised tonic-clonic seizures, nocturnal seizures, drug-resistant). Use plain language — "in rare cases, epilepsy can cause sudden death, and the best way to reduce the risk is seizure control, taking medication reliably, and having someone nearby at night". Night-time audio or movement monitors are available; their evidence base for SUDEP prevention is limited but patients may choose them. [1]
Withdrawal of AEDs
After a sustained seizure-free period (typically 2 years, longer for higher-risk patients), withdrawal of AEDs may be considered. The decision is shared: [1]
- Favourable factors: idiopathic generalised epilepsy, normal EEG and MRI, short epilepsy duration, single AED, childhood-onset epilepsy.
- Unfavourable factors: structural cause (especially hippocampal sclerosis), developmental delay, abnormal EEG with IEDs, multiple seizure types, JME (usually lifelong).
- Method: slow taper over 3 to 6 months; abrupt withdrawal provokes status epilepticus.
- Driving: must not drive during withdrawal and for 6 months after (Australia). [1]
DCE long-case approach
Opening statement (SASPOP)
"Ms Davies is a 28-year-old woman who works as a primary school teacher and presents to the epilepsy clinic with a 12-year history of juvenile myoclonic epilepsy. She currently takes sodium valproate 1000 mg twice daily and is seizure-free for 4 years. She is planning her first pregnancy and is concerned about her medication. She has no other significant medical history. Her mother had 'fainting turns' in her youth. [1]
Her main problems are:
- Juvenile myoclonic epilepsy, well-controlled on valproate — but valproate is contraindicated in pregnancy
- Preconception counselling required — drug switch, folic acid, seizure-control balance
- Driving and occupational considerations as a teacher
- Family history of probable epilepsy — possible genetic counselling
- Long-term prognosis and the question of lifelong therapy" [1]
Integrated management plan
- Switch AED preconception. Gradually transition from valproate to levetiracetam (or lamotrigine) over 4-8 weeks, ideally achieving seizure freedom on the new agent for at least 6 months before conception. Counsel explicitly that the new regimen may be less effective — accept a slightly higher seizure risk in exchange for fetal safety.
- Folic acid 5 mg daily. Start now, continue through the first trimester. [1]3. Contraception. Continue effective contraception (levonorgestrel IUD or copper IUD preferred; OCP unreliable if she is on an enzyme-inducing agent — levetiracetam and lamotrigine do not reduce OCP efficacy, but lamotrigine levels fall with OCP use).
- Lamotrigine levels in pregnancy. If lamotrigine is chosen, anticipate the 50-70 per cent fall in levels in second and third trimesters — check trough levels trimesterly and titrate.
- Driving. Must remain seizure-free. Switch carries a small risk of breakthrough seizure — she should not drive during the switch or for the regulatory period after any recurrence.
- Family history. Consider genetic counselling — JME is polygenic with a heritability of about 50 per cent.
- Long-term prognosis. JME is lifelong in most patients; AEDs are usually required for life. Counsel her accordingly.
- Breastfeeding. Plan to breastfeed — safe on levetiracetam or lamotrigine. [1]
DCE examiner probing questions you must anticipate:
- "Why is valproate so dangerous in pregnancy?" → 30-40 per cent major congenital malformation rate (neural tube, cardiac, cleft); consistent neurodevelopmental harm including IQ reduction and autism.
- "How would you counsel her on the risk-benefit of switching?" → Seizure control is the dominant maternal and fetal risk; accept some increase in seizure risk to avoid teratogenicity.
- "What if she has a generalised tonic-clonic seizure during pregnancy?" → Do not abandon the new regimen — optimise the dose, check levels, address adherence and provoking factors (sleep deprivation). Intravenous benzodiazepine if prolonged (status).
- "What about SUDEP?" → Risk is highest in uncontrolled generalised tonic-clonic seizures; the switch itself does not raise SUDEP risk if seizures remain controlled. Counsel honestly. [1]
DCE short-case approach: neurological examination in epilepsy
Instruction: "Examine this patient's neurological system. They have a long-standing seizure disorder." [1]
Systematic routine
- General inspection and observations: Stigmata of a neurocutaneous syndrome (ash-leaf macules, adenoma sebaceum, shagreen patches — tuberous sclerosis; port-wine stain in V1 distribution — Sturge-Weber; café-au-lait macules and neurofibromas — NF1). Signs of drug toxicity (gum hypertrophy — phenytoin; ataxia and nystagmus — phenytoin, carbamazepine; hirsutism — phenytoin; tremor — valproate). Surgical scar (temporal craniotomy). Cognitive state — is there intellectual disability suggesting a syndromic cause?
- Cranial nerves: Visual fields (homonymous hemianopia may indicate occipital lesion; quadrantanopia suggests temporal or parietal lesion). Fundoscopy (papilloedema in raised intracranial pressure). Facial asymmetry (postictal Todd's facial weakness resolves; permanent hemiparesis suggests structural lesion).
- Motor: Tone, power, reflexes, plantar response. Focal weakness or hyperreflexia lateralises the lesion. Cerebellar signs (ataxia, dysmetria, intention tremor) — chronic phenytoin toxicity causes cerebellar degeneration.
- Sensory: Cortical sensory testing (graphaesthesia, stereognosis, two-point discrimination) may reveal parietal involvement.
- Coordination and gait: Cerebellar ataxia from AED toxicity; hemiparetic gait from structural lesion.
- Speech and cognitive: Naming difficulty, fluency — dominant temporal lobe. Memory testing — mesial temporal lobe epilepsy often produces material-specific memory deficit (verbal in left, visuospatial in right). [1]
Presentation template
"I examined Ms Davies' neurological system. She is a 28-year-old woman with no cutaneous stigmata of a neurocutaneous syndrome. There is no gum hypertrophy, tremor, or ataxia to suggest drug toxicity. There is no surgical scar. [1]
Cranial nerves are intact — visual fields full to confrontation, pupils equal and reactive, fundi normal, facial sensation and power symmetrical, tongue and palate symmetrical. [1]
Motor examination reveals normal tone, power 5/5 in all four limbs, and symmetrical reflexes with flexor plantar responses. Sensation is intact to all modalities. Coordination is intact. Gait is normal. [1]
Her speech is fluent with normal comprehension and naming. Orientation and short-term recall are intact. [1]
In summary, the neurological examination is normal. The absence of focal signs is consistent with idiopathic generalised epilepsy, in which there is no structural lateralised lesion. The absence of drug toxicity signs indicates that the current valproate dose is well tolerated." [1]
DCE short-case trap: Examiners want you to specifically look for stigmata of drug toxicity and neurocutaneous syndromes in the epilepsy short case. Missing tuberous sclerosis or phenytoin toxicity is a viva failure. [1]
Key DWE MCQ patterns
- Drug choice by syndrome: A 19-year-old woman with morning myoclonic jerks and generalised tonic-clonic seizures on waking — the diagnosis is juvenile myoclonic epilepsy and the first-line agent is valproate (or levetiracetam/lamotrigine in women of childbearing age). Carbamazepine or oxcarbazepine would worsen myoclonus.
- Status epilepticus first-line: A patient convulsing with no IV access — give IM midazolam 10 mg (RAMPART). A patient still convulsing after IV lorazepam 4 mg — repeat lorazepam 4 mg then proceed to second-line. [1]3. Status epilepticus second-line: After two benzodiazepine doses — choose IV levetiracetam 60 mg/kg, fosphenytoin 20 mg PE/kg, or valproate 40 mg/kg (ESETT).
- First seizure treatment decision: A first unprovoked seizure with normal EEG and MRI — defer treatment unless recurrence risk factors present; discuss shared decision-making. A first seizure with a structural lesion on MRI — start an AED.
- Women of childbearing age on valproate: Always the wrong drug — prohibited without a Pregnancy Prevention Programme.
- Hypoglycaemia as a seizure mimic: Always check glucose at the bedside before imaging or treatment of suspected acute seizure.
- Lamotrigine in pregnancy: Anticipate the 50-70 per cent fall in levels in the second and third trimesters — check trough levels trimesterly.
- Drug-resistant focal epilepsy: After two failed AEDs — refer for surgical evaluation, do not continue to trial drugs indefinitely.
- Childhood absence epilepsy: Ethosuximide is first-line if absence seizures alone; add valproate or lamotrigine if tonic-clonic seizures coexist.
- Dravet syndrome (SCN1A): Avoid sodium-channel blockers (lamotrigine, carbamazepine, phenytoin) — they worsen seizures. [1]
References
[1] Fisher RS, Cross JH, French JA, et al. (ILAE 2017) — Operational classification of seizure types by the International League Against Epilepsy. Establishes the three-level framework: focal (aware, impaired awareness, focal to bilateral tonic-clonic, motor, non-motor), generalised (motor: tonic-clonic, myoclonic, atonic, tonic, clonic; non-motor: absence), and unknown onset. [2] Scheffer IE, Berkovic S, Capovilla G, et al. (ILAE 2017) — Classification of the epilepsies. Establishes the four levels (seizure type, epilepsy type, syndrome, aetiology) and the six aetiology categories (structural, genetic, infectious, metabolic, immune, unknown). [3] Trinka E, Cock H, Hesdorffer D, et al. (ILAE 2015) — Definition and classification of status epilepticus. Introduces the operational time points t1 (5 min, treatment threshold) and t2 (30 min, consequence threshold) for convulsive SE. [4] Marson AG, Al-Kharusi AM, Alwaidh M, et al. (SANAD 2007) — Unblinded RCT of carbamazepine, gabapentin, lamotrigine, oxcarbazepine, topiramate for partial epilepsy. Lamotrigine superior to carbamazepine for time to treatment failure; equivalent for seizure control. Supports lamotrigine as a first-line focal agent. [5] Marson AG, Appleton R, Baker GA, et al. (SANAD II 2021) — Open-label RCT of levetiracetam versus valproate for newly diagnosed generalised and unclassifiable epilepsy. Levetiracetam did not meet non-inferiority; valproate was superior. Reinforces valproate as the most effective generalised AED but the prohibition in women of childbearing potential remains. [6] Marson A, Jacoby A, Johnson A, et al. (MESS 2005) — RCT of immediate versus deferred AED treatment for early epilepsy and single seizures. Immediate treatment reduced early recurrence but did not alter long-term remission. Supports shared decision-making after a first seizure. [7] Kapur J, Elm J, Chamberlain JM, et al. (ESETT 2019) — Randomised comparative-effectiveness trial of levetiracetam, fosphenytoin, and valproate for benzodiazepine-refractory convulsive SE. All three were equivalent (about 45-47 per cent terminated SE at 60 min). [8] Silbergleit R, Durkalski V, Lowenstein D, et al. (RAMPART 2012) — Double-blind RCT of IM midazolam versus IV lorazepam for prehospital SE. IM midazolam was superior (73.4 per cent versus 63.4 per cent seizure-free on ED arrival) due to faster administration. [9] Wiebe S, Blume WT, Girvin JP, Eliasziw M (2001) — First RCT of surgery for temporal lobe epilepsy. 58 per cent seizure-free at 1 year with surgery versus 8 per cent with continued medical therapy. The most effective intervention in neurology. [10] Hancock EC, Osborne JP, Edwards SW (Cochrane 2013) — Treatment of infantile spasms (West syndrome). Hormonal therapy (ACTH or prednisolone) resolves spasms faster and in more infants than vigabatrin; vigabatrin is preferred when the cause is tuberous sclerosis.
NICE Guideline NG217 (Epilepsies in children, young people and adults, 2022); SIGN 143 (Diagnosis and management of epilepsy in adults); ILAE official classifications; TGA Sodium Valproate — not to be used in pregnancy; Epilepsy Foundation of Australia driving regulations; Austroads Assessing Fitness to Drive (2022). [1]
References
- [1]Fisher RS, Cross JH, French JA, et al. Operational classification of seizure types by the International League Against Epilepsy: Position Paper of the ILAE Commission for Classification and Terminology Epilepsia, 2017.PMID 28276060
- [2]Scheffer IE, Berkovic S, Capovilla G, et al. ILAE classification of the epilepsies: Position paper of the ILAE Commission for Classification and Terminology Epilepsia, 2017.PMID 28276062
- [3]Trinka E, Cock H, Hesdorffer D, et al. A definition and classification of status epilepticus--Report of the ILAE Task Force on Classification of Status Epilepticus Epilepsia, 2015.PMID 26336950
- [4]Marson AG, Al-Kharusi AM, Alwaidh M, et al. The SANAD study of effectiveness of carbamazepine, gabapentin, lamotrigine, oxcarbazepine, or topiramate for treatment of partial epilepsy: an unblinded randomised controlled trial Lancet, 2007.PMID 17382827
- [5]Marson AG, Appleton R, Baker GA, et al. The SANAD II study of the effectiveness and cost-effectiveness of valproate versus levetiracetam for newly diagnosed generalised and unclassifiable epilepsy: an open-label, non-inferiority, multicentre, phase 4, randomised controlled trial Lancet, 2021.PMID 33838758
- [6]Marson A, Jacoby A, Johnson A, Kim L, Gamble C, Chadwick D Immediate versus deferred antiepileptic drug treatment for early epilepsy and single seizures: a randomised controlled trial Lancet, 2005.PMID 15950714
- [7]Kapur J, Elm J, Chamberlain JM, et al. Randomized Trial of Three Anticonvulsant Medications for Status Epilepticus N Engl J Med, 2019.PMID 31774955
- [8]Silbergleit R, Durkalski V, Lowenstein D, et al. Intramuscular versus intravenous therapy for prehospital status epilepticus N Engl J Med, 2012.PMID 22335736
- [9]Wiebe S, Blume WT, Girvin JP, Eliasziw M A randomized, controlled trial of surgery for temporal-lobe epilepsy N Engl J Med, 2001.PMID 11484687
- [10]Hancock EC, Osborne JP, Edwards SW Treatment of infantile spasms Cochrane Database Syst Rev, 2013.PMID 23740534